
Nitro-, Cyano-, and Methylfuroxans, and Their Bis-Derivatives: From Green Primary to Melt-Cast Explosives

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
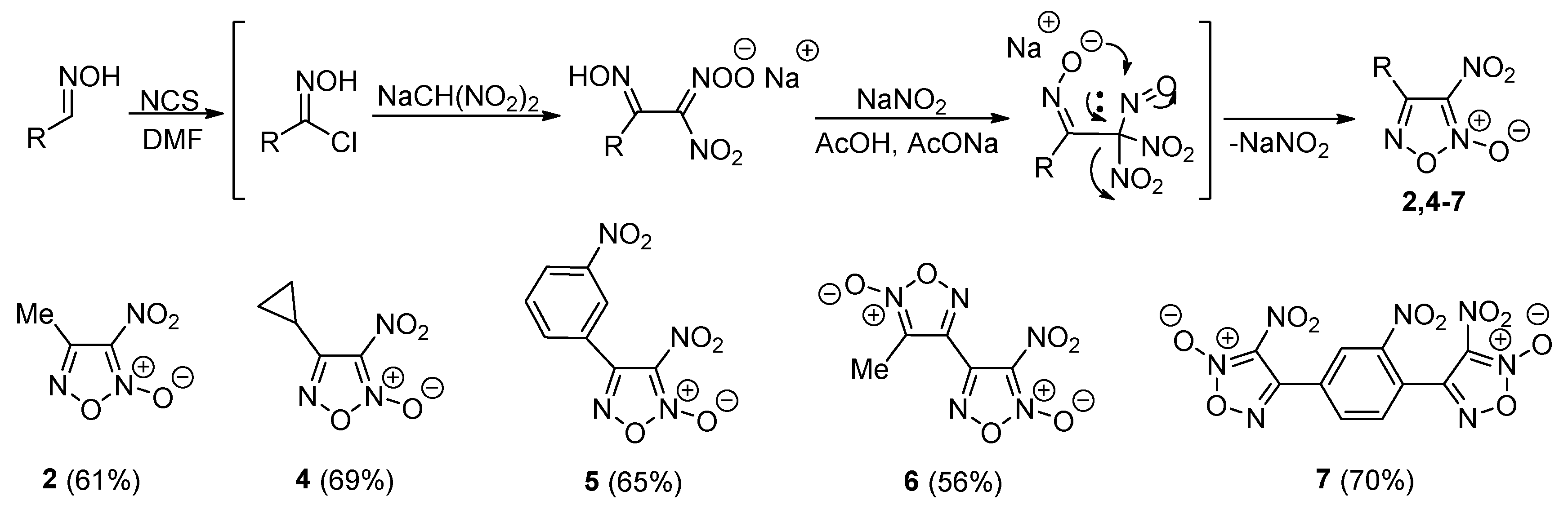
2.1. Synthesis of Furoxan Derivatives
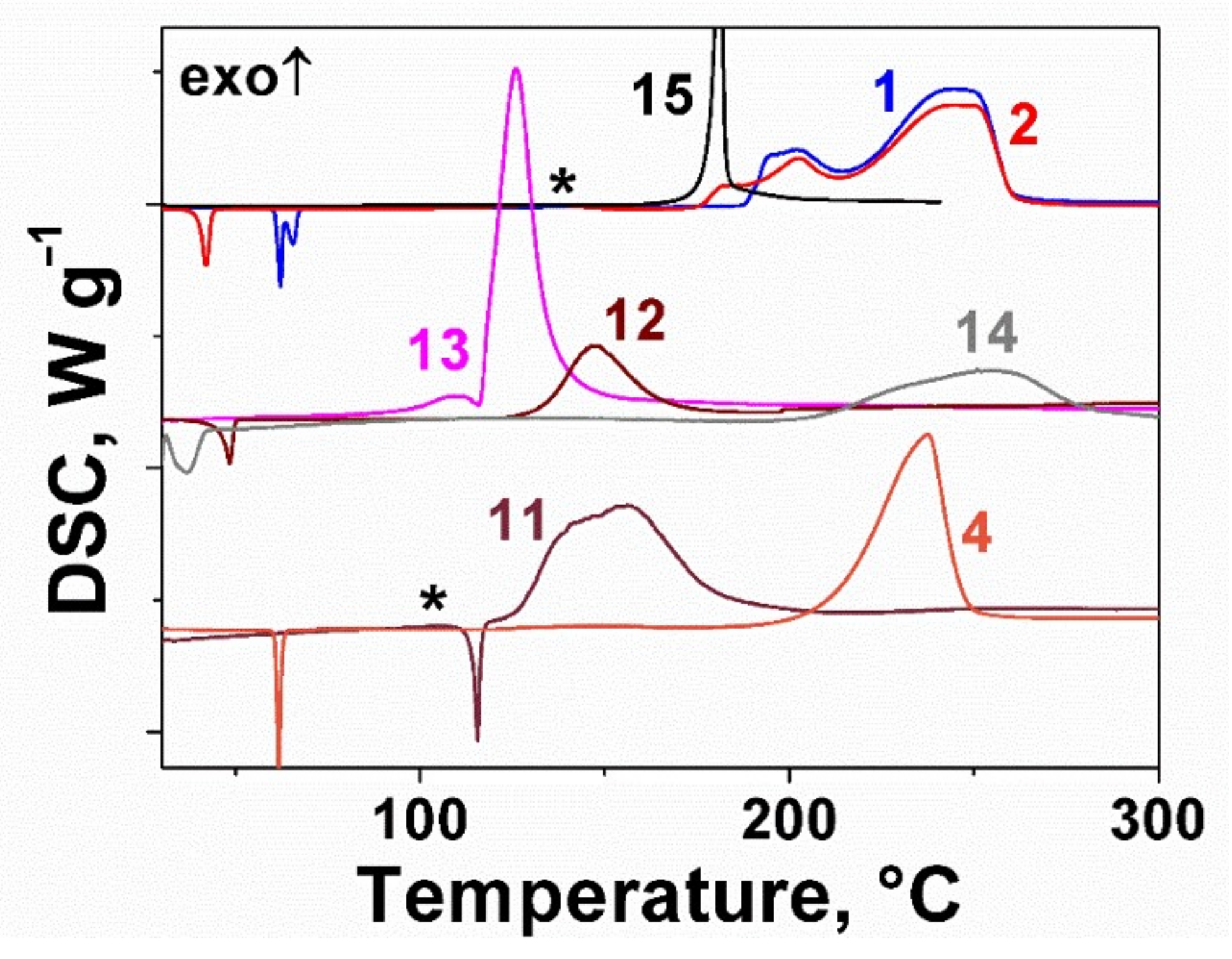
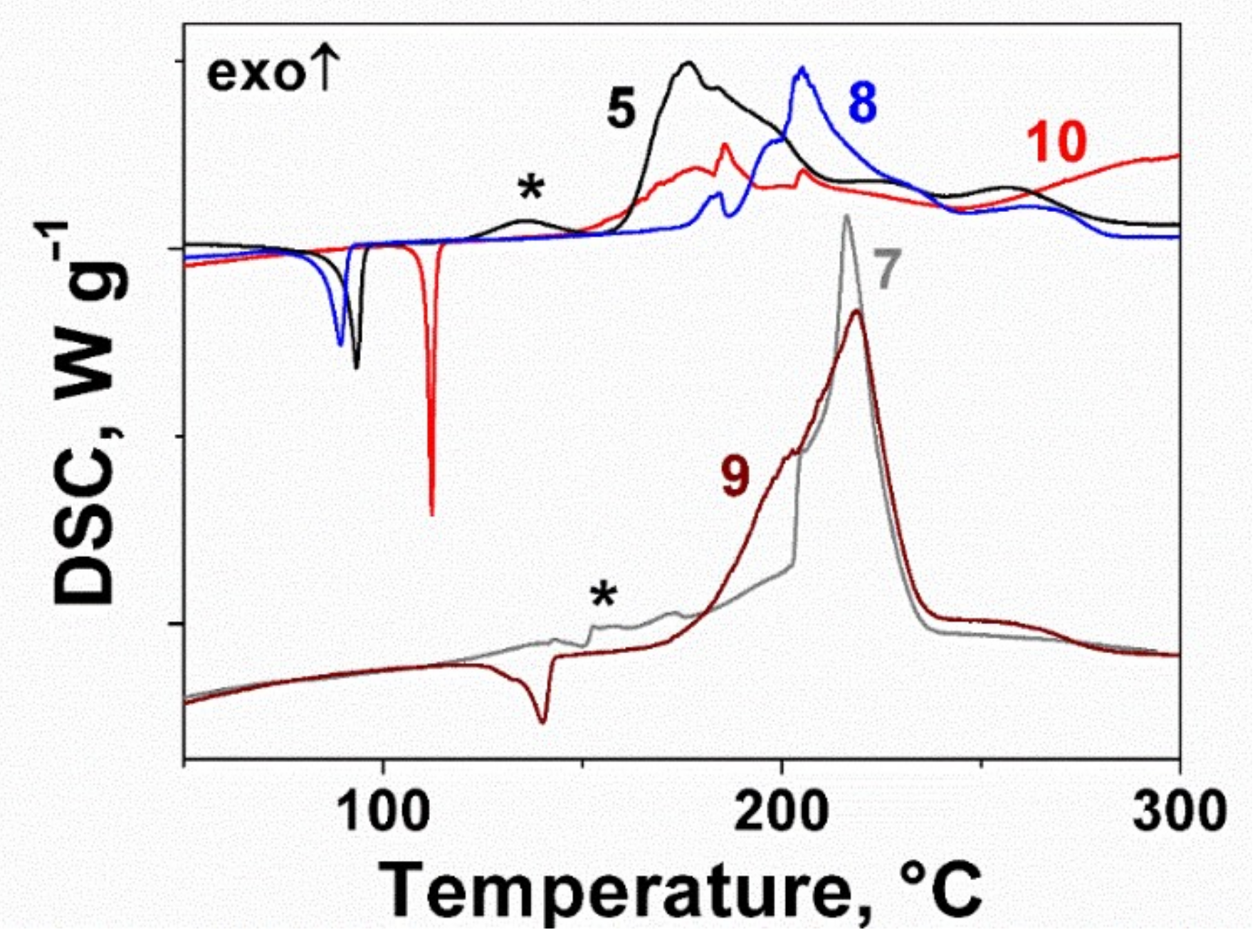
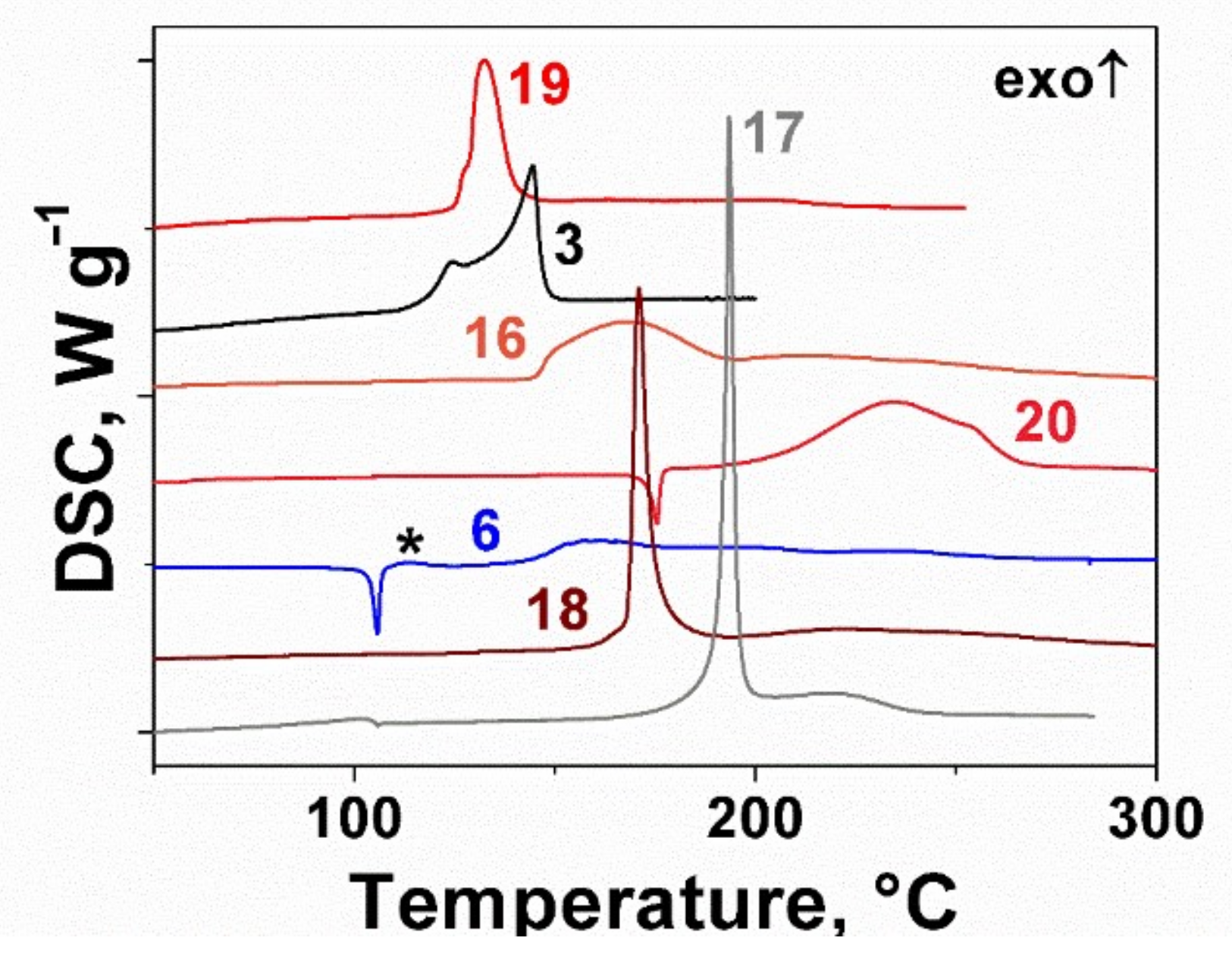
2.2. Thermal Behavior of the Furoxan Derivatives: Melting and Decomposition
2.3. Thermal Behavior of the Furoxan Derivatives: Vaporization
2.4. Gas-Phase Formation Enthalpies of Furoxan, Its Monocyclic, and Bis-Derivatives
2.5. Detonation Performance
2.6. Sensitivity to Mechanical Stimuli
3. Experimental and Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, L.; Yin, P.; Zhao, G.; He, C.; Imler, G.H.; Parrish, D.A.; Gao, H.; Shreeve, J.M. Conjugated Energetic Salts Based on Fused Rings: Insensitive and Highly Dense Materials. J. Am. Chem. Soc. 2018, 140, 15001–15007. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.M.; Edwards, J.T.; Johnson, E.C.; Bukowski, E.J.; Sausa, R.C.; Byrd, E.F.C.; Orlicki, J.A.; Sabatini, J.J.; Baran, P.S. Impact of Stereo- and Regiochemistry on Energetic Materials. J. Am. Chem. Soc. 2019, 141, 12531–12535. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, C.; Jiang, C.; Yang, C.; Du, Y.; Zhao, Y.; Hu, B.; Zheng, Z.; Christe, K.O. Synthesis of AgN5 and its extended 3D energetic framework. Nat. Commun. 2018, 9, 1269. [Google Scholar] [CrossRef]
- O’Sullivan, O.T.; Zdilla, M.J. Properties and Promise of Catenated Nitrogen Systems as High-Energy-Density Materials. Chem. Rev. 2020, 120, 5682–5744. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.; Ronen, Z. Biodegradation of the Explosives TNT, RDX and HMX. In Microbial Degradation of Xenobiotics; Singh, S.N., Ed.; Environmental Science and Engineering; Springer: Berlin/Heidelberg, Germany, 2012; pp. 135–176. ISBN 978-3-642-23788-1. [Google Scholar]
- Kuchurov, I.V.; Zharkov, M.N.; Fershtat, L.L.; Makhova, N.N.; Zlotin, S.G. Prospective Symbiosis of Green Chemistry and Energetic Materials. ChemSusChem 2017, 10, 3914–3946. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M. Chemistry of High-Energy Materials, 2nd ed.; De Gruyter: Berlin, Germany, 2012; ISBN 978-3-11-027359-5. [Google Scholar]
- Gao, H.; Shreeve, J.M. Azole-Based Energetic Salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef]
- Shreeve, J.M.; Hu, L.; Gao, H. Challenging the Limits of Nitrogen and Oxygen Content of Fused Rings. J. Mater. Chem. A 2020. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Makhova, N.N. 1,2,5-Oxadiazole-Based High-Energy-Density Materials: Synthesis and Performance. ChemPlusChem 2020, 85, 13–42. [Google Scholar] [CrossRef]
- Xiong, H.; Yang, H.; Lei, C.; Yang, P.; Hu, W.; Cheng, G. Combinations of furoxan and 1,2,4-oxadiazole for the generation of high performance energetic materials. Dalton Trans. 2019, 48, 14705–14711. [Google Scholar] [CrossRef]
- He, C.; Gao, H.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Boosting energetic performance by trimerizing furoxan. J. Mater. Chem. A 2018, 6, 9391–9396. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Witkowski, T.G. Nitrogen-Rich Energetic 1,2,5-Oxadiazole-Tetrazole—Based Energetic Materials. Propellants Explos. Pyrotech. 2015, 40, 366–373. [Google Scholar] [CrossRef]
- Wu, B.; Yang, H.; Lin, Q.; Wang, Z.; Lu, C.; Cheng, G. New thermally stable energetic materials: Synthesis and characterization of guanylhydrazone substituted furoxan energetic derivatives. New J. Chem. 2015, 39, 179–186. [Google Scholar] [CrossRef]
- Johnson, E.C.; Sabatini, J.J.; Chavez, D.E.; Wells, L.A.; Banning, J.E.; Sausa, R.C.; Byrd, E.F.C.; Orlicki, J.A. Bis(Nitroxymethylisoxazolyl) Furoxan: A Promising Standalone Melt-Castable Explosive. ChemPlusChem 2020, 85, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, A.I.; Dasko, D.V.; Astrat’ev, A.A. 3,4-Bis(4′-nitrofurazan-3′-yl)furoxan: A Melt Cast Powerful Explosive and a Valuable Building Block in 1,2,5-Oxadiazole Chemistry. Cent. Eur. J. Energ. Mater. 2012, 9, 329–342. [Google Scholar]
- He, C.; Tang, Y.; Mitchell, L.A.; Parrish, D.A.; Shreeve, J.M. N-Oxides light up energetic performances: Synthesis and characterization of dinitraminobisfuroxans and their salts. J. Mater. Chem. A 2016, 4, 8969–8973. [Google Scholar] [CrossRef]
- Pepekin, V.I.; Korsunskii, B.L.; Matyushin, Y.N. Explosive properties of furoxanes. Combust. Explos. Shock Waves 2008, 44, 110–114. [Google Scholar] [CrossRef]
- Zhai, L.; Bi, F.; Luo, Y.; Wang, N.; Zhang, J.; Wang, B. New Strategy for Enhancing Energetic Properties by Regulating Trifuroxan Configuration: 3,4-Bis(3-nitrofuroxan-4-yl)furoxan. Sci. Rep. 2019, 9, 4321. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Ovchinnikov, I.V.; Epishina, M.A.; Romanova, A.A.; Lempert, D.B.; Muravyev, N.V.; Makhova, N.N. Assembly of Nitrofurazan and Nitrofuroxan Frameworks for High-Performance Energetic Materials. ChemPlusChem 2017, 82, 1315–1319. [Google Scholar] [CrossRef]
- Larin, A.A.; Shaferov, A.V.; Epishina, M.A.; Melnikov, I.N.; Muravyev, N.V.; Ananyev, I.V.; Fershtat, L.L.; Makhova, N.N. Pushing the Energy-Sensitivity Balance with High-Performance Bifuroxans. ACS Appl. Energy Mater. 2020, 3, 7764–7771. [Google Scholar] [CrossRef]
- Zhai, L.; Bi, F.; Luo, Y.; Sun, L.; Huo, H.; Zhang, J.; Zhang, J.; Wang, B.; Chen, S. Exploring the highly dense energetic materials via regiochemical modulation: A comparative study of two fluorodinitromethyl-functionalized herringbone trifuroxans. Chem. Eng. J. 2020, 391, 123573. [Google Scholar] [CrossRef]
- Ravi, P.; Badgujar, D.M.; Gore, G.M.; Tewari, S.P.; Sikder, A.K. Review on Melt Cast Explosives. Propellants Explos. Pyrotech. 2011, 36, 393–403. [Google Scholar] [CrossRef]
- Nikolaeva, A.D.; Matyushin, Y.N.; Pepekin, V.I.; Smelov, V.S.; Bulidorov, V.V.; Bulidorova, T.I.; Apin, A.Y. Synthesis and study of detonation properties of 3-methyl-4-nitrofuroxan. Bull. Acad. Sci. USSR Div. Chem. Sci. 1972, 21, 927–928. [Google Scholar] [CrossRef]
- Hobbs, M.L.; Baer, M. Calibrating the BKW-EOS with a large product species data base and measured C-J properties. In Proceedings of the Tenth Symposium (International) on Detonation, Boston, MA, USA, 12–16 July 1993; pp. 409–418. [Google Scholar]
- Stepanov, R.S.; Kruglyakova, L.A.; Astakhov, A.M. Kinetics and thermolysis mechanism of some 4-substituted 3-methylfuroxans. Russ. J. Gen. Chem. 2009, 79, 336–337. [Google Scholar] [CrossRef]
- Thermal Decomposition and Combustion of Explosives and Propellants; Manelis, G.B., Nazin, G.M., Rubtsov, Y.I., Strunin, V.A., Eds.; Taylor & Francis: London, UK, 2003; ISBN 978-0-415-29984-8. [Google Scholar]
- Ovchinnikov, I.V.; Makhova, N.N.; Khmel’nitskii, L.I.; Kuz’min, V.S.; Akimova, L.N.; Pepekin, V.I. Dinitrodiazenofuroxan as a new energetic explosive. Dokl. Chem. 1998, 39, 67–70. [Google Scholar]
- Gottfried, J.L.; Klapötke, T.M.; Witkowski, T.G. Estimated Detonation Velocities for TKX-50, MAD-X1, BDNAPM, BTNPM, TKX-55, and DAAF using the Laser-induced Air Shock from Energetic Materials Technique. Propellants Explos. Pyrotech. 2017, 42, 353–359. [Google Scholar] [CrossRef]
- Pepekin, V.I.; Matyushin, Y.N.; Gubina, T.V. Enthalpy of formation and explosive properties of 5,6-(3,4-furazano)-1,2,3,4-tetrazine-1,3-dioxide. Russ. J. Phys. Chem. B 2011, 5, 97–100. [Google Scholar] [CrossRef]
- Afanas’ev, G.T.; Pepekin, V.I. Mechanical sensitivity and detonation ability of the solid explosives. Russ. J. Phys. Chem. 2005, 24, 70–77. [Google Scholar]
- Lobbecke, S.; Schuppler, H.; Schweikert, W. Thermal properties of different substituted energetic furoxanes. In Proceedings of the 33th International Conference of the Fraunhofer ICT, Karlsruhe, Germany, 25–28 June 2002; p. P115/1-12. [Google Scholar]
- Guo, T.; Liu, M.; Huang, X.-C.; Wang, Z.; Qiu, S.; Ge, Z.; Meng, Z. Efficient preparation and comprehensive properties of thermal decomposition and detonation for 4,4′-dinitro-3,3′-azofuroxan. J. Anal. Appl. Pyrolysis 2017, 128, 451–458. [Google Scholar] [CrossRef]
- Fischer, D.; Klapötke, T.M.; Stierstorfer, J. Synthesis and Characterization of Diaminobisfuroxane: Synthesis and Characterization of Diaminobisfuroxane. Eur. J. Inorg. Chem. 2014, 2014, 5808–5811. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Butrico, M.S.; Fuge, M.D.; Chung, P.W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 2018, 8, 9059. [Google Scholar] [CrossRef]
- Casey, A.D.; Son, S.F.; Bilionis, I.; Barnes, B.C. Prediction of Energetic Material Properties from Electronic Structure Using 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fershtat, L.L.; Struchkova, M.I.; Goloveshkin, A.S.; Bushmarinov, I.S.; Makhova, N.N. Dinitrogen Trioxide-Mediated Domino Process for the Regioselective Construction of 4-Nitrofuroxans from Acrylic Acids: Dinitrogen Trioxide-Mediated Domino Process for the Regioselective Construction of 4-Nitrofuroxans. Heteroat. Chem. 2014, 25, 226–237. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Larin, A.A.; Epishina, M.A.; Kulikov, A.S.; Ovchinnikov, I.V.; Ananyev, I.V.; Makhova, N.N. Regioselective synthesis of bifuroxanyl systems with the 3-nitrobifuroxanyl core via a one-pot acylation/nitrosation/cyclization cascade. Tetrahedron Lett. 2016, 57, 4268–4272. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Epishina, M.A.; Ovchinnikov, I.V.; Struchkova, M.I.; Romanova, A.A.; Ananyev, I.V.; Makhova, N.N. Side-chain prototropic tautomerism of 4-hydroxyfuroxans in methylation reactions. Tetrahedron Lett. 2016, 57, 5685–5689. [Google Scholar] [CrossRef]
- Fershtat, L.; Bystrov, D.; Zhilin, E.; Makhova, N. N-Oxide-Controlled Chemoselective Reduction of Nitrofuroxans. Synthesis 2019, 51, 747–756. [Google Scholar] [CrossRef]
- Finogenov, A.O.; Epishina, M.A.; Ovchinnikov, I.V.; Kulikov, A.S.; Anan’ev, I.V.; Makhova, N.N. Synthesis of isomeric 1,3- and 1,4-bis[3(4)-nitrofuroxan-4(3)-yl]nitrobenzenes by nitration of the corresponding isomeric 1,3- and 1,4-bis[3(4)-nitrofuroxan-4(3)-yl]benzenes. Russ. Chem. Bull. 2011, 60, 339–344. [Google Scholar] [CrossRef]
- Andrianov, V.G. Synthesis and properties of derivatives of 4-aminofuroxan-3-carboxylic acid. Chem. Heterocycl. Compd. 1997, 33, 973–976. [Google Scholar] [CrossRef]
- Kulikov, A.S.; Ovchinnikov, I.V.; Molotov, S.I.; Makhova, N.N. Synthesis of furoxan derivatives based on 4-aminofuroxan-3-carboxylic acid azide. Russ. Chem. Bull. 2003, 52, 1822–1828. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Epishina, M.A.; Kulikov, A.S.; Ovchinnikov, I.V.; Ananyev, I.V.; Makhova, N.N. An efficient access to (1H-tetrazol-5-yl)furoxan ammonium salts via a two-step dehydration/[3+2]-cycloaddition strategy. Tetrahedron 2015, 71, 6764–6775. [Google Scholar] [CrossRef]
- Johnson, E.C.; Bukowski, E.J.; Sausa, R.C.; Sabatini, J.J. Safer and Convenient Synthesis of 3,4-Dicyanofuroxan. Org. Process Res. Dev. 2019, 23, 1275–1279. [Google Scholar] [CrossRef]
- Blinnikov, A.N.; Makhova, N.N. Novel synthesis of 3-monosubstituted furoxans. Mendeleev Commun. 1999, 9, 13–14. [Google Scholar] [CrossRef]
- Epishina, M.A.; Finogenov, A.O.; Kulikov, A.S.; Makhova, N.N.; Anan’ev, I.V.; Tartakovsky, V.A. Synthesis and nitration of 3-R-4-(2,2,2-trinitroethyl)aminofuroxans. Russ. Chem. Bull. 2012, 61, 1575–1581. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Monogarov, K.A.; Bragin, A.A.; Fomenkov, I.V.; Pivkina, A.N. HP-DSC study of energetic materials. Part I. Overview of pressure influence on thermal behavior. Thermochim. Acta 2016, 631, 1–7. [Google Scholar] [CrossRef]
- Gorn, M.V.; Monogarov, K.A.; Dalinger, I.L.; Melnikov, I.N.; Kiselev, V.G.; Muravyev, N.V. Pressure DSC for energetic materials. Part 2. Switching between evaporation and thermal decomposition of 3,5-dinitropyrazole. Thermochim. Acta 2020, 690, 178697. [Google Scholar] [CrossRef]
- Siitsman, C.; Oja, V. Extension of the DSC method to measuring vapor pressures of narrow boiling range oil cuts. Thermochim. Acta 2015, 622, 31–37. [Google Scholar] [CrossRef]
- Belyaev, A.F.; Yusephovich, N.A. Vliyanie temperaturi kipeniya vzrivchatikh vechestv na kharakter teplovoi vspychki. Dokl. Akad. Nauk USSR 1940, 27, 133–135. [Google Scholar]
- Belyakov, A.V.; Oskorbin, A.A.; Losev, V.A.; Rykov, A.N.; Shishkov, I.F.; Fershtat, L.L.; Larin, A.A.; Kuznetsov, V.V.; Makhova, N.N. The equilibrium molecular structure of 3-methyl-4-nitro- and 4-methyl-3-nitrofuroxans by gas-phase electron diffraction and coupled cluster calculations. J. Mol. Struct. 2020, 1222, 128856. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Gritsan, N.P. Theoretical Study of the Nitroalkane Thermolysis. 1. Computation of the Formation Enthalpy of the Nitroalkanes, Their Isomers and Radical Products. J. Phys. Chem. A 2008, 112, 4458–4464. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Goldsmith, C.F. Accurate Prediction of Bond Dissociation Energies and Barrier Heights for High-Energy Caged Nitro and Nitroamino Compounds Using a Coupled Cluster Theory. J. Phys. Chem. A 2019, 123, 4883–4890. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Goldsmith, C.F. Accurate Thermochemistry of Novel Energetic Fused Tricyclic 1,2,3,4-Tetrazine Nitro Derivatives from Local Coupled Cluster Methods. J. Phys. Chem. A 2019, 123, 9818–9827. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N.; Schleyer, P.V.R.; Allen, W.D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. [Google Scholar] [CrossRef] [PubMed]
- Westwell, M.S.; Searle, M.S.; Wales, D.J.; Williams, D.H. Empirical Correlations between Thermodynamic Properties and Intermolecular Forces. J. Am. Chem. Soc. 1995, 117, 5013–5015. [Google Scholar] [CrossRef]
- Kempa, P.B.; Bathelt, H.; Volk, F.; Weindel, M. ICT Database of Thermochemical Values; Fraunhofer Institut für Chemische Technologie: Pfintztal, Germany, 2004. [Google Scholar]
- Muravyev, N.V.; Pivkina, A.N.; Kiselev, V.G. Comment on “Studies on Thermodynamic Properties of FOX-7 and Its Five Closed-Loop Derivatives”. J. Chem. Eng. Data 2017, 62, 575–576. [Google Scholar] [CrossRef]
- Matyushin, Y.N.; Lebedev, V.P. Thermochemical Properties of Azoles and Oxadiazoles. In Proceedings of the 28th International Annual Conference of the Fraunhofer ICT, Karlsruhe, Germany, 24–27 June 1997; Fraunhofer ICT: Pfinztal, Germany, 1997; p. 98/1-10. [Google Scholar]
- Kemp, M.D.; Goldhagen, S.; Zihlman, F.A. Vapor Pressures and Cryoscopic Data for Some Aliphatic Dinitroxy and Trinitroxy Compounds. J. Phys. Chem. 1957, 61, 240–242. [Google Scholar] [CrossRef]
- Altenburg, T.; Klapötke, T.M.; Penger, A. Primary Nitramines Related to Nitroglycerine: 1-Nitramino-2,3-dinitroxypropane and 1,2,3-Trinitraminopropane. Cent. Eur. J. Energ. Mater. 2009, 6, 255–275. [Google Scholar]
- Smirnov, A.S.; Voronko, O.; Korsunskii, B.L.; Pivina, T.S. Impact and Friction Sensitivity of Energetic Materials: Methodical Evaluation of Technological Safety Features. Chin. J. Explos. Propellants 2015, 38, 1–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, Version 2010.1, a Package of ab Initio Programs; Molpro: Cardiff, UK, 2010. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0: Software update. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Karton, A.; Martin, J.M.L. Explicitly correlated Wn theory: W1-F12 and W2-F12. J. Chem. Phys. 2012, 136, 124114. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Redfern, P.C.; Pople, J.A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063–1079. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M.L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [Google Scholar] [CrossRef]
- Karton, A.; Schreiner, P.R.; Martin, J.M.L. Heats of formation of platonic hydrocarbon cages by means of high-level thermochemical procedures. J. Comput. Chem. 2016, 37, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Karton, A. A computational chemist’s guide to accurate thermochemistry for organic molecules: A computational chemist’s guide to accurate thermochemistry for organic molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 292–310. [Google Scholar] [CrossRef]
- Allison, T. JANAF Thermochemical Tables, NIST Standard Reference Database 13; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1996. [Google Scholar]
- Liakos, D.G.; Sparta, M.; Kesharwani, M.K.; Martin, J.M.L.; Neese, F. Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Kossmann, S.; Neese, F. Comparison of two efficient approximate Hartee–Fock approaches. Chem. Phys. Lett. 2009, 481, 240–243. [Google Scholar] [CrossRef]
- STANAG 4489. Explosives, Impact Sensitivity Tests; NATO: Brussels, Belgium, 1999; p. 1. [Google Scholar]
- STANAG 4487. Explosives, Friction Sensitivity Tests; NATO: Brussels, Belgium, 2002; p. 1. [Google Scholar]
- Pepekin, V.I.; Lebedev, Y.A. Criteria of the detonation parameters estimation for explosive. Dokl. Akad. Nauk USSR 1977, 234, 1391–1394. [Google Scholar]
- Bruker. APEX-III; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
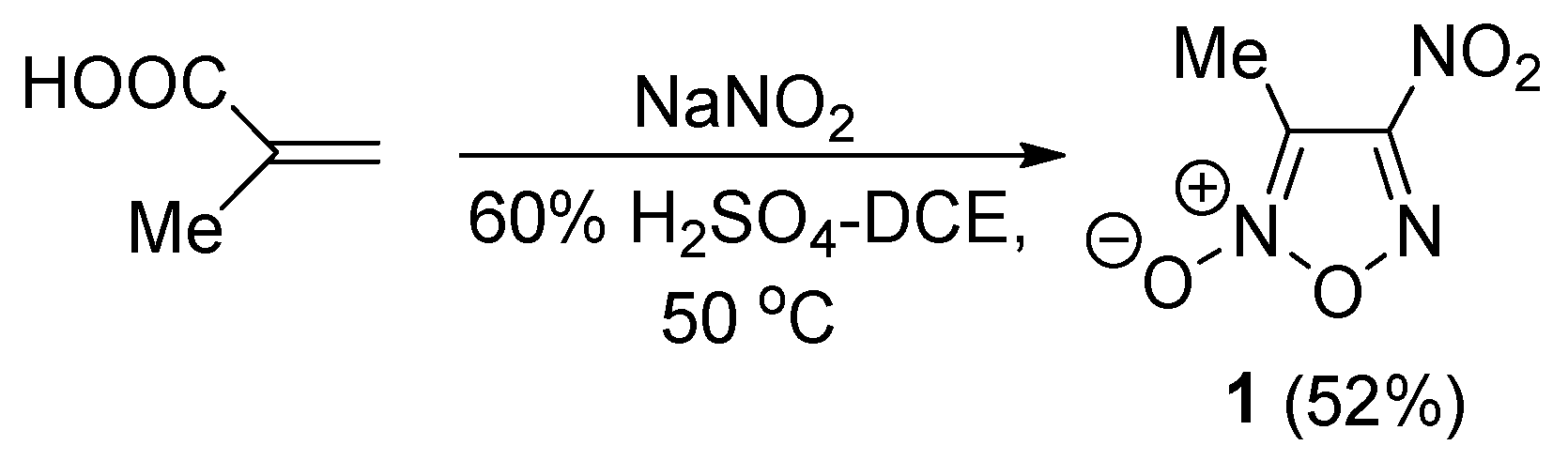{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔfH0(g), kJ mol−1 | Source |
|---|---|---|
| 1 | 186.4 | [a] |
| 2 | 194.7 | [a] |
| 3 | 800.4 | [b] |
| 4 | 306.1 | [b] |
| 5 | 335.3 | [c] |
| 6 | 437.5 | [d] |
| 7 | 615.8 | [c] |
| 8 | 324.9 | [c] |
| 9 | 594.6 | [c] |
| 10 | 328.9 | [c] |
| 11 | 66.4 | [a] |
| 12 | 421.3 | [a] |
| 13 | 360.8 | [a] |
| 14 | 554.0 | [a] |
| 15 | 158.6 | [a] |
| 16 | 588.4 | [b] |
| 17 | 341.1 | [b] |
| 18 | 1053.2 | [b] |
| 19 | 479.9 | [e] |
| 20 | 552.6 | [b] |
| 21 | 205.2 | [a] |
| 22 | 246.6 | [a] |
| 23 | 237.1 | [a] |
| 24 | 418.2 | [a] |
| 25 | 425.6 | [a] |
| 26 | 697.3 | [a] |
| 27 | 699.5 | [a] |
| # | Stoichiometry | OB a | ρ, g cm−3 | ΔfH0(s), kJ mol−1 | tm, °C | tdec, °C | IS, J | FS, N | VD, km s−1 | Qex, kJ g−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C3H3N3O4 | 0.53 | 1.66 | 101 c | 65 | 188 | 5 | 270 | 7.8 | 5.4 |
| 2 | C3H3N3O4 | 0.53 | 1.68 | 136 | 40 | 176 | 7 | 180 | 7.9 | 5.6 |
| 3 | C4N8O8 | 1.00 | 1.98 b | 668 d | – | 117 | 0.7 | 6 | 10.0 | 7.4 |
| 4 | C5H5N3O4 | 0.32 | 1.62 | 243 | 61 | 211 | 23 | 170 | 7.3 | 5.4 |
| 5 | C8H4N4O6 | 0.33 | 1.60 | 267 | 90 | 162 | 2 | >360 | 6.9 | 4.6 |
| 6 | C5H3N5O6 | 0.52 | 1.76 | 365 | 103 | 129 | 3 | 70 | 8.2 | 5.8 |
| 7 | C10H3N7O10 | 0.47 | 1.70 | 527 | – | 199 | 5 | 150 | 7.6 | 5.3 |
| 8 | C8H4N4O6 | 0.33 | 1.58 | 258 | 83 | 175 | 8 | >360 | 6.8 | 4.5 |
| 9 | C10H3N7O10 | 0.47 | 1.77 | 518 | 133 | 173 | 3 | 150 | 7.8 | 5.3 |
| 10 | C8H3N5O8 | 0.46 | 1.75 | 257 | 111 | 153 | 4 | 190 | 7.6 | 5.1 |
| 11 | C3H2N4O5 | 0.71 | 1.85 | −6 | 114 | 114 | 18 | 180 | 8.4 | 5.1 |
| 12 | C3N4O4 | 0.67 | 1.87 | 381 d | 46 | 133 | 5 | 130 | 9.1 | 6.4 |
| 13 | C3H2N4O2 | 0.29 | 1.90 | 288 | 117 | 96 | 45 | 90 | 8.5 | 4.9 |
| 14 | C4N4O2 | 0.25 | 1.59 | 465 d | 39 | 204 | >100 | 280 | 7.4 | 4.3 |
| 15 | C3H4N2O2 | 0.25 | 1.24 | 70 e | <RT | 180 | >100 | – | 6.1 | 4.1 |
| 16 | C6H6N6O4 | 0.27 | 1.52 | 510 | – | 145 | 13 | 160 | 7.2 | 5.0 |
| 17 | C6H4N8O6 | 0.43 | 1.88 | 254 | – | 188 | 4 | 120 | 8.3 | 4.5 |
| 18 | C6N8O4 | 0.33 | 1.72 | 970 | – | 168 | 3 | 90 | 8.3 | 5.2 |
| 19 | C7H8N8O8 | 0.44 | 1.68 | 405 | – | 125 | 3 | 110 | 7.9 | 5.4 |
| 20 | C6H6N6O5 | 0.33 | 1.58 | 469 | 172 | 172 | 4 | 210 | 7.5 | 5.2 |
| NG | C3H5N3O9 | 1.06 | 1.59 | −370 | 13 f | 143 g | <0.3 | – | 7.8 | 5.9 |
| TNT | C7H5N3O6 | 0.36 | 1.64 | −75 | 81 | 306 | 30 | >360 | 6.8 | 4.2 |
| PETN | C5H8N4O12 | 0.86 | 1.78 | −539 | 141 | 181 | 3 | 70 | 8.4 | 5.7 |
| RDX | C3H6N6O6 | 0.67 | 1.8 | 62 | 204 | 204 | 8 | 140 | 8.9 | 5.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larin, A.A.; Bystrov, D.M.; Fershtat, L.L.; Konnov, A.A.; Makhova, N.N.; Monogarov, K.A.; Meerov, D.B.; Melnikov, I.N.; Pivkina, A.N.; Kiselev, V.G.; et al. Nitro-, Cyano-, and Methylfuroxans, and Their Bis-Derivatives: From Green Primary to Melt-Cast Explosives. Molecules 2020, 25, 5836. https://doi.org/10.3390/molecules25245836
Larin AA, Bystrov DM, Fershtat LL, Konnov AA, Makhova NN, Monogarov KA, Meerov DB, Melnikov IN, Pivkina AN, Kiselev VG, et al. Nitro-, Cyano-, and Methylfuroxans, and Their Bis-Derivatives: From Green Primary to Melt-Cast Explosives. Molecules. 2020; 25(24):5836. https://doi.org/10.3390/molecules25245836
Chicago/Turabian StyleLarin, Alexander A., Dmitry M. Bystrov, Leonid L. Fershtat, Alexey A. Konnov, Nina N. Makhova, Konstantin A. Monogarov, Dmitry B. Meerov, Igor N. Melnikov, Alla N. Pivkina, Vitaly G. Kiselev, and et al. 2020. "Nitro-, Cyano-, and Methylfuroxans, and Their Bis-Derivatives: From Green Primary to Melt-Cast Explosives" Molecules 25, no. 24: 5836. https://doi.org/10.3390/molecules25245836
APA StyleLarin, A. A., Bystrov, D. M., Fershtat, L. L., Konnov, A. A., Makhova, N. N., Monogarov, K. A., Meerov, D. B., Melnikov, I. N., Pivkina, A. N., Kiselev, V. G., & Muravyev, N. V. (2020). Nitro-, Cyano-, and Methylfuroxans, and Their Bis-Derivatives: From Green Primary to Melt-Cast Explosives. Molecules, 25(24), 5836. https://doi.org/10.3390/molecules25245836