
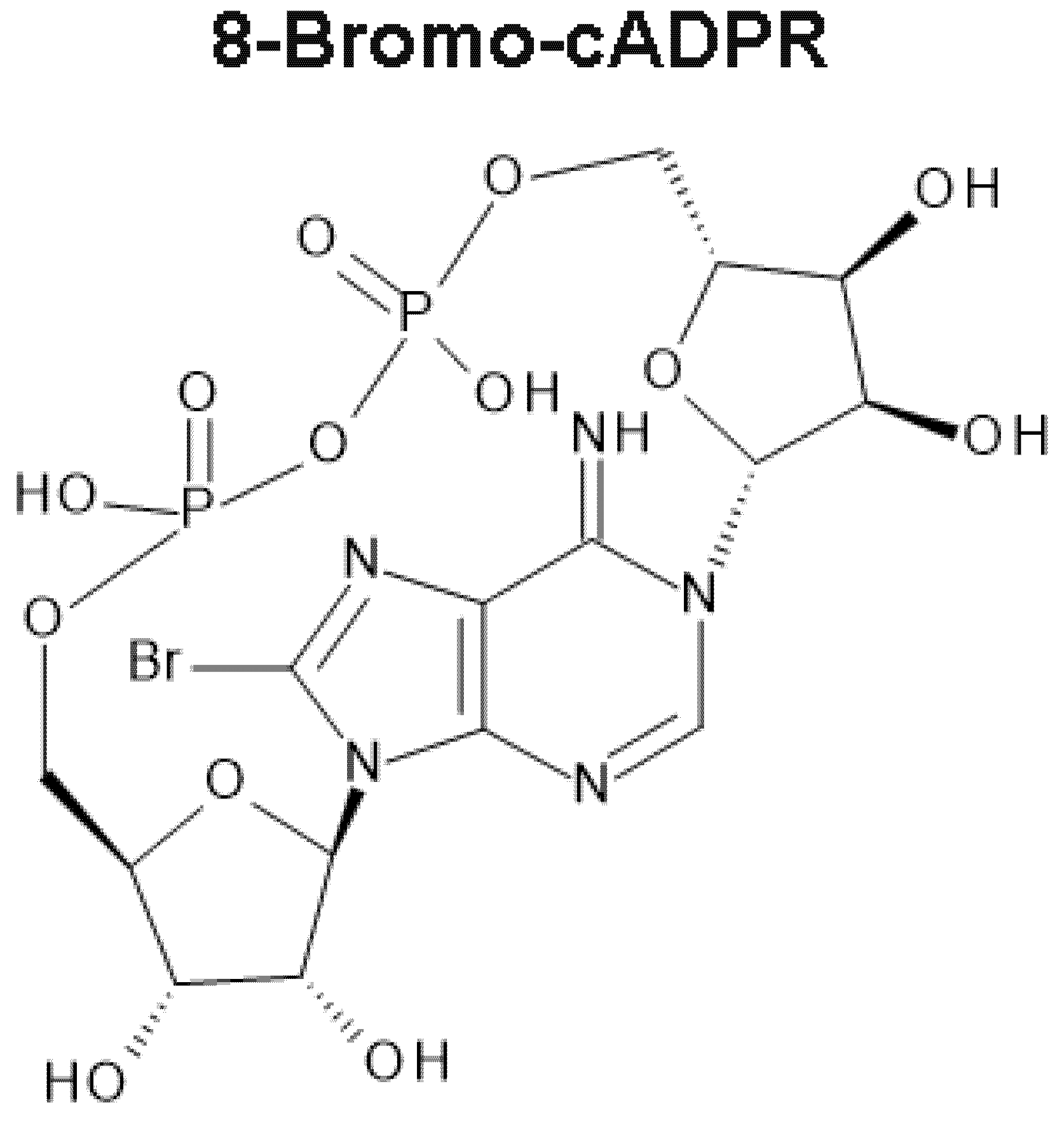
On a Magical Mystery Tour with 8-Bromo-Cyclic ADP-Ribose: From All-or-None Block to Nanojunctions and the Cell-Wide Web

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A Tale of Two Channels and Perhaps a Few Dollars More
3. KN or Not KN That Was the Question
4. Two for Tea: Another Awkward Moment in Time
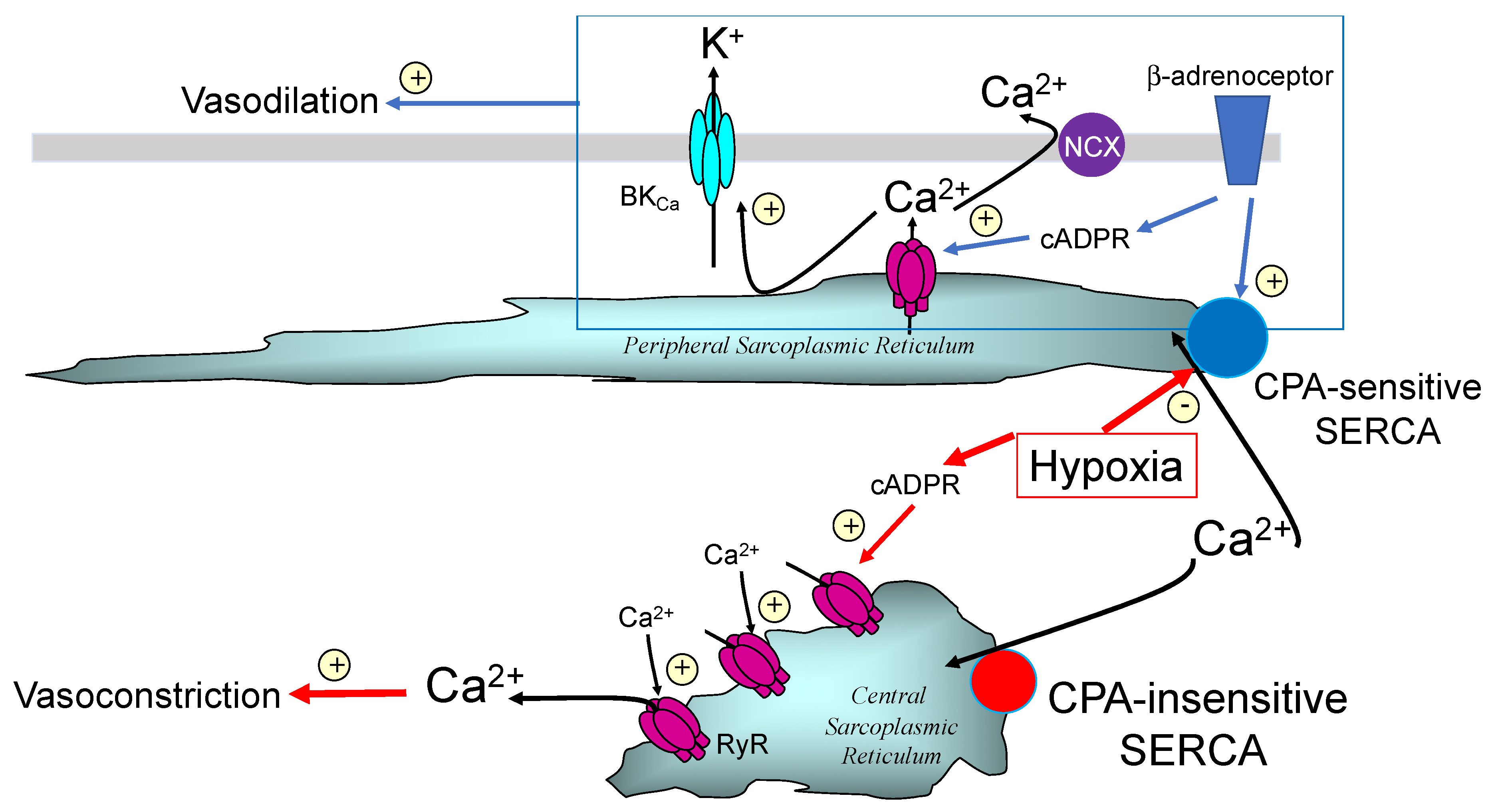
5. Hey Hey CPA Say: Had Me a Store but One Ran Away
6. 8-Bromo-cADPR and the Magical Mystery Tour Takes Off
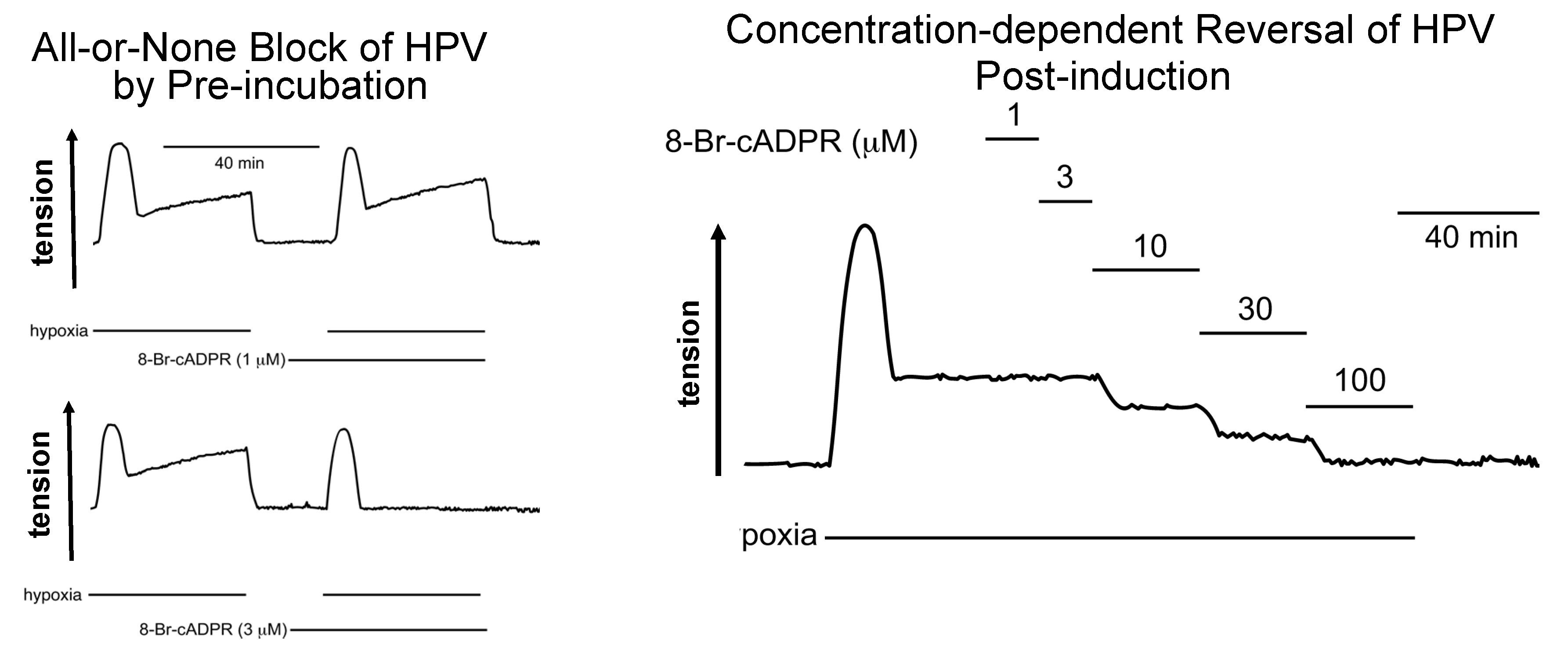
7. 8-Bromo-cADPR Action: On the Face of It All or Nothing
8. Location, Location: Dispense with the Wax and Shine Some Light on Here
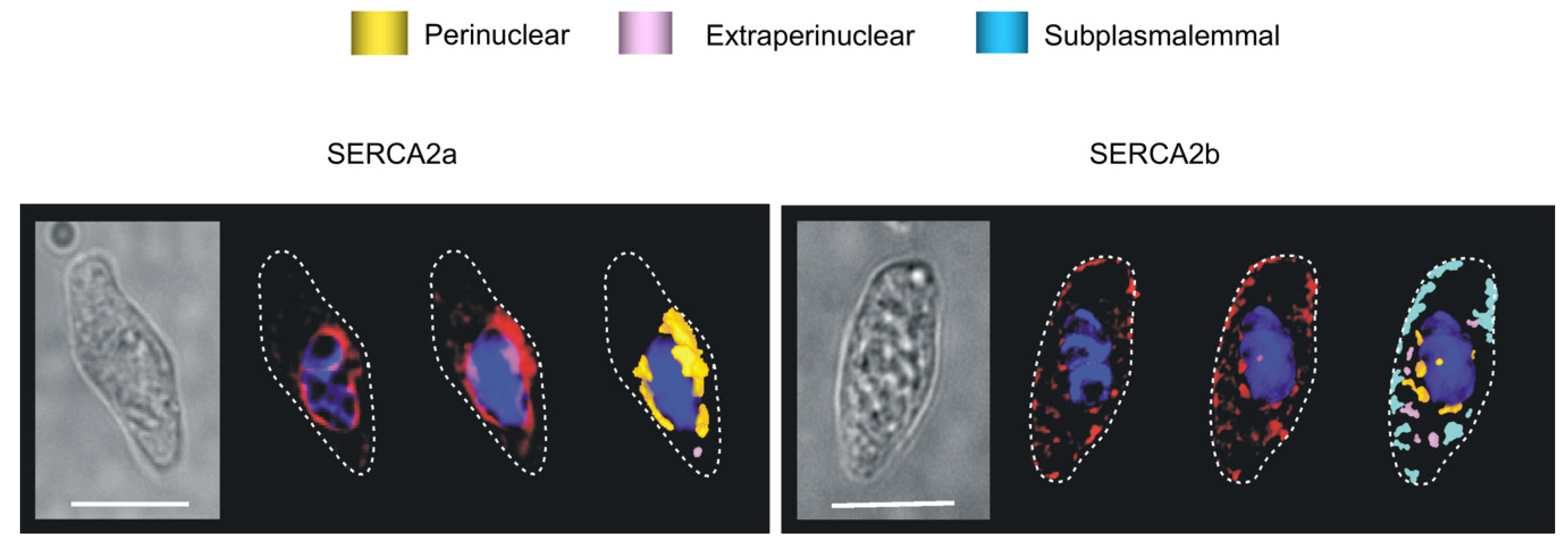
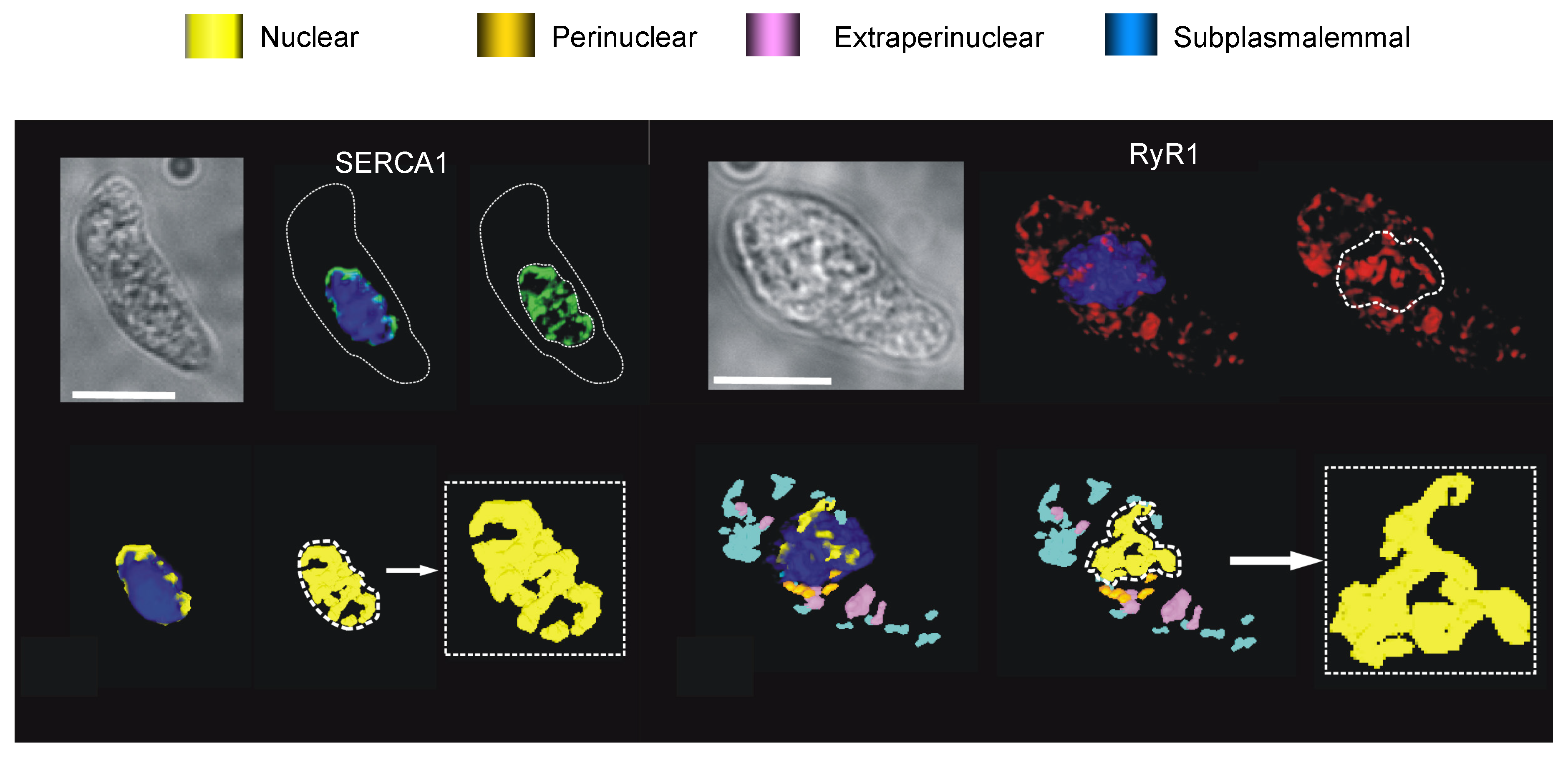
8.1. The SERCA Circus
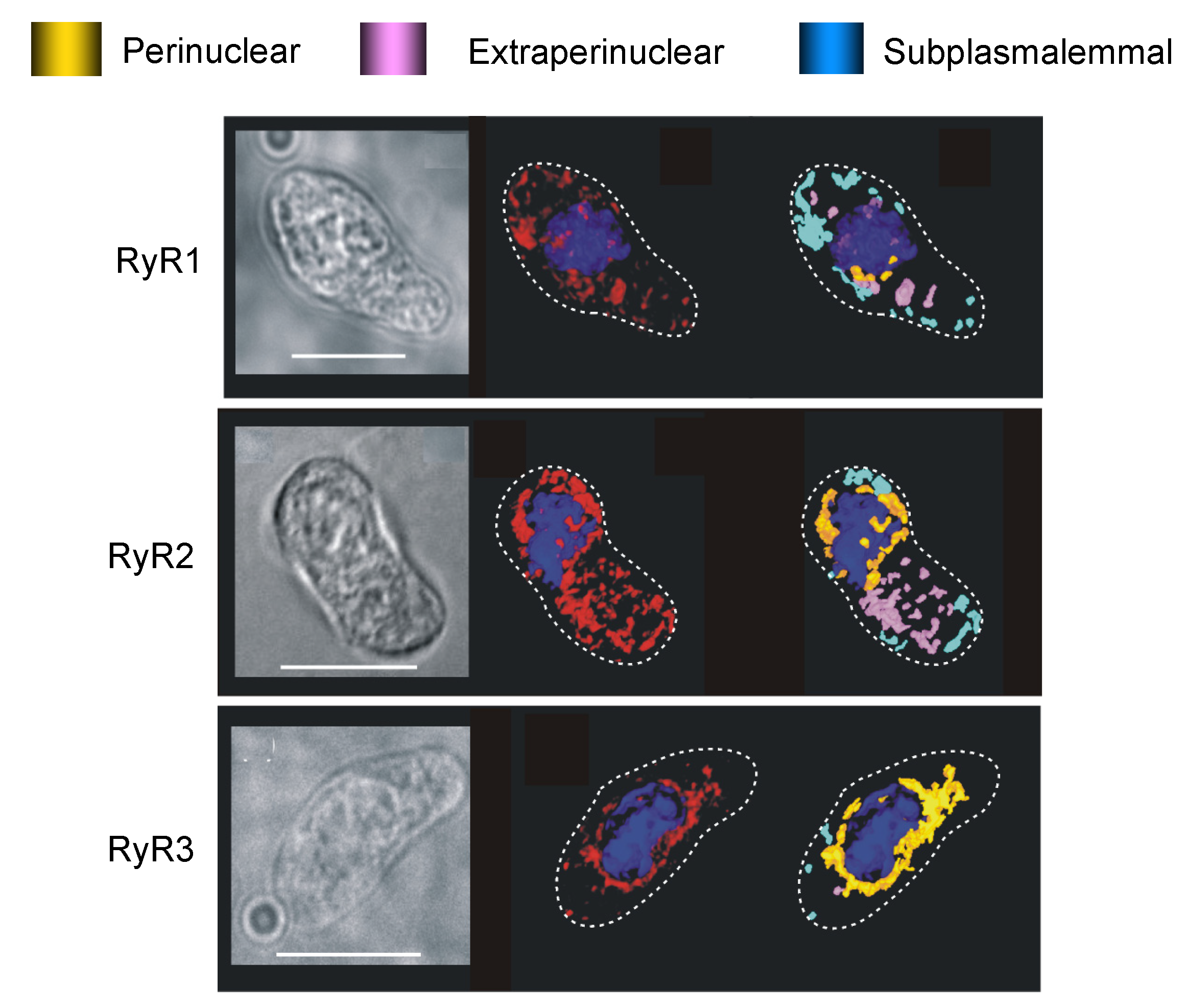
8.2. To Be Three RyRs
9. Another Twist in the Tale
10. We Will Be Fine, Its Nano Time: Even though “NAScA” Say We Out of Line
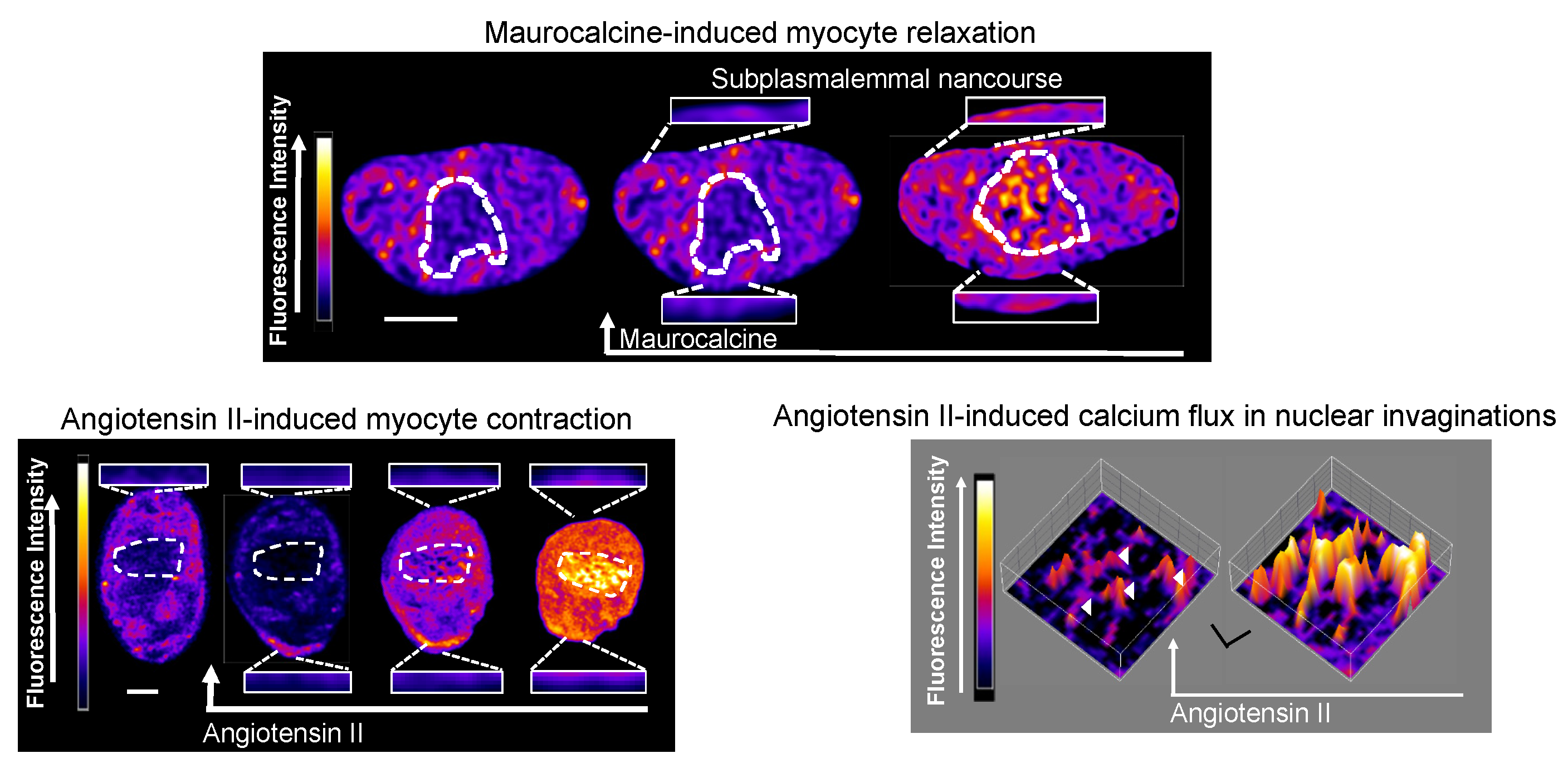
11. PM-SR Nanojunctions of Vascular Smooth Muscles
12. China Girl: How the Cell-Wide Web Was Weaved
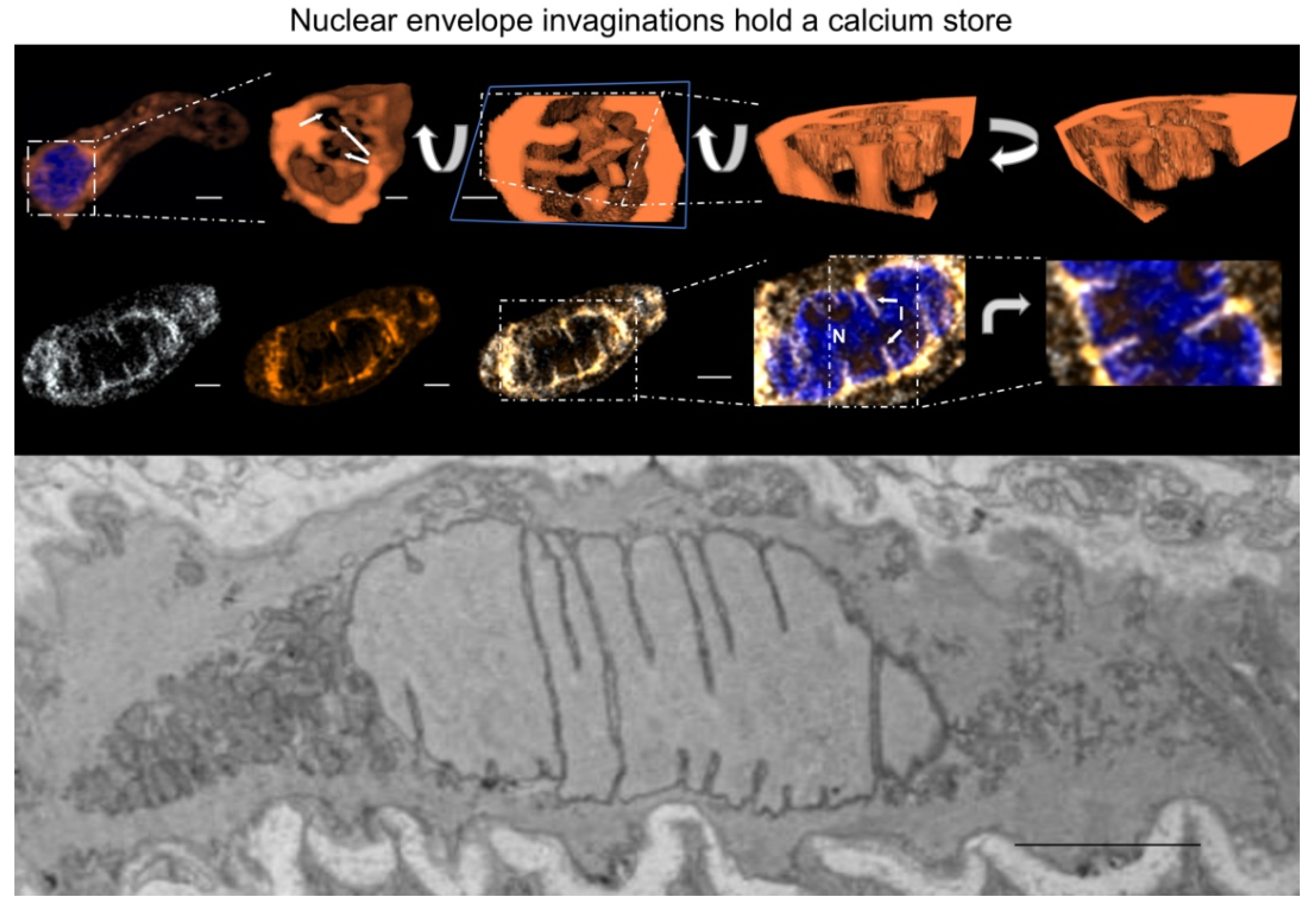
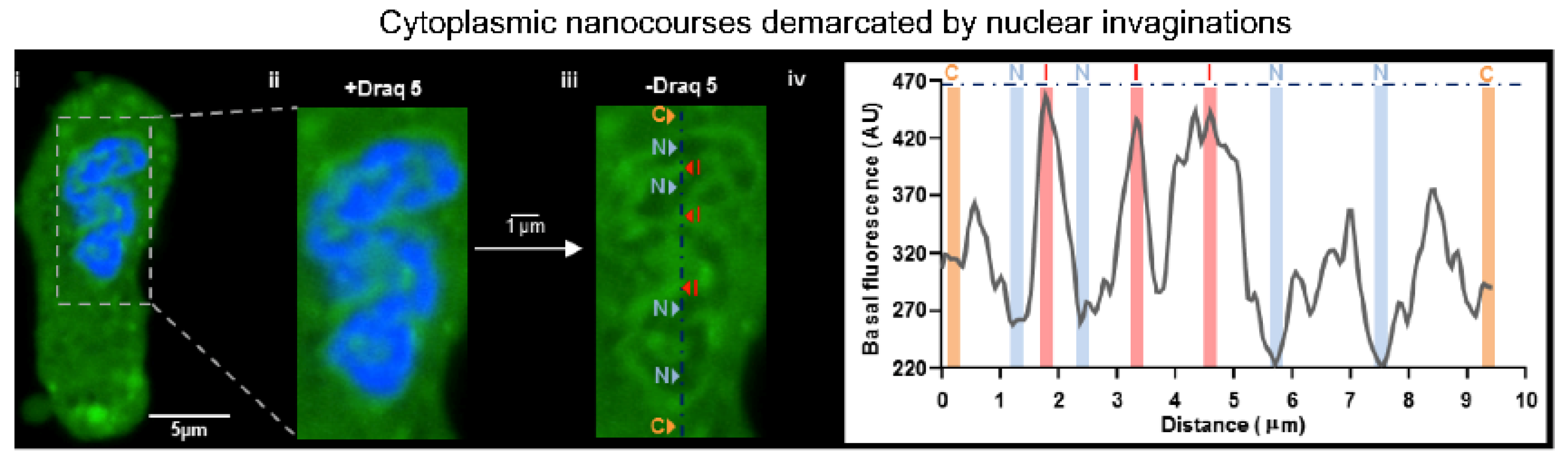
13. Nuclear Invaginations: A Road for El Dorado
14. More Than This: Lost in Proliferation
15. Reviewing the Situation: Got to Pick a Pocket or Two Boys
Funding
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Clapham, D.E. Evolutionary genomics reveals lineage-specific gene loss and rapid evolution of a sperm-specific ion channel complex: Catspers and catsperbeta. PLoS ONE 2008, 3, e3569. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Wang, X.; Clapham, D.E. Early evolution of the eukaryotic Ca2+ signaling machinery: Conservation of the catsper channel complex. Mol. Biol. Evol. 2014, 31, 2735–2740. [Google Scholar] [CrossRef] [Green Version]
- Brochet, D.X.; Xie, W.; Yang, D.; Cheng, H.; Lederer, W.J. Quarky calcium release in the heart. Circ. Res. 2011, 108, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Kong, C.H.T.; Hancox, J.C.; Cannell, M.B. Late Ca2+ sparks and ripples during the systolic Ca2+ transient in heart muscle cells. Circ. Res. 2018, 122, 473–478. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ikegaya, Y. Locally sequential synaptic reactivation during hippocampal ripples. Sci. Adv. 2020, 6, eaay1492. [Google Scholar] [CrossRef] [Green Version]
- Vierra, N.C.; Kirmiz, M.; van der List, D.; Santana, L.F.; Trimmer, J.S. Kv2.1 mediates spatial and functional coupling of l-type calcium channels and ryanodine receptors in mammalian neurons. Elife 2019, 8, e49953. [Google Scholar] [CrossRef]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef]
- Asada, Y.; Yamazawa, T.; Hirose, K.; Takasaka, T.; Iino, M. Dynamic Ca2+ signalling in rat arterial smooth muscle cells under the control of local renin-angiotensin system. J. Physiol. 1999, 521 Pt 2, 497–505. [Google Scholar] [CrossRef]
- Evans, A.M. Nanojunctions of the sarcoplasmic reticulum deliver site- and function-specific calcium signaling in vascular smooth muscles. Adv. Pharm. 2017, 78, 1–47. [Google Scholar]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plattner, H.; Verkhratsky, A. Evolution of calcium signalling. Cell Calcium 2015, 57, 121–122. [Google Scholar] [CrossRef]
- King, C.M.; Bohmbach, K.; Minge, D.; Delekate, A.; Zheng, K.; Reynolds, J.; Rakers, C.; Zeug, A.; Petzold, G.C.; Rusakov, D.A.; et al. Local resting ca(2+) controls the scale of astroglial Ca2+ signals. Cell Rep. 2020, 30, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, M.; Queisser, G.; Eder, A.; Wiegert, J.S.; Bengtson, C.P.; Hellwig, A.; Wittum, G.; Bading, H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: Interplay between nuclear structure, histone h3 phosphorylation, and nuclear calcium signaling. J. Neurosci. 2009, 29, 14687–14700. [Google Scholar] [CrossRef]
- Queisser, G.; Wiegert, S.; Bading, H. Structural dynamics of the cell nucleus: Basis for morphology modulation of nuclear calcium signaling and gene transcription. Nucleus 2011, 2, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Bootman, M.D.; Fearnley, C.; Smyrnias, I.; MacDonald, F.; Roderick, H.L. An update on nuclear calcium signalling. J. Cell Sci. 2009, 122, 2337–2350. [Google Scholar] [CrossRef] [Green Version]
- Eder, A.; Bading, H. Calcium signals can freely cross the nuclear envelope in hippocampal neurons: Somatic calcium increases generate nuclear calcium transients. BMC Neurosci. 2007, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Sethi, J.K.; Empson, R.M.; Bailey, V.C.; Potter, B.V.; Galione, A. 7-deaza-8-bromo-cyclic adp-ribose, the first membrane-permeant, hydrolysis-resistant cyclic adp-ribose antagonist. J. Biol. Chem. 1997, 272, 16358–16363. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Aarhus, R. Adp-ribosyl cyclase: An enzyme that cyclizes nad+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Van Breemen, C.; Fameli, N.; Evans, A.M. Pan-junctional sarcoplasmic reticulum in vascular smooth muscle: Nanospace Ca2+ transport for site- and function-specific Ca2+ signalling. J. Physiol. 2013, 591, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Wyatt, C.N.; Kinnear, N.P.; Clark, J.H.; Blanco, E.A. Pyridine nucleotides and calcium signalling in arterial smooth muscle: From cell physiology to pharmacology. Pharm. Ther. 2005, 107, 286–313. [Google Scholar] [CrossRef] [PubMed]
- Dipp, M.; Evans, A.M. Cyclic adp-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ. Res. 2001, 89, 77–83. [Google Scholar] [CrossRef] [Green Version]
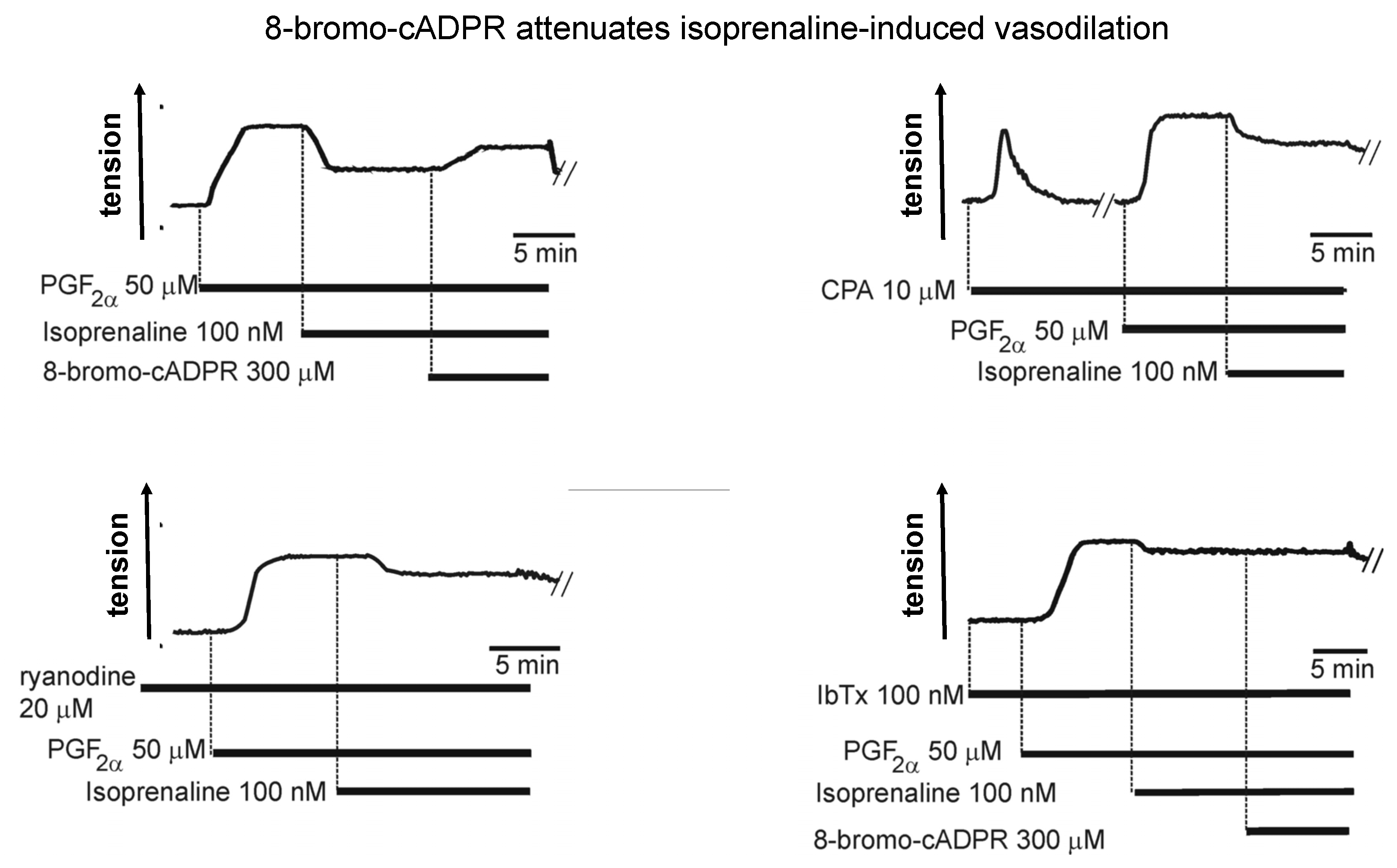
- Boittin, F.X.; Dipp, M.; Kinnear, N.P.; Galione, A.; Evans, A.M. Vasodilation by the calcium-mobilizing messenger cyclic adp-ribose. J. Biol. Chem. 2003, 278, 9602–9608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.M.; Osipenko, O.N.; Haworth, S.G.; Gurney, A.M. Resting potentials and potassium currents during development of pulmonary artery smooth muscle cells. Am. J. Physiol. 1998, 275, 887–899. [Google Scholar] [CrossRef]
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 1992, 262, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Wang, J.; Juhaszova, M.; Golovina, V.A.; Rubin, L.J. Molecular basis and function of voltage-gated k+ channels in pulmonary arterial smooth muscle cells. Am. J. Physiol. 1998, 274, 621–635. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based o2 sensor in rat pulmonary vasculature. Circ. Res. 1993, 73, 1100–1112. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am. J. Physiol. 1993, 264, 116–123. [Google Scholar] [CrossRef]
- Osipenko, O.N.; Evans, A.M.; Gurney, A.M. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, o2-sensing potassium current. Br. J. Pharm. 1997, 120, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.M.; Osipenko, O.N.; Gurney, A.M. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J. Physiol. 1996, 496 Pt 2, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Casteels, R.; Kitamura, K.; Kuriyama, H.; Suzuki, H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J. Physiol. 1977, 271, 41–61. [Google Scholar] [CrossRef]
- Byron, K.L.; Brueggemann, L.I. Kv7 potassium channels as signal transduction intermediates in the control of microvascular tone. Microcirculation 2018, 25, 12419. [Google Scholar] [CrossRef] [PubMed]
- Barrese, V.; Stott, J.B.; Greenwood, I.A. Kcnq-encoded potassium channels as therapeutic targets. Annu. Rev. Pharm. Toxicol. 2018, 58, 625–648. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Yu, Y.; Remillard, C.V.; Yuan, J.X. Acute hypoxia selectively inhibits kcna5 channels in pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 290, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Von Euler, U.S.; Liljestrand, G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol. Scand. 1946, 12, 301–320. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Dean, H.P. The pulmonary circulation. J. Physiol. 1894, 16, 34–158. [Google Scholar] [CrossRef]
- Bergofsky, E.H.; Haas, F.; Porcelli, R. Determination of the sensitive vascular sites from which hypoxia and hypercapnia elicit rises in pulmonary arterial pressure. Fed. Proc. 1968, 27, 1420–1425. [Google Scholar]
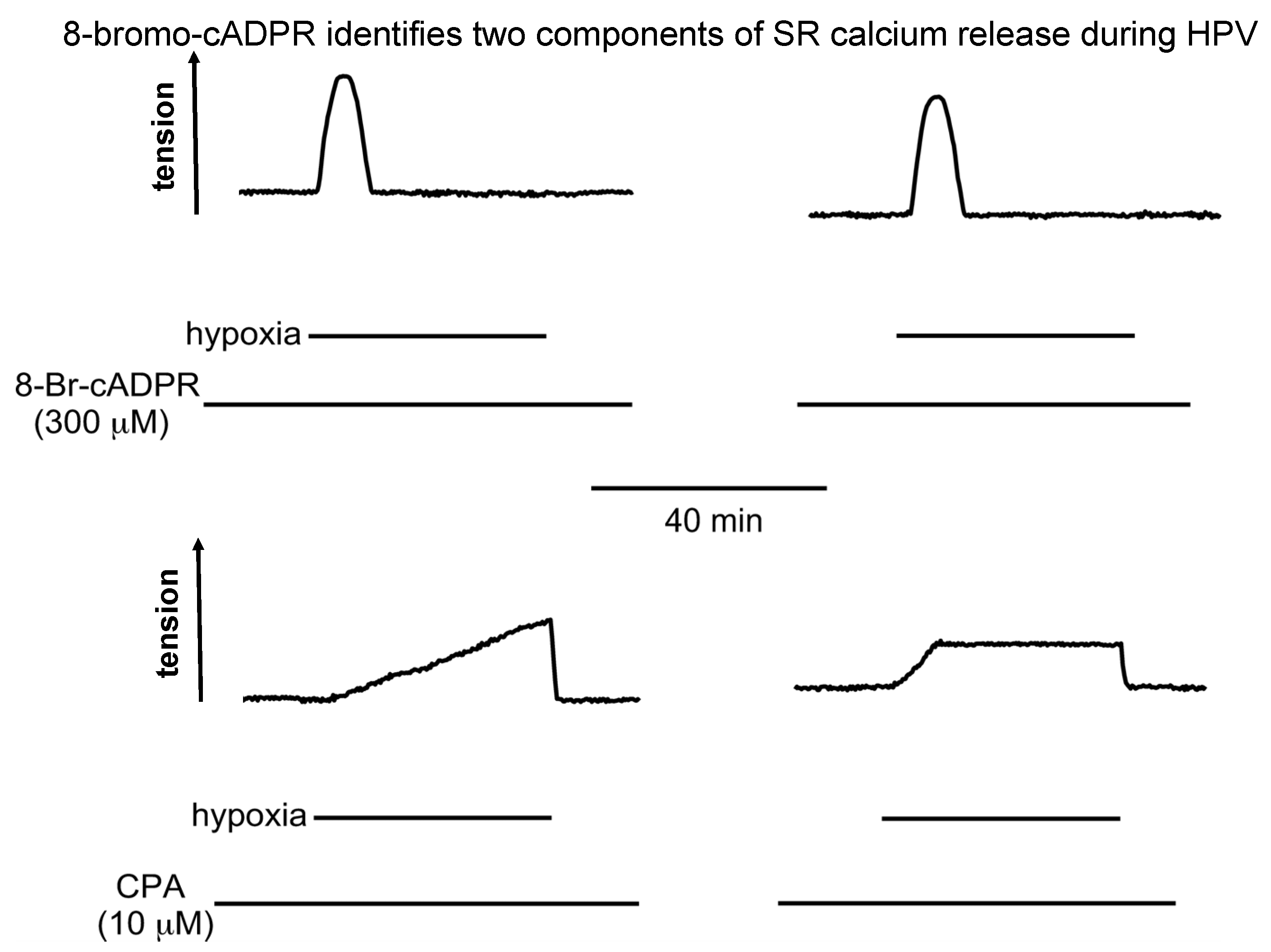
- Dipp, M.; Nye, P.C.; Evans, A.M. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L318–L325. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.P.; Robbins, P.A.; Dorrington, K.L. Release by hypoxia of a soluble vasoconstrictor from rabbit small pulmonary arteries. Br. J. Anaesth. 2003, 91, 592–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, T.P.; Ward, J.P.; Aaronson, P.I. Hypoxia induces the release of a pulmonary-selective, Ca2+-sensitising, vasoconstrictor from the perfused rat lung. Cardiovasc. Res. 2001, 50, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Gaine, S.P.; Hales, M.A.; Flavahan, N.A. Hypoxic pulmonary endothelial cells release a diffusible contractile factor distinct from endothelin. Am. J. Physiol. 1998, 274, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Lejeune, P.; Leeman, M.; Melot, C.; Closset, J. Pulmonary vascular responses to surgical chemodenervation and chemical sympathectomy in dogs. J. Appl. Physiol. 1989, 66, 42–50. [Google Scholar] [CrossRef]
- Robin, E.D.; Theodore, J.; Burke, C.M.; Oesterle, S.N.; Fowler, M.B.; Jamieson, S.W.; Baldwin, J.C.; Morris, A.J.; Hunt, S.A.; Vankessel, A.; et al. Hypoxic pulmonary vasoconstriction persists in the human transplanted lung. Clin. Sci. 1987, 72, 283–287. [Google Scholar] [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial complex IV subunit 4 isoform 2 is essential for acute pulmonary oxygen sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef]
- Evans, A.M.; Hardie, D.G. Ampk and the need to breathe and feed: What’s the matter with oxygen? Int. J. Mol. Sci. 2020, 21, 3518. [Google Scholar] [CrossRef]
- Evans, A.M.; Mustard, K.J.; Wyatt, C.N.; Peers, C.; Dipp, M.; Kumar, P.; Kinnear, N.P.; Hardie, D.G. Does amp-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in o2-sensing cells? J. Biol. Chem. 2005, 280, 41504–41511. [Google Scholar] [CrossRef] [Green Version]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The lkb1-ampk-alpha1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci. Signal. 2018, 11, eaau0296. [Google Scholar] [CrossRef] [Green Version]
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. Amp-activated protein kinase inhibits kv 1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J. Physiol. 2016, 594, 4901–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, T.P.; Aaronson, P.I.; Ward, J.P. Hypoxic vasoconstriction and intracellular ca2+ in pulmonary arteries: Evidence for pkc-independent Ca2+ sensitization. Am. J. Physiol. 1995, 268, 301–307. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Harabin, A.L.; Peake, M.D.; Frank, R.S. Vasodilator and constrictor responses to hypoxia in isolated pig lungs. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1980, 49, 820–825. [Google Scholar] [CrossRef] [PubMed]
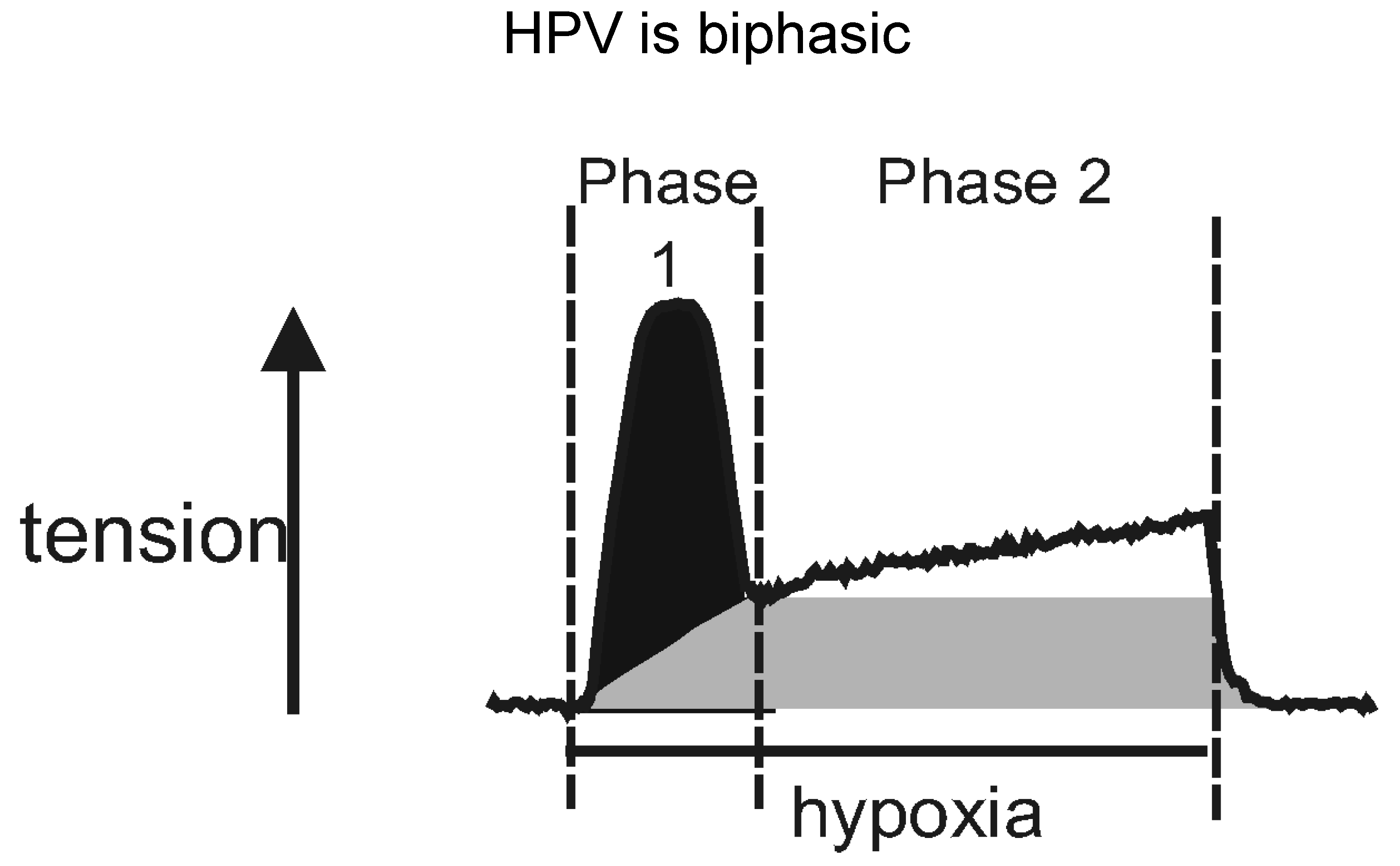
- Bennie, R.E.; Packer, C.S.; Powell, D.R.; Jin, N.; Rhoades, R.A. Biphasic contractile response of pulmonary artery to hypoxia. Am. J. Physiol. 1991, 261, 156–163. [Google Scholar] [CrossRef]
- Evans, A.M.; Dipp, M. Hypoxic pulmonary vasoconstriction: Cyclic adenosine diphosphate-ribose, smooth muscle Ca2+ stores and the endothelium. Respir. Physiol. Neurobiol. 2002, 132, 3–15. [Google Scholar] [CrossRef]
- Rubanyi, G.M.; Vanhoutte, P.M. Hypoxia releases a vasoconstrictor substance from the canine vascular endothelium. J. Physiol. 1985, 364, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Dunham-Snary, K.J.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R.A.; Archer, S.L. Ndufs2, a core subunit of mitochondrial complex i, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 2019, 124, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef]
- Connolly, M.J.; Prieto-Lloret, J.; Becker, S.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in the absence of pretone: Essential role for intracellular Ca2+ release. J. Physiol. 2013, 591, 4473–4498. [Google Scholar] [CrossRef]
- Reyes, R.V.; Castillo-Galan, S.; Hernandez, I.; Herrera, E.A.; Ebensperger, G.; Llanos, A.J. Revisiting the role of trp, orai, and asic channels in the pulmonary arterial response to hypoxia. Front. Physiol. 2018, 9, 486. [Google Scholar] [CrossRef] [Green Version]
- Salvaterra, C.G.; Goldman, W.F. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. Am. J. Physiol. 1993, 264, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Gelband, C.H.; Gelband, H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation 1997, 96, 3647–3654. [Google Scholar] [CrossRef]
- Jabr, R.I.; Toland, H.; Gelband, C.H.; Wang, X.X.; Hume, J.R. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. Br. J. Pharm. 1997, 122, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, Y.; Obara, H.; Kusunoki, M.; Fujii, Y.; Iwai, S. Hypoxic contractile response in isolated human pulmonary artery: Role of calcium ion. J. Appl. Physiol. 1988, 65, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, J.P.; Jones, L.R.; Hathaway, D.R.; Henry, B.G.; Watanabe, A.M. Beta-adrenergic stimulation of phospholamban phosphorylation and ca2+-atpase activity in guinea pig ventricles. J. Biol. Chem. 1983, 258, 464–471. [Google Scholar]
- Raeymaekers, L.; Eggermont, J.A.; Wuytack, F.; Casteels, R. Effects of cyclic nucleotide dependent protein kinases on the endoplasmic reticulum ca2+ pump of bovine pulmonary artery. Cell Calcium 1990, 11, 261–268. [Google Scholar] [CrossRef]
- Janiak, R.; Wilson, S.M.; Montague, S.; Hume, J.R. Heterogeneity of calcium stores and elementary release events in canine pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2001, 280, 22–33. [Google Scholar] [CrossRef]
- Li, Y.; Kranias, E.G.; Mignery, G.A.; Bers, D.M. Protein kinase a phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2002, 90, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.; Lehmann, G.; Serebryakov, V.N.; Mewes, H.; Hopp, H.H. Camp-dependent protein kinase is in an active state in rat small arteries possessing a myogenic tone. Am. J. Physiol. 1999, 277, 1145–1155. [Google Scholar] [CrossRef]
- Bolton, T.B.; Imaizumi, Y. Spontaneous transient outward currents in smooth muscle cells. Cell Calcium 1996, 20, 141–152. [Google Scholar] [CrossRef]
- Nazer, M.A.; Van Breemen, C. A role for the sarcoplasmic reticulum in Ca2+ extrusion from rabbit inferior vena cava smooth muscle. Am. J. Physiol. 1998, 274, 123–131. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Olson, M.L. A single luminally continuous sarcoplasmic reticulum with apparently separate ca2+ stores in smooth muscle. J. Biol. Chem. 2008, 283, 7206–7218. [Google Scholar] [CrossRef] [Green Version]
- Verboomen, H.; Wuytack, F.; De Smedt, H.; Himpens, B.; Casteels, R. Functional difference between serca2a and serca2b Ca2+ pumps and their modulation by phospholamban. Biochem. J. 1992, 286, 591–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.M.; Kessler, P.D.; Sagara, Y.; Inesi, G.; Fambrough, D.M. Nucleotide sequences of avian cardiac and brain sr/er Ca2+-atpases and functional comparisons with fast twitch Ca2+-atpase. Calcium affinities and inhibitor effects. J. Biol. Chem. 1991, 266, 16050–16055. [Google Scholar] [PubMed]
- Iino, M.; Kobayashi, T.; Endo, M. Use of ryanodine for functional removal of the calcium store in smooth muscle cells of the guinea-pig. Biochem. Biophys. Res. Commun. 1988, 152, 417–422. [Google Scholar] [CrossRef]
- Shima, H.; Blaustein, M.P. Modulation of evoked contractions in rat arteries by ryanodine, thapsigargin, and cyclopiazonic acid. Circ. Res. 1992, 70, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Golovina, V.A.; Blaustein, M.P. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science 1997, 275, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Tribe, R.M.; Borin, M.L.; Blaustein, M.P. Functionally and spatially distinct ca2+ stores are revealed in cultured vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 1994, 91, 5908–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovina, V.A.; Bambrick, L.L.; Yarowsky, P.J.; Krueger, B.K.; Blaustein, M.P. Modulation of two functionally distinct ca2+ stores in astrocytes: Role of the plasmalemmal na/ca exchanger. Glia 1996, 16, 296–305. [Google Scholar] [CrossRef]
- Thompson, B.T.; Hassoun, P.M.; Kradin, R.L.; Hales, C.A. Acute and chronic hypoxic pulmonary hypertension in guinea pigs. J. Appl. Physiol. 1989, 66, 920–928. [Google Scholar] [CrossRef]
- Ethier, M.F.; Yamaguchi, H.; Madison, J.M. Effects of cyclopiazonic acid on cytosolic calcium in bovine airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Kajita, J.; Madison, J.M. Isoproterenol increases peripheral [Ca2+]i and decreases inner [Ca2+]i in single airway smooth muscle cells. Am. J. Physiol. 1995, 268, 771–779. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar]
- Walseth, T.F.; Aarhus, R.; Zeleznikar, R.J., Jr.; Lee, H.C. Determination of endogenous levels of cyclic adp-ribose in rat tissues. Biochim. Biophys. Acta 1991, 1094, 113–120. [Google Scholar] [CrossRef]
- Galione, A.; Lee, H.C.; Busa, W.B. Ca2+-induced ca2+ release in sea urchin egg homogenates: Modulation by cyclic adp-ribose. Science 1991, 253, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Sitsapesan, R.; McGarry, S.J.; Williams, A.J. Cyclic adp-ribose competes with atp for the adenine nucleotide binding site on the cardiac ryanodine receptor Ca2+-release channel. Circ. Res. 1994, 75, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, E.N.; Dousa, T.P. Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (naadp), a Ca2+-releasing agonist, in rat tissues. Biochem. Biophys. Res. Commun. 1995, 209, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.S.; Fenton, A.M.; Prakash, Y.S.; Sieck, G.C. Cyclic adp-ribose stimulates sarcoplasmic reticulum calcium release in porcine coronary artery smooth muscle. Am. J. Physiol. 1996, 270, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, L.G.; Bak, J.; Chu, A. Cyclic adp-ribose as an endogenous regulator of the non-skeletal type ryanodine receptor ca2+ channel. Nature 1993, 364, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, S.; Galione, A.; Ashamu, G.A.; Potter, B.V.; Terrar, D.A. A specific cyclic adp-ribose antagonist inhibits cardiac excitation-contraction coupling. Curr. Biol. 1996, 6, 989–996. [Google Scholar] [CrossRef]
- Wilson, H.L.; Dipp, M.; Thomas, J.M.; Lad, C.; Galione, A.; Evans, A.M. Adp-ribosyl cyclase and cyclic adp-ribose hydrolase act as a redox sensor. A primary role for cyclic adp-ribose in hypoxic pulmonary vasoconstriction. J. Biol. Chem. 2001, 276, 11180–11188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Barnes, A.; Katz, R.L. Neuromuscular sensitivity to tubocurarine. A comparison of 10 parameters. Br. J. Anaesth. 1976, 48, 1045–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Chen, D.; Katz, R.L. Characteristics of nondepolarizing neuromuscular block: (i) post-junctional block by alpha-bungarotoxin. Can. Anaesth. Soc. J. 1977, 24, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, B.; Miledi, R. A re-examination of curare action at the motor endplate. Proc. R. Soc. Lond. B. Biol. Sci. 1978, 203, 119–133. [Google Scholar]
- Del Castillo, J.; Katz, B. Quantal components of the end-plate potential. J. Physiol. 1954, 124, 560–573. [Google Scholar] [CrossRef]
- Colquhoun, D.; Dreyer, F.; Sheridan, R.E. The actions of tubocurarine at the frog neuromuscular junction. J. Physiol. 1979, 293, 247–284. [Google Scholar] [CrossRef]
- Del Castillo, J.; Katz, B. Localization of active spots within the neuromuscular junction of the frog. J. Physiol. 1956, 132, 630–649. [Google Scholar] [CrossRef]
- Fatt, P.; Katz, B. Membrane potentials at the motor end-plate. J. Physiol. 1950, 111, 46–47. [Google Scholar]
- Fatt, P.; Katz, B. An analysis of the end-plate potential recorded with an intracellular electrode. J. Physiol. 1951, 115, 320–370. [Google Scholar] [CrossRef]
- Eggermont, J.A.; Wuytack, F.; Verbist, J.; Casteels, R. Expression of endoplasmic-reticulum Ca2+-pump isoforms and of phospholamban in pig smooth-muscle tissues. Biochem. J. 1990, 271, 649–653. [Google Scholar] [CrossRef] [Green Version]
- Herrmann-Frank, A.; Darling, E.; Meissner, G. Functional characterization of the Ca2+-gated Ca2+ release channel of vascular smooth muscle sarcoplasmic reticulum. Pflug. Arch. 1991, 418, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Neylon, C.B.; Richards, S.M.; Larsen, M.A.; Agrotis, A.; Bobik, A. Multiple types of ryanodine receptor/Ca2+ release channels are expressed in vascular smooth muscle. Biochem. Biophys. Res. Commun. 1995, 215, 814–821. [Google Scholar] [CrossRef]
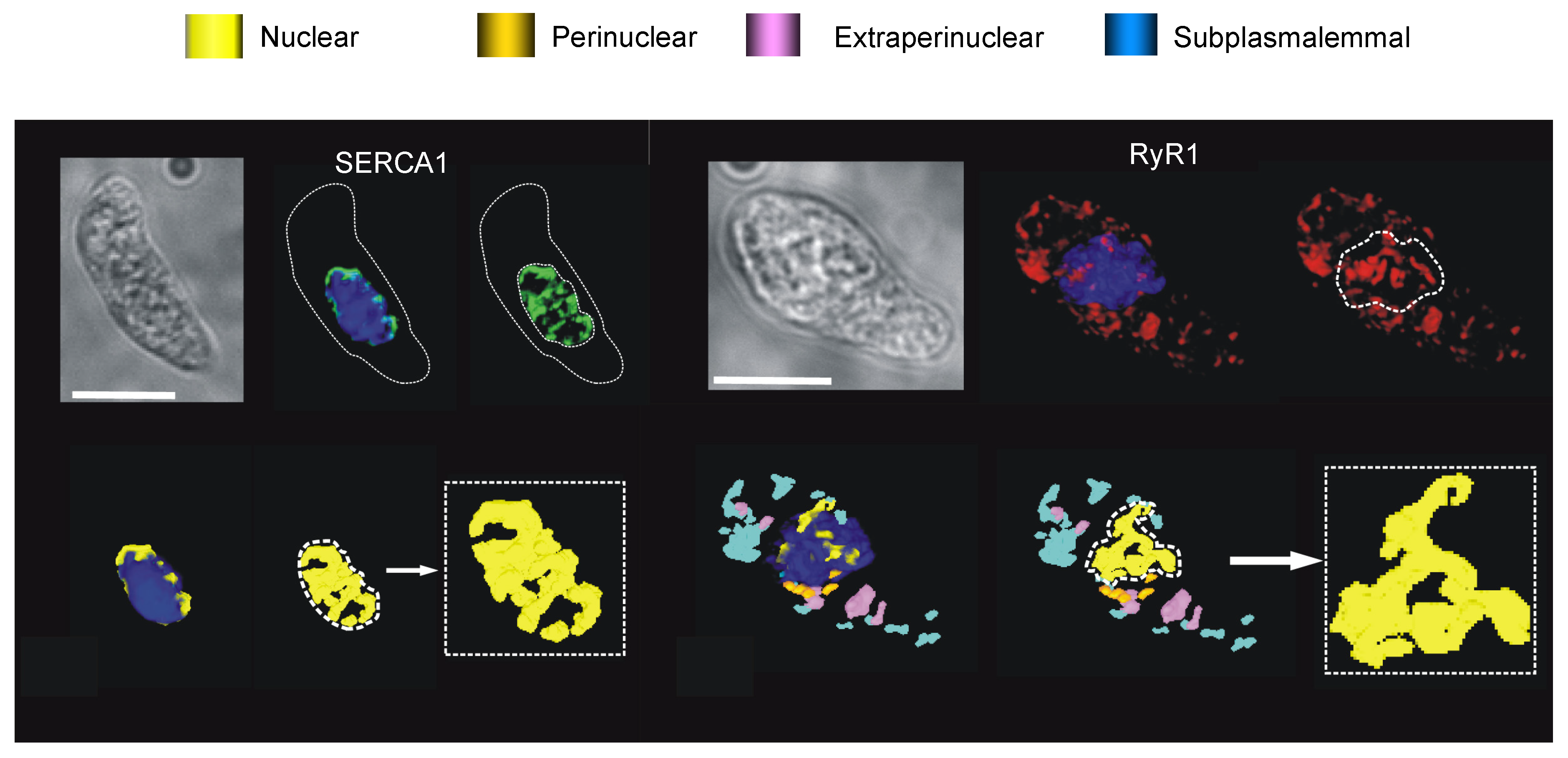
- Clark, J.H.; Kinnear, N.P.; Kalujnaiab, S.; Cramb, G.; Fleischer, S.; Jeyakumar, L.H.; Wuytack, F.; Evans, A.M. Identification of functionally segregated sarcoplasmic reticulum calcium stores in pulmonary arterial smooth muscle. J. Biol. Chem. 2010, 285, 13542–13549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, R.C., Jr.; Banerjee, A.; Hahn, A.R.; Agrafojo, J.; Fullerton, D.A. Selective inhibition of cyclic adenosine monophosphate-mediated pulmonary vasodilation by acute hypoxia. Surgery 1995, 117, 314–318. [Google Scholar] [CrossRef]
- Morio, Y.; McMurtry, I.F. Ca2+ release from ryanodine-sensitive store contributes to mechanism of hypoxic vasoconstriction in rat lungs. J. Appl. Physiol. 2002, 92, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, T.P.; Mustard, K.J.; Lewis, T.H.; Clark, J.H.; Wyatt, C.N.; Blanco, E.A.; Peers, C.; Hardie, D.G.; Evans, A.M. Amp-activated protein kinase and hypoxic pulmonary vasoconstriction. Eur. J. Pharm. 2008, 595, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.R.; Lin, M.J.; Yip, K.P.; Jeyakumar, L.H.; Fleischer, S.; Leung, G.P.; Sham, J.S. Multiple ryanodine receptor subtypes and heterogeneous ryanodine receptor-gated Ca2+ stores in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, 338–348. [Google Scholar] [CrossRef] [Green Version]
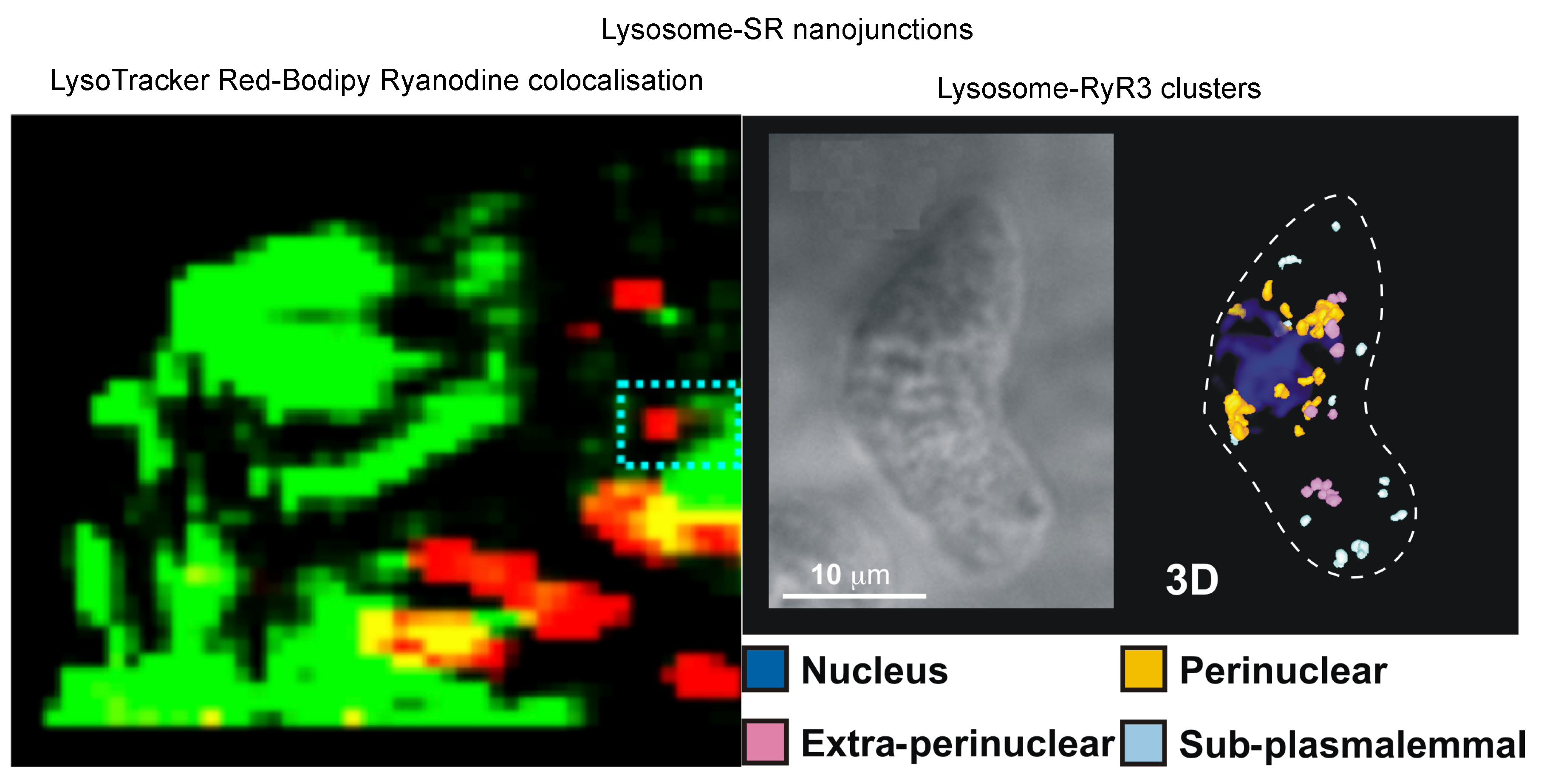
- Kinnear, N.P.; Wyatt, C.N.; Clark, J.H.; Calcraft, P.J.; Fleischer, S.; Jeyakumar, L.H.; Nixon, G.F.; Evans, A.M. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by naadp in rat pulmonary arterial smooth muscle. Cell Calcium 2008, 44, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Ogunbayo, O.A.; Zhu, Y.; Rossi, D.; Sorrentino, V.; Ma, J.; Zhu, M.X.; Evans, A.M. Cyclic adenosine diphosphate ribose activates ryanodine receptors, whereas naadp activates two-pore domain channels. J. Biol. Chem. 2011, 286, 9136–9140. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Galione, A.; Terrar, D.A. Effects of photoreleased cadp-ribose on calcium transients and calcium sparks in myocytes isolated from guinea-pig and rat ventricle. Biochem. J. 1999, 342, 269–273. [Google Scholar] [CrossRef]
- Li, P.; Chen, S.R. Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. 2001, 118, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.R.; Li, X.; Ebisawa, K.; Zhang, L. Functional characterization of the recombinant type 3 ca2+ release channel (ryanodine receptor) expressed in hek293 cells. J. Biol. Chem. 1997, 272, 24234–24246. [Google Scholar] [CrossRef] [Green Version]
- Rios, E. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 2018, 150, 521–537. [Google Scholar] [CrossRef] [Green Version]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeller, C.; Crossman, D.; Gilbert, R.; Cannell, M.B. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 14958–14963. [Google Scholar] [CrossRef] [Green Version]
- Kinnear, N.P.; Boittin, F.X.; Thomas, J.M.; Galione, A.; Evans, A.M. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004, 279, 54319–54326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boittin, F.X.; Galione, A.; Evans, A.M. Nicotinic acid adenine dinucleotide phosphate mediates Ca2+ signals and contraction in arterial smooth muscle via a two-pool mechanism. Circ. Res. 2002, 91, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Churchill, G.C.; Galione, A. Naadp induces Ca2+ oscillations via a two-pool mechanism by priming ip3- and cadpr-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. Naadp mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Aarhus, R. A derivative of nadp mobilizes calcium stores insensitive to inositol trisphosphate and cyclic adp-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Calcraft, P.J.; Ogunbayo, O.; Ma, J.; Galione, A.; Churchill, G.C.; Zhu, M.X.; Evans, A.M. Does nicotinic acid adenine dinucletoide phosphate elicit Ca2+ release via two-pore channel 2 in rat pulmonary arterial smooth muscle cells. Proc. Physiol. Soc. 2008, 13, 18. [Google Scholar]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. Naadp mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.X.; Ma, J.; Parrington, J.; Calcraft, P.J.; Galione, A.; Evans, A.M. Calcium signaling via two-pore channels: Local or global, that is the question. Am. J. Physiol. Cell Physiol. 2010, 298, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Galione, A. Naadp receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035071. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in naadp-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Gordienko, D.V.; Greenwood, I.A.; Bolton, T.B. Direct visualization of sarcoplasmic reticulum regions discharging Ca2+ sparks in vascular myocytes. Cell Calcium 2001, 29, 13–28. [Google Scholar] [CrossRef]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mtorc2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paddenberg, R.; Stieger, P.; von Lilien, A.L.; Faulhammer, P.; Goldenberg, A.; Tillmanns, H.H.; Kummer, W.; Braun-Dullaeus, R.C. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir. Res. 2007, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Thakore, P.; Pritchard, H.A.T.; Griffin, C.S.; Yamasaki, E.; Drumm, B.T.; Lane, C.; Sanders, K.M.; Feng Earley, Y.; Earley, S. Trpml1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci. Signal. 2020, 13, eaba1015. [Google Scholar] [CrossRef]
- Bhat, O.M.; Li, G.; Yuan, X.; Huang, D.; Gulbins, E.; Kukreja, R.C.; Li, P.L. Arterial medial calcification through enhanced small extracellular vesicle release in smooth muscle-specific asah1 gene knockout mice. Sci. Rep. 2020, 10, 1645. [Google Scholar] [CrossRef]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. Trpml1: The ca(2+) retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular trpml1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lu, W.; Du, J. Architecture of the trpm2 channel and its activation mechanism by adp-ribose and calcium. Nature 2018, 562, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Penner, R. Trpm2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. Mtorc1 controls lysosomal Ca2+ release through the two-pore channel tpc2. Sci. Signal. 2018, 11, eaao5775. [Google Scholar] [CrossRef] [Green Version]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Diaz, M.C.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis iv Ca2+ channel trpml1 (mcoln1) is regulated by the tor kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.P.; Quignard, J.F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Lifshitz, L.M.; Carmichael, J.D.; Lai, F.A.; Sorrentino, V.; Bellve, K.; Fogarty, K.E.; ZhuGe, R. Spatial organization of ryrs and bk channels underlying the activation of stocs by Ca2+ sparks in airway myocytes. J. Gen. Physiol. 2011, 138, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Vaithianathan, T.; Narayanan, D.; Asuncion-Chin, M.T.; Jeyakumar, L.H.; Liu, J.; Fleischer, S.; Jaggar, J.H.; Dopico, A.M. Subtype identification and functional characterization of ryanodine receptors in rat cerebral artery myocytes. Am. J. Physiol. Cell Physiol. 2010, 299, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Frazier, M.; McMahon, T.J.; Eu, J.P. Redox activation of intracellular calcium release channels (ryanodine receptors) in the sustained phase of hypoxia-induced pulmonary vasoconstriction. Chest 2005, 128, 556–558. [Google Scholar] [CrossRef]
- Li, X.Q.; Zheng, Y.M.; Rathore, R.; Ma, J.; Takeshima, H.; Wang, Y.X. Genetic evidence for functional role of ryanodine receptor 1 in pulmonary artery smooth muscle cells. Pflug. Arch. 2009, 457, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Song, T.; Truong, L.; Reyes-Garcia, J.; Wang, L.; Zheng, Y.M.; Wang, Y.X. Important role of sarcoplasmic reticulum Ca2+ release via ryanodine receptor-2 channel in hypoxia-induced rieske iron-sulfur protein-mediated mitochondrial reactive oxygen species generation in pulmonary artery smooth muscle cells. Antioxid. Redox. Signal. 2020, 32, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.M.; Wang, Q.S.; Rathore, R.; Zhang, W.H.; Mazurkiewicz, J.E.; Sorrentino, V.; Singer, H.A.; Kotlikoff, M.I.; Wang, Y.X. Type-3 ryanodine receptors mediate hypoxia-, but not neurotransmitter-induced calcium release and contraction in pulmonary artery smooth muscle cells. J. Gen. Physiol. 2005, 125, 427–440. [Google Scholar] [CrossRef] [Green Version]
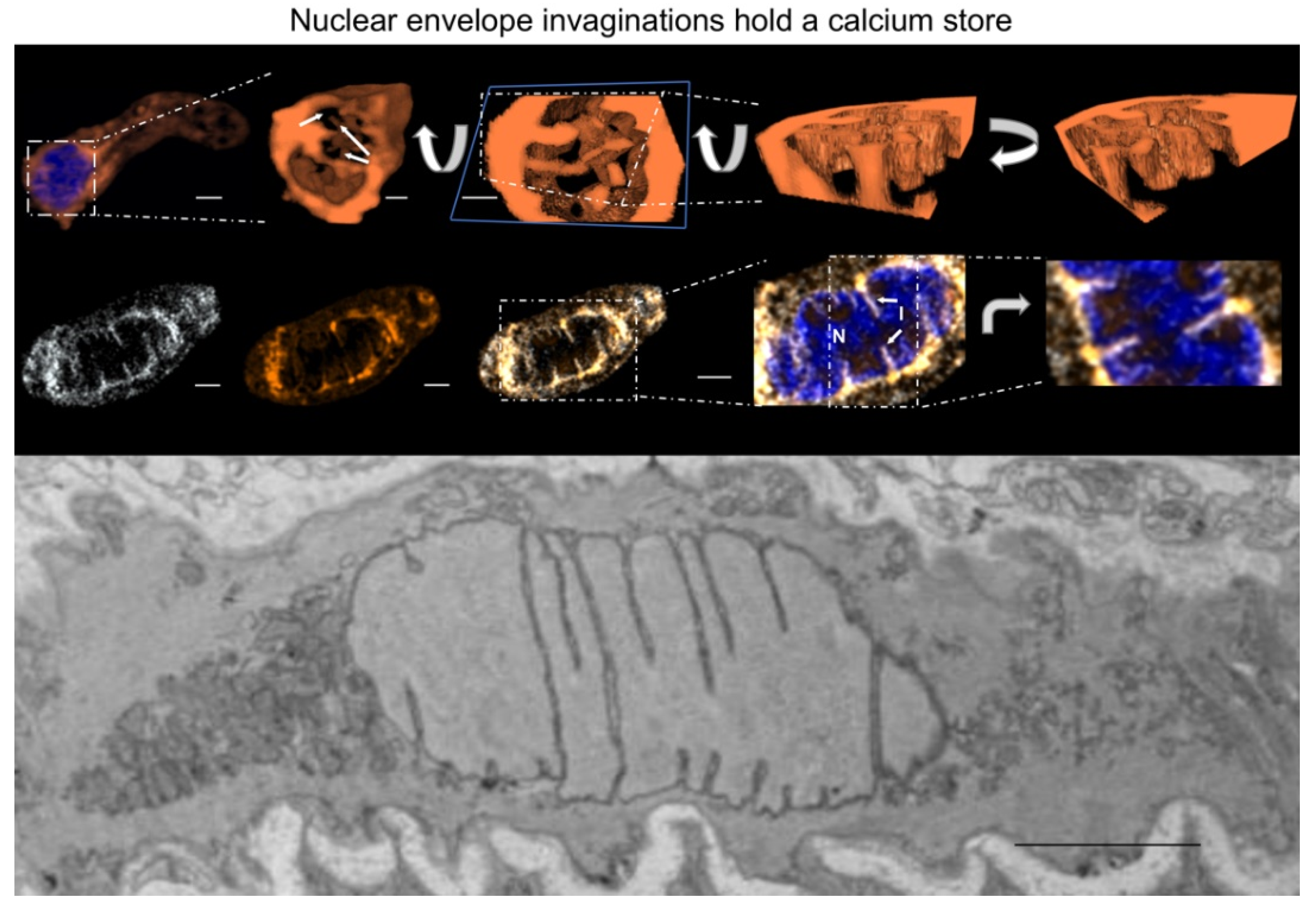
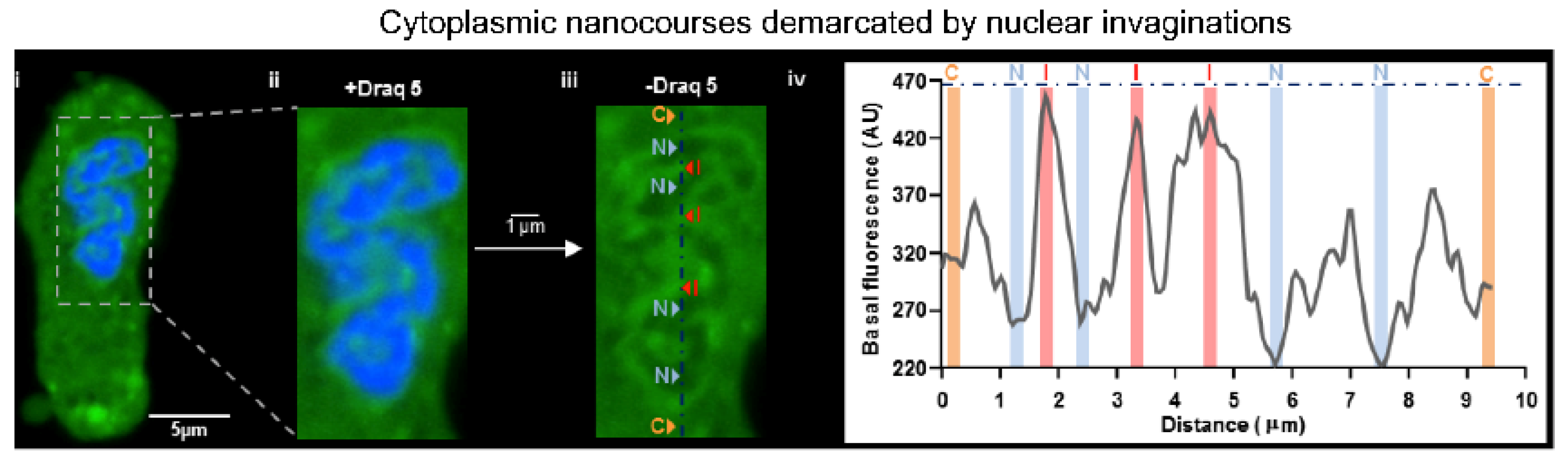
- Duan, J.; Navarro-Dorado, J.; Clark, J.H.; Kinnear, N.P.; Meinke, P.; Schirmer, E.C.; Evans, A.M. The cell-wide web coordinates cellular processes by directing site-specific Ca2+ flux across cytoplasmic nanocourses. Nat. Commun. 2019, 10, 2299. [Google Scholar] [CrossRef] [Green Version]
- Van Breemen, C.; Chen, Q.; Laher, I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharm. Sci. 1995, 16, 98–105. [Google Scholar] [CrossRef]
- Poburko, D.; Kuo, K.H.; Dai, J.; Lee, C.H.; van Breemen, C. Organellar junctions promote targeted ca2+ signaling in smooth muscle: Why two membranes are better than one. Trends Pharm. Sci. 2004, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Fameli, N.; Ogunbayo, O.A.; van Breemen, C.; Evans, A.M. Cytoplasmic nanojunctions between lysosomes and sarcoplasmic reticulum are required for specific calcium signaling. F1000Research 2014, 3, 93. [Google Scholar] [CrossRef]
- Fameli, N.; van Breemen, C.; Kuo, K.H. A quantitative model for linking Na+/Ca2+ exchanger to serca during refilling of the sarcoplasmic reticulum to sustain [Ca2+] oscillations in vascular smooth muscle. Cell Calcium 2007, 42, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Smooth muscle cell calcium activation mechanisms. J. Physiol. 2008, 586, 5047–5061. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Hirose, K.; Kadowaki, S.; Iino, M. Allosteric regulation by cytoplasmic Ca2+ and ip3 of the gating of ip3 receptors in permeabilized guinea-pig vascular smooth muscle cells. J. Physiol. 1998, 506 Pt 2, 407–414. [Google Scholar] [CrossRef]
- Grayson, T.H.; Haddock, R.E.; Murray, T.P.; Wojcikiewicz, R.J.; Hill, C.E. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium 2004, 36, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Westcott, E.B.; Goodwin, E.L.; Segal, S.S.; Jackson, W.F. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J. Physiol. 2012, 590, 1849–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C. Multiplicity of Ca2+ messengers and Ca2+ stores: A perspective from cyclic adp-ribose and naadp. Curr. Mol. Med. 2004, 4, 227–237. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Ma, J.; Zhu, M.X.; Evans, A.M. Lysosome-er coupling supported by two pore channel 2 is required for nicotinic acid adenine dinucleotide phosphate-induced global calcium waves in pulmonary arterial myocytes. Proc. Physiol. Soc. 2015, 34, 134. [Google Scholar]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. Tpc proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. Mtor regulates lysosomal atp-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Saunter, C.; Wilson, C.; Girkin, J.M.; Chalmers, S. Mitochondria structure and position in the local control of calcium signals in smooth muscle cells. In Signal Transduction and Smooth Muscle; Trebak, M., Earley, S., Eds.; Taylor & Francis Group: Abingdon, UK, 2018; pp. 173–190. [Google Scholar]
- Zhao, G.; Neeb, Z.P.; Leo, M.D.; Pachuau, J.; Adebiyi, A.; Ouyang, K.; Chen, J.; Jaggar, J.H. Type 1 ip3 receptors activate bkca channels via local molecular coupling in arterial smooth muscle cells. J. Gen. Physiol. 2010, 136, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Dabertrand, F.; Nelson, M.T.; Brayden, J.E. Acidosis dilates brain parenchymal arterioles by conversion of calcium waves to sparks to activate bk channels. Circ. Res. 2012, 110, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Tumelty, J.; Scholfield, N.; Stewart, M.; Curtis, T.; McGeown, G. Ca2+-sparks constitute elementary building blocks for global Ca2+-signals in myocytes of retinal arterioles. Cell Calcium 2007, 41, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Kur, J.; Bankhead, P.; Scholfield, C.N.; Curtis, T.M.; McGeown, J.G. Ca2+ sparks promote myogenic tone in retinal arterioles. Br. J. Pharm. 2013, 168, 1675–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzini-Armstrong, C.; Ferguson, D.G. Density and disposition of Ca2+-atpase in sarcoplasmic reticulum membrane as determined by shadowing techniques. Biophys. J. 1985, 48, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Franzini-Armstrong, C.; Protasi, F. Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol. Rev. 1997, 77, 699–729. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, V.; Sharma, V.K.; Sheu, S.S.; Franzini-Armstrong, C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann. N. Y. Acad. Sci. 1998, 853, 341–344. [Google Scholar] [CrossRef]
- Rosenbluth, J. Subsurface cisterns and their relationship to the neuronal plasma membrane. J. Cell Biol. 1962, 13, 405–421. [Google Scholar] [CrossRef]
- Feldmeyer, D.; Melzer, W.; Pohl, B.; Zollner, P. Fast gating kinetics of the slow Ca2+ current in cut skeletal muscle fibres of the frog. J. Physiol. 1990, 425, 347–367. [Google Scholar] [CrossRef]
- Ma, J.; Gonzalez, A.; Chen, R. Fast activation of dihydropyridine-sensitive calcium channels of skeletal muscle. Multiple pathways of channel gating. J. Gen. Physiol. 1996, 108, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Nakai, J.; Dirksen, R.T.; Nguyen, H.T.; Pessah, I.N.; Beam, K.G.; Allen, P.D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 1996, 380, 72–75. [Google Scholar] [CrossRef]
- Fameli, N.; Kuo, K.H.; van Breemen, C. A model for the generation of localized transient [Na+] elevations in vascular smooth muscle. Biochem. Biophys. Res. Commun. 2009, 389, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabella, G. Caveolae intracellulares and sarcoplasmic reticulum in smooth muscle. J. Cell Sci. 1971, 8, 601–609. [Google Scholar] [PubMed]
- Devine, C.E.; Somlyo, A.V.; Somlyo, A.P. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. J. Cell Biol. 1972, 52, 690–718. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, C.; Saida, K. Cellular mechanisms regulating [Ca2+]i smooth muscle. Annu. Rev. Physiol. 1989, 51, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, C. Calcium requirement for activation of intact aortic smooth muscle. J. Physiol. 1977, 272, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deth, R.; van Breemen, C. Agonist induced release of intracellular Ca2+ in the rabbit aorta. J. Membr. Biol. 1977, 30, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Rembold, C.M.; Chen, X.L. The buffer barrier hypothesis, [Ca2+]i homogeneity, and sarcoplasmic reticulum function in swine carotid artery. J. Physiol. 1998, 513 Pt 2, 477–492. [Google Scholar] [CrossRef]
- White, C.; McGeown, J.G. Ca2+ uptake by the sarcoplasmic reticulum decreases the amplitude of depolarization-dependent [Ca2+]i transients in rat gastric myocytes. Pflug. Arch. 2000, 440, 488–495. [Google Scholar] [CrossRef]
- Yoshikawa, A.; van Breemen, C.; Isenberg, G. Buffering of plasmalemmal Ca2+ current by sarcoplasmic reticulum of guinea pig urinary bladder myocytes. Am. J. Physiol. 1996, 271, 833–841. [Google Scholar] [CrossRef]
- Young, R.C.; Schumann, R.; Zhang, P. Intracellular calcium gradients in cultured human uterine smooth muscle: A functionally important subplasmalemmal space. Cell Calcium 2001, 29, 183–189. [Google Scholar] [CrossRef]
- Shmigol, A.V.; Eisner, D.A.; Wray, S. The role of the sarcoplasmic reticulum as a ca2+ sink in rat uterine smooth muscle cells. J. Physiol. 1999, 520 Pt 1, 153–163. [Google Scholar] [CrossRef]
- Nazer, M.A.; van Breemen, C. Functional linkage of na(+)-Ca2+ exchange and sarcoplasmic reticulum ca2+ release mediates Ca2+ cycling in vascular smooth muscle. Cell Calcium 1998, 24, 275–283. [Google Scholar] [CrossRef]
- Spinelli, A.M.; Trebak, M. Orai channel-mediated ca2+ signals in vascular and airway smooth muscle. Am. J. Physiol. Cell Physiol. 2016, 310, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, M.; Hollinshead, M.; White, N.; Vaux, D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J. Cell Biol. 1997, 136, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, B.D.; Eng, D.L.; Kocsis, J.D. Intranuclear Ca2+ transients during neurite regeneration of an adult mammalian neuron. Proc. Natl. Acad. Sci. USA 1992, 89, 7978–7982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bkaily, G.; Nader, M.; Avedanian, L.; Choufani, S.; Jacques, D.; D’Orleans-Juste, P.; Gobeil, F.; Chemtob, S.; Al-Khoury, J. G-protein-coupled receptors, channels, and na+-h+ exchanger in nuclear membranes of heart, hepatic, vascular endothelial, and smooth muscle cells. Can. J. Physiol. Pharm. 2006, 84, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef]
- Gordienko, D.V.; Bolton, T.B.; Cannell, M.B. Variability in spontaneous subcellular calcium release in guinea-pig ileum smooth muscle cells. J. Physiol. 1998, 507 Pt 3, 707–720. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, J. Highly cooperative and hysteretic response of the skeletal muscle ryanodine receptor to changes in proton concentrations. Biophys. J. 1994, 67, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.; Saunter, C.; Wilson, C.; Coats, P.; Girkin, J.M.; McCarron, J.G. Mitochondrial motility and vascular smooth muscle proliferation. Arter. Thromb. Vasc. Biol. 2012, 32, 3000–3011. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.C.; Sweeney, M.; Jones, C.J.; Zhang, H.; O’Neill, S.C.; Prior, I.; Taggart, M.J. Three-dimensional electron microscopic reconstruction of intracellular organellar arrangements in vascular smooth muscle—Further evidence of nanospaces and contacts. J. Cell Mol. Med. 2009, 13, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Gurrola, G.B.; Zhang, J.; Valdivia, C.R.; SanMartin, M.; Zamudio, F.Z.; Zhang, L.; Possani, L.D.; Valdivia, H.H. Structure-function relationships of peptides forming the calcin family of ryanodine receptor ligands. J. Gen. Physiol. 2016, 147, 375–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.Q.; Wei, C.; Zhao, G.; Brochet, D.X.; Shen, J.; Song, L.S.; Wang, W.; Yang, D.; Cheng, H. Imaging microdomain Ca2+ in muscle cells. Circ. Res. 2004, 94, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Bradley, K.N.; Craig, J.W.; Muir, T.C.; McCarron, J.G. The sarcoplasmic reticulum and sarcolemma together form a passive Ca2+ trap in colonic smooth muscle. Cell Calcium 2004, 36, 29–41. [Google Scholar] [CrossRef]
- Poburko, D.; Fameli, N.; Kuo, K.H.; van Breemen, C. Ca2+ signaling in smooth muscle: Trpc6, ncx and lnats in nanodomains. Channels 2008, 2, 10–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X. Upregulated expression of stim2, trpc6, and orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Cell Physiol. 2015, 308, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.H.; Sun, H.; Paudel, O.; Lin, M.J.; Sham, J.S. Conformation of ryanodine receptor-2 gates store-operated calcium entry in rat pulmonary arterial myocytes. Cardiovasc. Res. 2016, 111, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Beard, N.A.; Sakowska, M.M.; Dulhunty, A.F.; Laver, D.R. Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys. J. 2002, 82, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Gyorke, S.; Terentyev, D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 2008, 77, 245–255. [Google Scholar] [CrossRef]
- Wang, Q.; Michalak, M. Calsequestrin. Structure, function, and evolution. Cell Calcium 2020, 90, 102242. [Google Scholar] [CrossRef]
- Gilchrist, J.S.; Belcastro, A.N.; Katz, S. Intraluminal ca2+ dependence of Ca2+ and ryanodine-mediated regulation of skeletal muscle sarcoplasmic reticulum ca2+ release. J. Biol. Chem. 1992, 267, 20850–20856. [Google Scholar] [PubMed]
- Protasi, F.; Paolini, C.; Canato, M.; Reggiani, C.; Quarta, M. Lessons from calsequestrin-1 ablation in vivo: Much more than a Ca2+ buffer after all. J. Muscle Res. Cell Motil. 2012, 32, 257–270. [Google Scholar] [CrossRef]
- Park, D.R.; Nam, T.S.; Kim, Y.W.; Lee, S.H.; Kim, U.H. Cd38-cadpr-serca signaling axis determines skeletal muscle contractile force in response to beta-adrenergic stimulation. Cell. Physiol. Biochem. 2018, 46, 2017–2030. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki-Mann, M.; Demuro, A.; Parker, I. Cadpr stimulates serca activity in xenopus oocytes. Cell Calcium 2009, 45, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki-Mann, M.; Demuro, A.; Parker, I. Modulation of er Ca2+ store filling by cadpr promotes ip3-evoked ca2+ signals. J. Biol. Chem. 2010, in press. [Google Scholar] [CrossRef]
- Moore, E.D.; Voigt, T.; Kobayashi, Y.M.; Isenberg, G.; Fay, F.S.; Gallitelli, M.F.; Franzini-Armstrong, C. Organization of ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys. J. 2004, 87, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Poburko, D.; Kuo, K.H.; Seow, C.Y.; van Breemen, C. Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.P.; Sutherland, C.; Walsh, M.P. Ca2+ activation of smooth muscle contraction: Evidence for the involvement of calmodulin that is bound to the triton insoluble fraction even in the absence of ca2+. J. Biol. Chem. 2002, 277, 2186–2192. [Google Scholar] [CrossRef] [Green Version]
- Boittin, F.X.; Macrez, N.; Halet, G.; Mironneau, J. Norepinephrine-induced Ca2+ waves depend on insp(3) and ryanodine receptor activation in vascular myocytes. Am. J. Physiol. 1999, 277, 139–151. [Google Scholar] [CrossRef]
- Boittin, F.X.; Coussin, F.; Macrez, N.; Mironneau, C.; Mironneau, J. Inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channel-dependent Ca2+ signalling in rat portal vein myocytes. Cell Calcium 1998, 23, 303–311. [Google Scholar] [CrossRef]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. Emerin and histone deacetylase 3 (hdac3) cooperatively regulate expression and nuclear positions of myod, myf5, and pax7 genes during myogenesis. Chromosome Res. 2013, 21, 765–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattout, A.; Cabianca, D.S.; Gasser, S.M. Chromatin states and nuclear organization in development—A view from the nuclear lamina. Genome Biol. 2015, 16, 174. [Google Scholar] [CrossRef] [Green Version]
- Luperchio, T.R.; Wong, X.; Reddy, K.L. Genome regulation at the peripheral zone: Lamina associated domains in development and disease. Curr. Opin. Genet. Dev. 2014, 25, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Cheedipudi, S.; Puri, D.; Saleh, A.; Gala, H.P.; Rumman, M.; Pillai, M.S.; Sreenivas, P.; Arora, R.; Sellathurai, J.; Schroder, H.D.; et al. A fine balance: Epigenetic control of cellular quiescence by the tumor suppressor prdm2/riz at a bivalent domain in the cyclin a gene. Nucleic Acids Res. 2015, 43, 6236–6256. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone h3 lysine 9 through histone methyltransferase g9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Zhou, X.; Chen, H.; Li, Q.; Costa, M. Modulation of histone methylation and mlh1 gene silencing by hexavalent chromium. Toxicol. Appl. Pharm. 2009, 237, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takama, H.; Sugiura, K.; Ogawa, Y.; Muro, Y.; Akiyama, M. Possible roles of barrier-to-autointegration factor 1 in regulation of keratinocyte differentiation and proliferation. J. Derm. Sci. 2013, 71, 100–106. [Google Scholar] [CrossRef]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.M.; Han, X.J.; Duan, T.T.; Xu, Y.D.; Wang, Y.; Ulloa, L.; Yang, Y.Q. Decreased s100a9 expression promoted rat airway smooth muscle cell proliferation by stimulating ros generation and inhibiting p38 mapk. Can. Respir J. 2016, 2016, 1462563. [Google Scholar] [CrossRef]
- Pindyurin, A.V.; Moorman, C.; de Wit, E.; Belyakin, S.N.; Belyaeva, E.S.; Christophides, G.K.; Kafatos, F.C.; van Steensel, B.; Zhimulev, I.F. Suur joins separate subsets of pcg, hp1 and b-type lamin targets in drosophila. J. Cell Sci. 2007, 120, 2344–2351. [Google Scholar] [CrossRef] [Green Version]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. Lbr and lamin a/c sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [Green Version]
- Al-Mohanna, F.A.; Caddy, K.W.; Bolsover, S.R. The nucleus is insulated from large cytosolic calcium ion changes. Nature 1994, 367, 745–750. [Google Scholar] [CrossRef]
- Qi, Y.X.; Yao, Q.P.; Huang, K.; Shi, Q.; Zhang, P.; Wang, G.L.; Han, Y.; Bao, H.; Wang, L.; Li, H.P.; et al. Nuclear envelope proteins modulate proliferation of vascular smooth muscle cells during cyclic stretch application. Proc. Natl. Acad. Sci. USA 2016, 113, 5293–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berra-Romani, R.; Mazzocco-Spezzia, A.; Pulina, M.V.; Golovina, V.A. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am. J. Physiol. Cell Physiol. 2008, 295, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, H.; Hoshijima, M.; Song, L.S. Ca2+ microdomains organized by junctophilins. Cell Calcium 2015, 58, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshima, H.; Komazaki, S.; Nishi, M.; Iino, M.; Kangawa, K. Junctophilins: A novel family of junctional membrane complex proteins. Mol. Cell 2000, 6, 11–22. [Google Scholar]
- Saeki, T.; Suzuki, Y.; Yamamura, H.; Takeshima, H.; Imaizumi, Y. A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and bkca channels in the ca(2+) microdomain of vascular smooth muscle. J. Biol. Chem. 2019, 294, 13093–13105. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Srikanth, S.; Nishi, M.; Ping, P.; Takeshima, H.; Gwack, Y. Junctophilin-4, a component of the endoplasmic reticulum-plasma membrane junctions, regulates ca2+ dynamics in t cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2762–2767. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, H.A.T.; Gonzales, A.L.; Pires, P.W.; Drumm, B.T.; Ko, E.A.; Sanders, K.M.; Hennig, G.W.; Earley, S. Microtubule structures underlying the sarcoplasmic reticulum support peripheral coupling sites to regulate smooth muscle contractility. Sci. Signal. 2017, 10, eaan2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, H.A.T.; Griffin, C.S.; Yamasaki, E.; Thakore, P.; Lane, C.; Greenstein, A.S.; Earley, S. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc. Natl. Acad. Sci. USA 2019, 116, 21874–21881. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Zorec, R. Astroglial signalling in health and disease. Neurosci. Lett. 2019, 689, 1–4. [Google Scholar] [CrossRef] [PubMed]
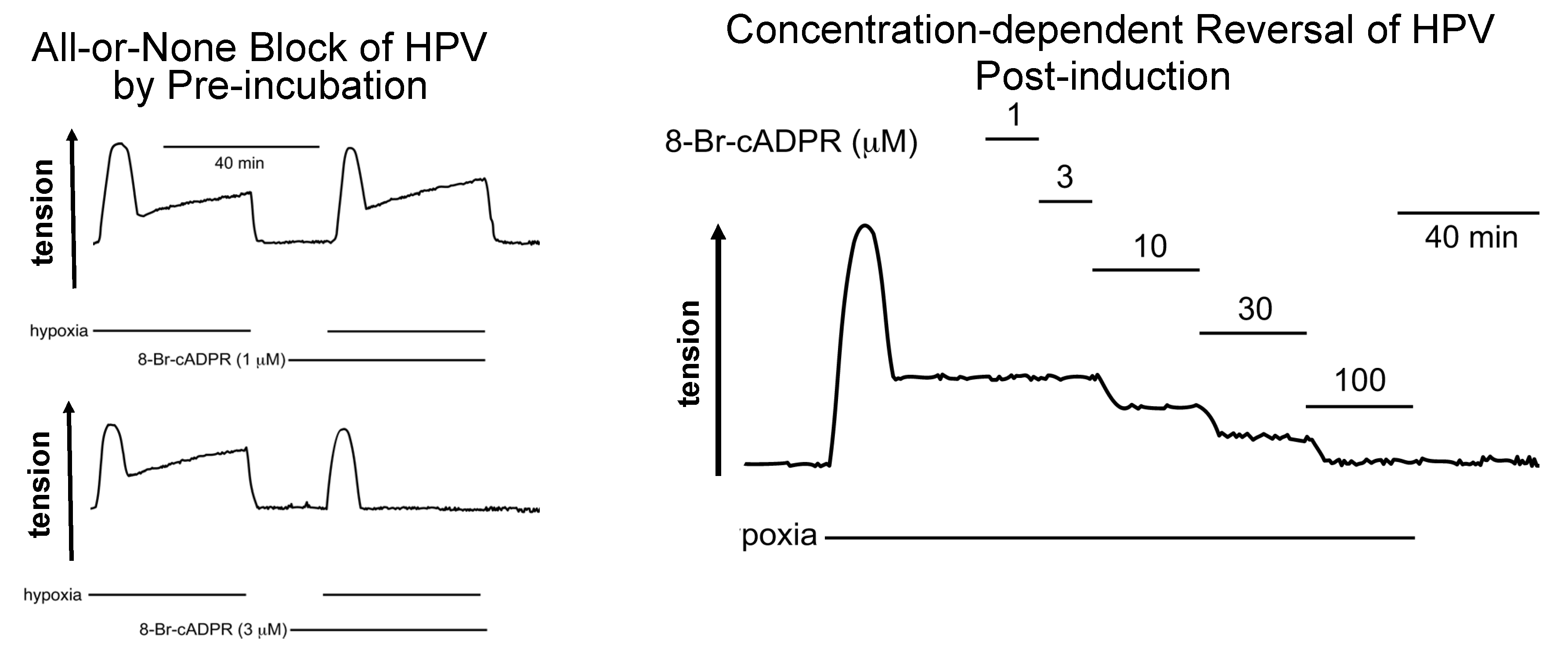
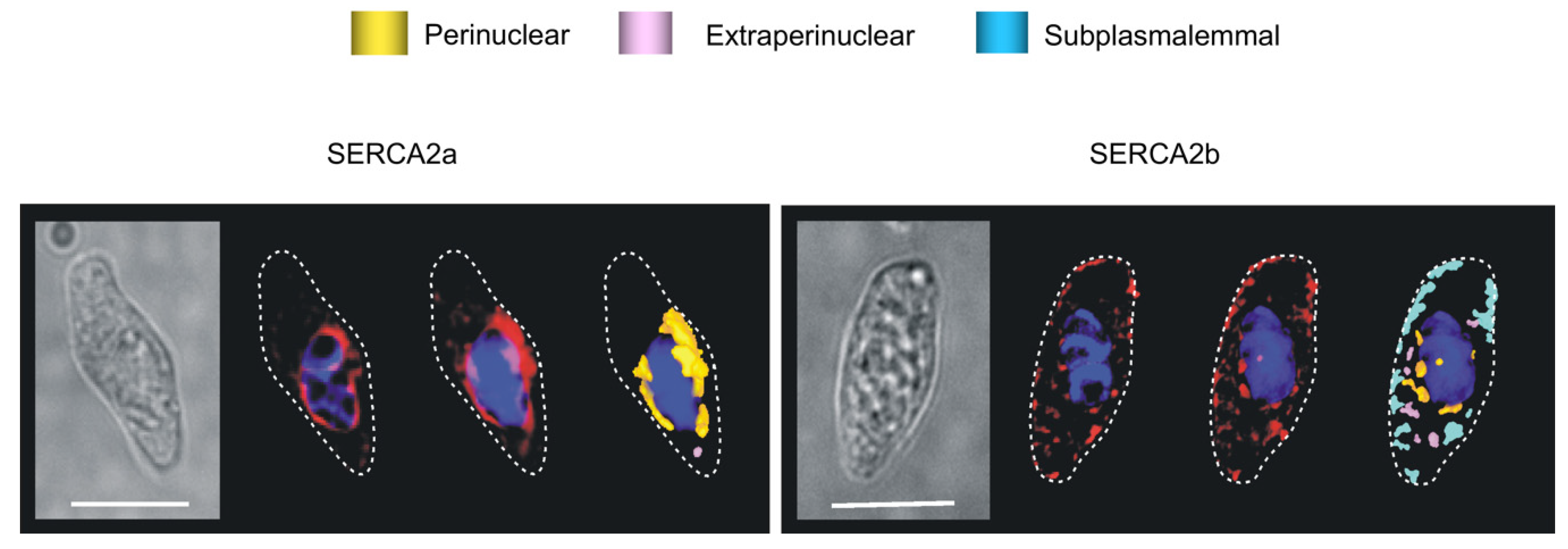
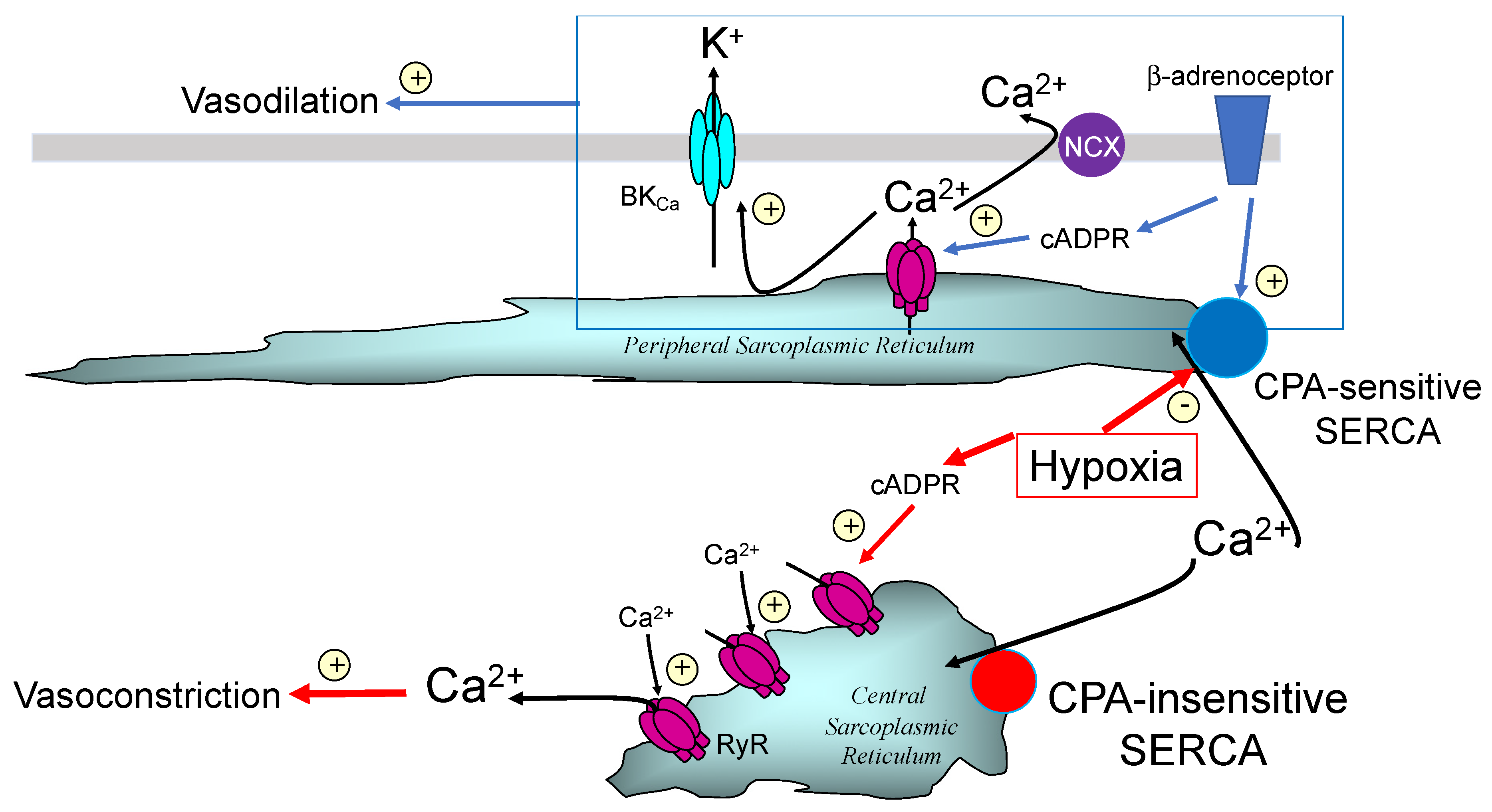

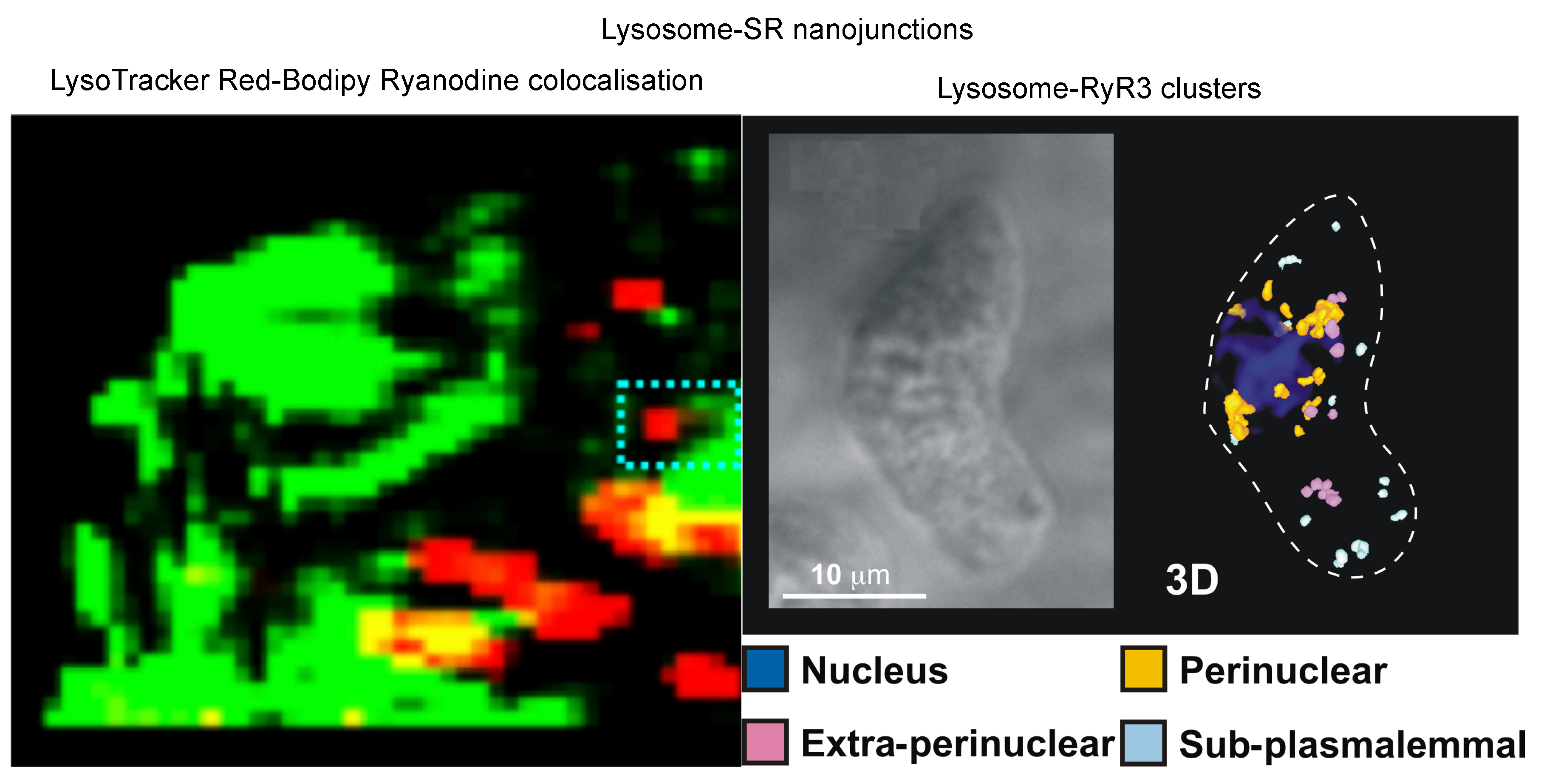


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, A.M. On a Magical Mystery Tour with 8-Bromo-Cyclic ADP-Ribose: From All-or-None Block to Nanojunctions and the Cell-Wide Web. Molecules 2020, 25, 4768. https://doi.org/10.3390/molecules25204768
Evans AM. On a Magical Mystery Tour with 8-Bromo-Cyclic ADP-Ribose: From All-or-None Block to Nanojunctions and the Cell-Wide Web. Molecules. 2020; 25(20):4768. https://doi.org/10.3390/molecules25204768
Chicago/Turabian StyleEvans, A. Mark. 2020. "On a Magical Mystery Tour with 8-Bromo-Cyclic ADP-Ribose: From All-or-None Block to Nanojunctions and the Cell-Wide Web" Molecules 25, no. 20: 4768. https://doi.org/10.3390/molecules25204768
APA StyleEvans, A. M. (2020). On a Magical Mystery Tour with 8-Bromo-Cyclic ADP-Ribose: From All-or-None Block to Nanojunctions and the Cell-Wide Web. Molecules, 25(20), 4768. https://doi.org/10.3390/molecules25204768