Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor for redox enzymes, but also moonlights as a regulator for ion channels, the same as its metabolites. Ca2+ homeostasis is dysregulated in cancer cells and affects processes such as tumorigenesis, angiogenesis, autophagy, progression, and metastasis. Herein, we summarize the regulation of the most common calcium channels (TRPM2, TPCs, RyRs, and TRPML1) by NAD+ and its metabolites, with a particular focus on their roles in cancers. Although the mechanisms of NAD+ metabolites in these pathological processes are yet to be clearly elucidated, these ion channels are emerging as potential candidates of alternative targets for anticancer therapy.
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) is an essential biomolecule involved in many critical processes, especially in energy metabolism and electron transfer. Most organisms synthesize NAD+ via two major pathways: (1) synthesis from tryptophan (Trp) (the de novo pathway); (2) synthesis from various extracellular and intracellular precursors including nicotinic acid (NA), NA riboside (NAR), nicotinamide (NAM), and NAM riboside (NR) (the salvage pathway) [1]. In particular, in the de novo pathway, synthesis of NAD+ begins from the conversion of dietary precursor Trp to quinolinic acid (QA), which is converted to nicotinamide adenine mononucleotide (NAMN). NAMN is also synthesized in the salvage pathway via NAR by nicotinamide riboside kinases 1 and 2 (NRK1/2) or indirectly via NA by nicotinate phosphoribosyltransferase (NAPRT). NAMN is adenylated to nicotinic acid adenine dinucleotide (NAAD+) via nicotinamide mononucleotide adenyltransferases 1–3 (NMNATs) before NAD+ synthesis via NAD synthetase (NADS). Moreover, NAD+ can also be generated from nicotinamide mononucleotide (NMN) via NMNATs, and NMN is synthesized in a salvage pathway via NR by NRK1/2 or NAM by nicotinamide phosphoribosyltransferase (NAMPT). In the metabolome of NAD+, it is reduced by tricarboxylic acid (TCA) cycle enzymes to NADH, and can be regenerated from NADH. Besides, NAD+ can be converted to NADP+ by NAD+ kinase (NADK). The corresponding phosphorylated redox pair NADP/NADPH is a crucial TCA cycle intermediate that provides reducing equivalents for endogenous antioxidant defense systems to maintain redox homeostasis. Then NAD+ can be consumed by poly (ADP-ribose) polymerases (PARPs) and sirtuins (SIRTs), or be degraded by membrane-bound glycohydrolases CD38/CD157 or sterile α and TIR motif-containing 1 (SARM1) to cyclic ADP-ribose (cADPR) and ADPR [2,3,4,5,6]. In this enzymatic conversion of NAD+ to cADPR, the nicotinamide group of NAD+ is cleaved, and the N1 of the adenine is linked back to the terminal ribose, forming a head to tail circled molecule, which had been documented as a natural metabolite in a wide range of endogenous systems. On the other hand, NADP+ can also be converted to nicotinic acid adenine dinucleotide phosphate (NAADP) under the functions of the same set of enzymes, in which the structural change is the conversion of the amide group of the nicotinamide in NADP+ to a carboxyl group. Taken together, these pyridine nucleotides (NAAD+, NAD+, NADH, NADP+, NADPH, cADPR, ADPR, and NAADP) derived from NAD+ constitute a regulatory system of energy metabolism (Figure 1), which plays an important role in a variety of physiological and pathological processes [7].
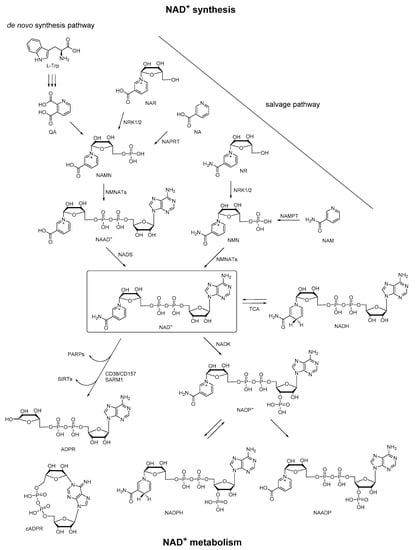
Figure 1.
Pathways of NAD+ synthesis and metabolism.
As for energy metabolism, all living organisms need to trap or liberate energy or to synthesize essential cell constituents and metabolites. These processes are usually realized by oxidation–reduction reactions, which are accomplished by specialized biomolecules that package and shuttle energy between different processes. As we know, ATP is the universal energy currency of cells. However, to generate ATP, energy must be extracted from nutrients by a series of coupled catabolic reactions. This process requires specialized electron carriers that can deliver energy to the mitochondrial electron-transport chain. NAD+ and its metabolites would be one of the most important systems that accomplish the shuttling of electrons between different reactions, by which the redox homeostasis and bioenergetics of organisms are maintained [7]. As the soluble electron carriers, NAD(H) is recognized by enzymes that catalyze catabolic reactions of glycolysis and by components of the electron transport chain, and NADP(H) is recognized by enzymes that are involved primarily in anabolic (reductive) reactions, such as lipid or cholesterol synthesis or fatty acid chain elongation [8]. Generally, a high NAD+:NADH ratio is maintained to readily accept electrons generated by catabolic reactions, whereas the low NADP+:NADPH ratio reflects a state of readiness to donate electrons to biosynthetic reactions or antioxidant defense [9]. As a result, the availability and the redox state of NAD+ and its metabolites regulate the activity of the processes involved in the intermediary metabolism, biosynthesis, and antioxidant defense.
Cancer is a multistep progression, and its critical hallmark is the reprogramming of the energy metabolism, mainly reflected in the altered mitochondrial bioenergetic and biosynthetic state of the cancer cells (excessive proliferation, impaired cell death signaling, and deregulated metabolism) [10]. In normal cells, energy transduction eventually leads to the oxidation of nutrients via oxidative phosphorylation. Glycolysis continuously generates pyruvate (Pyr), which is preferentially transported into mitochondria and further metabolized via the TCA cycle; and the ratio of NAD+ to NADH is balanced in favor of NAD+ in normal cells. While in cancer cells, a high rate of glycolysis is observed with a highly increased glucose uptake. Lactate dehydrogenase isoform A (LDHA) preferentially converts accumulating Pyr to lactate, thereby regenerating NAD+ from NADH to maintain glycolysis. Excess lactate is secreted and contributes to an extracellular environment that promotes tumor progression [11]. Meanwhile, the accumulation of lactate in tumors implies an increase in NADH relative to NAD+. For example, the intracellular NADH level in the breast cancer cell line (Hs578T) has been quantified to be approximately 1.8-fold higher than in breast normal cells (Hs578Bst) [12]. As mentioned above, The NAD+:NADH ratio plays an important role in regulating the intracellular redox state and several enzymes involved in the regulation of the metabolism. It has been reported that changes in NAD+ concentration and/or the NAD+:NADH ratio can induce DNA repair and increase cell defense, by regulating diverse signaling pathways and transcriptional events, and thus plays an important role in cancer progression [13,14]. For instance, NAD+ has been proved to regulate DNA damage repair, cell cycle progression, and epithelial-to-mesenchymal transition (EMT) via PARP-mediated ADP-ribosylation and SIRT-mediated deacetylation [15,16,17]. In addition, NAD+ can be metabolically converted to cADPR, a specialized signaling molecule that regulates multiple aspects of cancer biology, including cell survival, apoptosis, and inflammation [18]. Besides the NAD+/NADH system, another distinctive biochemical characteristic in carcinogenesis is the increased availability of the anabolic coenzyme NADPH. Cancer cells adapt their metabolism to fulfill their increased demand for energy, biosynthetic intermediates, and to counter aerobic respiration-induced oxidative stress by diverting glycolysis to pentose phosphate pathway (PPP). During this process, NADPH is produced to counteract reactive oxygen species (ROS) and to act as a cofactor for the synthesis of nucleotides, proteins, and fatty acids [19]. It has also been proved that NADK had a cancer-promoting role that converted cytosolic and mitochondrial NAD+ to NADP+, which could be further reduced to NADPH in PPP [20]. Taken together, in cancerous cells, any changes in the concentrations of these metabolites will break their synthesis and consumption homeostasis, thereby affecting the functions of their associated proteins and signaling pathways to participate in multiple processes in cancers to modulate cell metabolism, survival, progression, and invasion. To elucidate these molecular mechanisms will be of great significance for the treatment of cancer targeting NAD+ and its metabolites.
The mechanisms of these pyridine nucleotides to act as essential cofactors in redox reactions, or substrates in ribosyl transfer reactions, have been recently reviewed in detail [21]. Moreover, in addition to these regulatory functions, recent work has shown that NAD+ and its metabolites also regulate the activity of ion channels, including the Slo K+ channels, voltage-gated potassium channels and sodium channels, ATP-regulated K+ channels, and some specific types of calcium channels [8]. Considering the important role of Ca2+ homeostasis in malignant transformation, tumor progression, and response to treatment [22], we review in this article the evidence implicating NAD+ and its metabolites as regulators of calcium channels, and the function of these ion channels in cancer, aiming to shed light on the mechanisms of NAD+ metabolites related to calcium signaling in tumorigenesis, metastasis, and therapy. At the same time, this review is also to thank Professor Barry V. L. Potter for his great contribution in this research field over the years.
2. Transient Receptor Potential Melastatin 2 (TRPM2) Channel
The TRPM2 channel is a Ca2+-permeable cation channel [23] that functions as a polymodal channel responding to warm temperature, pH, trace metal ions, as well as ROS [24,25,26,27,28]. It is abundant in the brain, spleen, liver, lung, heart, myeloid cells, and so on. Accumulating evidence indicates that the TRPM2 channel is a complex molecular machine crosslinked with several signaling pathways, uniquely linking the adenine nucleotide metabolic network to the intracellular redox status. So it is critical to clarify the TRPM2 gating mechanisms of endogenous ligands from the NAD+ metabolites. Each subunit of the TRPM2 tetramer contains an intracellular N-terminal MHR domain, the typical six transmembrane domains, and a large intracellular C terminus. A series of studies has demonstrated that the NUDT9 homology (NUDT9-H) domain in the C terminus was essential for binding of NAD+ metabolites and thereby for activating the channel [29,30,31,32]. Recent cryo-electron microscopy (cryo-EM) studies have reported structures of the full-length human TRPM2 (hsTRPM2) channel as well as the non-mammalian Nematostella vectensis and Danio rerio TRPM2 channels, including the ADPR-bound state with two ADPR densities in the cleft of the MHR1/2 domain and the NUDT9-H domain, respectively [33,34,35,36,37]. Although these structural studies suggest noticeable differences or even contradictions in ADPR binding and channel gating mechanisms, activation of this channel by NAD+ metabolites with a pyridine nucleoside structure is well recognized.
Early studies reported that NAD+ itself induced a large inward current through the TRPM2 channel in endogenous-expressed immunocytes and exogenous-transfected HEK293 cells [28,38]. However, later studies showed that there was not any effect when NAD+ was infused into TRPM2-transfected cells [39], and argued that the stimulation of the TRPM2 channel by NAD+ might be attributable to contamination of a trace level of ADPR in the commercially available NAD+ preparations [30]. Moreover, our recent study also indicated that NAD+ failed to bind to the NUDT9-H domain of the hsTRPM2 channel by using the SPR approach [40]. Although the regulation of the TRPM2 channel by NAD+ remains controversial, NAD+ is still able to regulate the hsTRPM2 channel by its endogenous metabolite ADPR, since stimulation of the TRPM2 channel was likely to occur after activation of CD38 to generate ADPR from the cleavage of NAD+ [41].
Among these NAD+ metabolites, ADPR is considered to be the most potent endogenous agonist of the TRPM2 channel, with EC50 values of 10–90 µM [23]. Since the NUDT9-H domain in the C terminal is homologous to the NUDT9 ADP-ribose pyrophosphatase (~50% similarity), the activation of the hsTRPM2 channel by ADPR was originally proposed to be mediated by an enzymatic process in which ADPR bound to NUDT9-H and was converted to AMP and ribose-5′-phophate [32,42]. However, this view had been refuted by later work [43,44], with a demonstration that the hsTRPM2 channel does not act as a chanzyme for the lack of an ADPR-hydrolase activity. Nevertheless, the binding of ADPR to the NUDT9-H domain is believed to be essential for hsTRPM2 channel opening, and could be impaired upon mutations in this pocket [45,46]. Our previous study identified the key residues for ADPR binding to the NUDT9-H domain by combining homology modeling, MD simulations with functional assays [47], some of which had been verified in the cryo-EM structures of the hsTRPM2 channel lately [34,37].
cADPR and NAADP are two other controversial TRPM2 channel activators. cADPR was well recognized to mediate Ca2+ signaling pathways by binding to FKBP12.6 and modulating the function of the ryanodine receptors (RyRs), which will be discussed later in this review. However, when RyRs were specifically blocked, cADPR was still able to induce the intracellular Ca2+ increase in rabbit skeletal muscles, indicating RyRs-independent mechanisms that contribute to cADPR-induced Ca2+ responses [48]. This cADPR-induced calcium flux, in addition to RyRs, was later identified by the contribution of the TRPM2 channel [29,30,31]. These studies also confirmed the activation of the TRPM2 channel by NAADP. However, later studies had challenged those views and suggested that those earlier results might be compromised by ADPR contamination in the commercial cADPR [49] and ADPR-2′-phosphate (ADPRP, a TRPM2 agonist) contamination in the commercial NAADP [50]. Nevertheless, through a combination of surface plasmon resonance (SPR), whole-cell and single-channel patch-clamp recordings with purified cADPR, one of our recent studies had clearly demonstrated that cADPR is a bona fide activator of the TRPM2 channel [40]. To further confirm whether these NAD+ metabolites directly activate the TRPM2 channel, it is required to provide the evidence of the high resolution structure information in future.
As we all know, oxidative stress results from an imbalance between the amount of ROS produced and antioxidant levels. Low levels of ROS can modulate cell survival and metabolic pathways to enhance cell proliferation, while high levels of ROS damage tissues through protein oxidation, lipid peroxidation, DNA oxidation and mutagenesis that further activates cell death pathways [51]. Elevated levels of ROS have been found in the majority of cancers and promote tumorigenesis through activation of transcription factors, signaling pathways and DNA damages [52]. Under such circumstances, cancer cells show increased oxidative stress. The ADPR level and NADH/NAD+ ratios are also altered [53], which thus activates the TRPM2 channel. For example, in pancreatic cancer cells, SIRT6 was observed to catalyze the NAD+-dependent deacetylation of target histones, thereby generating 2′-O-acetyl-ADPR (OAADPR) that can be subsequently hydrolyzed to ADPR, which in turn activates the TRPM2 channel, triggering Ca2+ influx, and further to induce the expression of IL-8 and TNF, and enhance cell migration [54].
In most of the nonmalignant cells, it is supported that a sustained increase in intracellular Ca2+ or Zn2+ may occur leading to cell death simultaneously with the TRPM2 activation by oxidative stress [55,56,57]. However, the data in cancer models mostly supports the conclusion that TRPM2 expression and function have an important role in preserving cancer cell viability and survival. Consistent with this view, the TRPM2 channel has been found to be highly expressed in numerous cancers including bladder, breast, head and neck, lung, pancreatic, prostate, melanoma, and neuroblastoma [51], among which most studies were focused on the neuroblastoma [58,59,60,61,62,63,64] (Figure 2). The higher levels of ROS in cancer cells impel the enhancement of their anti-oxidant capacity to detoxify ROS and preserve cells viability. The transcription factor nuclear factor (erythroid-derived 2)-related factor-2 (Nrf2) takes responsibility for expression of a series of genes to regulate enzymes or cofactors involved in the anti-oxidant response [65]. Nrf2 has been observed to be highly expressed in many malignant cells, and regulated by Ketch-like ECH-associated protein 1 (Keap1) via a Ca2+-dependent process [66,67]. Meanwhile, it has been found that with the inhibition of the TRPM2 channel, Ca2+ influx was reduced, which caused the reduction of Nrf2. Its downstream enzymes involved in GSH, NADPH, and NADH production were significantly decreased, which led to weakened antioxidant responses, increasing the susceptibility to chemotherapeutic agents and decreasing cell survival and tumor growth [68].
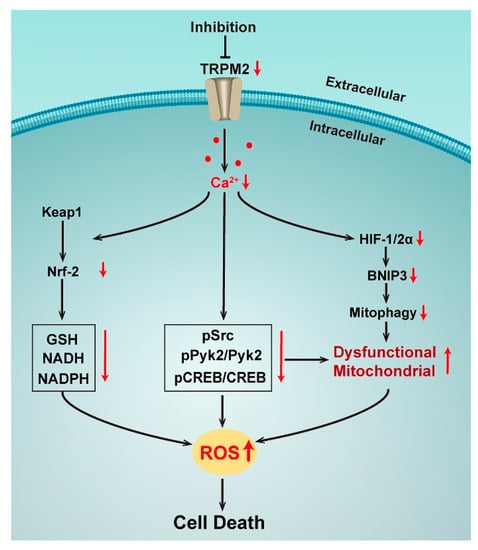
Figure 2.
Effects of TRPM2 inhibition in neuronblastoma.
In addition to Nrf2, the transcription factors HIF-1/2α and cyclic AMP-responsive element binding protein (CREB) are also regulated by TRPM2. HIF-1/2α was significantly reduced with the TRPM2 inhibition by expression of the negative short splice variant TRPM2-S in neuroblastoma cell lines. ROS thus increased as well as the accumulation of dysfunctional mitochondria with a reduced bioenergetic capacity by the down-regulation of autophagy/mitophagy via a decreasing mitochondrial membrane potential, and impairing Ca2+ uptake [59]. A study has further confirmed that re-expression of wild type TRPM2 in such a condition could rescue cell viability, mitochondrial function, and reduce ROS, demonstrating the critical role of TRPM2-mediated Ca2+ entry in the modulation of tumor growth, mitochondrial function, and cellular bioenergetics in neuroblastoma [60]. Interestingly, this mechanism was also found to be related to TRPM2-mediated CREB expression. CREB is a key transcription factor that regulates the genes involved in oncogenesis and cell survival. Ca2+ influx via TRPM2 results in the activation of phosphorylation of Pyk2, which regulates the cell survival and tumor growth of various cancers through the CREB pathway, leading to increased expression of phosphorylated and total CREB. When TRPM2 was inhibited, pPyk2, Pyk2, pCREB, and CREB were reduced and mitochondrial function as well as mitochondrial Ca2+ uptake were impaired, together with more mitochondrial and cellular ROS, reducing cells survival and tumor growth [62]. Taken together, all studies in neuroblastoma show the critical role of TRPM2 that modulates both ROS production and the antioxidant response through the Ca2+ entry via the channel activation. When the TRPM2 channel is inhibited, ROS are significantly increased by both mitochondrial dysfunction and reduced antioxidants, then reaching to a cytotoxic threshold of cell death.
In other types of cancers, a high expression of TRPM2 was also observed to increase the cancer cells survival and proliferation. The mechanisms might include minimizing DNA damage in breast adenocarcinoma cells [69,70]; increasing the migration/invasion of pancreatic ductal adenocarcinoma cells [71], gastric cancer cells [72], and tongue carcinoma SCC cells [73]; or inhibiting nuclear ADP-ribosylation in prostate cancer cells [74]. It has also been reported that the inhibition of TRPM2 could accelerate the cancer cells death by increasing the intracellular ROS in non-small cell lung (NSCLC) cells [75]; the impairment of autophagy through the JNK-signaling in gastric cancer cells [76]; or reducing the G2/M ratio in the proliferation cycle of leukemia cells [77] and NSCLC cells [75]. In addition, we also noticed that recent studies have reported a novel long non-coding RNA TRPM2-AS with a high expression correlated with a larger tumor size, advanced TNM stage, and poor patient outcomes in a variety of cancers [78,79,80,81,82,83,84,85,86,87]. Since its mechanisms in tumors are complicated and less related to the regulation of the signaling pathway by the ion channel, we did not review it here. Further work will be necessary to understand its impact on TRPM2 expression and function, as well as its role in tumorigenesis.
However, a few studies had found that TRPM2 high expression in certain types of cancer cells correlated with improved patient outcomes. For example, TRPM2 overexpression promoted apoptosis of T24 bladder cancer cells [88]; survival time was significantly longer in patients with higher TRPM2 levels than in those with lower TRPM2 levels [89]. These data suggest that the differential effects of the TRPM2 channel in cancers depend on the types of carcinomas. We believe that the oxidative stress balance regulated by the TRPM2 channel via NAD+ metabolites is the decisive factor of this channel to be a friend/foe. In any case, the NAD+ and its metabolite-activated TRPM2 channel is an exciting potential therapeutic target for a variety of cancers where the mechanisms in tumorigenesis, metastasis, and therapy need to be further uncovered.
3. Two-Pore Channels (TPCs)
Two-Pore Channels (TPCs, TPCN as the gene is named) are cation permeable channels located on endolysosomal membranes and act as important mediators of intracellular Ca2+ signaling. They are subdivided based on their structural similarity into three groups: TPC1, TPC2, and TPC3. These channels contain two putative pore-forming repeats, and each of these repeats contains six transmembrane segments and an intervening pore-loop, an architecture common to numerous voltage-gated ion channels. The transmembrane regions of TPCs are homologous to that of Nav or TRP channels [90]. However, unlike these related channels, TPCs are not expressed on plasma membranes. Only TPC1 and TPC2 are found in human and mouse cells. TPC1 is found in a range of endolysosomal organelles, and TPC2 is the predominant form expressed in late endosomes and lysosomes [91]. It has been observed that cells expressing the TPC2 channel showed a marked calcium release on intracellular application with NAADP, while genetic knockdown of this channel abolished NAADP-induced calcium release, indicating that TPC2 is an endogenous target of NAADP [92]. This study also proved that membranes enriched with TPC2 exhibited a high affinity for NAADP binding. Besides, it had proposed that NAADP was the most potent calcium regulator of NAD+ metabolites, since it stimulates calcium release at concentrations as low as 5 to 10 nM. However, intracellular dialysis of 1 mM NAADP failed to elicit a Ca2+ release, indicating homologous self-inactivation of the Ca2+ release process by NAADP, which prompted that a high-affinity binding site on the TPC2 may confer a channel opening, while a low-affinity site may confer inactivation/desensitization [93]. In addition to TPC2, NAADP also evokes endolysosomal cation release via TPC1 [94], and it has shown that arginine residues in the first S4–S5 linker were required to trigger Ca2+ signaling upon NAADP binding to TPC1 [95]. However, a recent study provided information that human TPC is, in fact, not directly activated by NAADP [96]. Although the 3D structures of mouse TPC1 and human TPC2 were recently determined by cryo-EM [97,98], the bound state of NAADP on these channels remains unknown, and further investigation is still required. There are also a series of studies developing a photoaffinity probe for the NAADP receptor, 5-N3-NAADP, which showed that NAADP did not bind to TPCs directly, but through NAADP-binding proteins [99]. It is worth mentioning that in addition to TPCs, NAADP also regulates RyRs and the TRP subtype mucolipin 1 (TRPML1) (this will be discussed later in this review) and the TRPM2 channel (see above). Therefore, it has been suggested that the responses of multiple NAADP targets are integrated such that the release of calcium by NAADP via TPCs is amplified by those neighboring receptors to generate well-orchestrated calcium oscillations [99,100].
The TPCs have been reported to be involved in various pathophysiological processes, including cell growth and development, metabolism, and cancer progression [101]. For instance, TPC2 was observed to be overexpressed in oral squamous cell carcinoma cell lines, raising intriguing questions regarding the role of TPC2 as a driver of oncogenesis [102]. In addition, TPCN2 was found to be significantly associated with survival in bladder cancer [103], and has also been reported to be one of the six gene signatures correlated with prostate cancer to predict postoperative biochemical recurrence [104]. Many studies on the association between TPCs and cancers have revealed the role of NAADP/TPC/Ca2+ signaling (Figure 3).
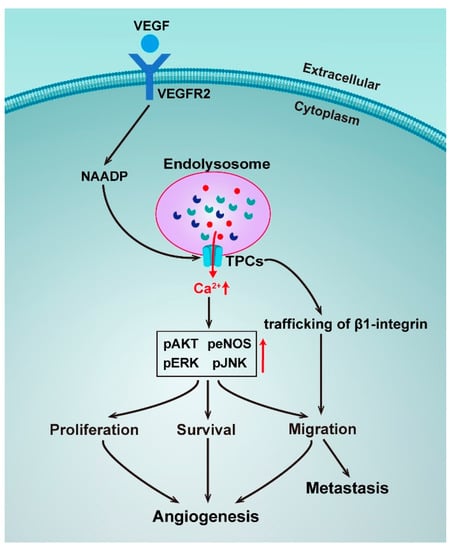
Figure 3.
Schematic representation of the roles of TPCs in angiogenesis and metastasis.
Angiogenesis is a process that is crucial for cancer progression and a key step in the transition of a tumor’s state from benign to malignant. Vascular endothelial growth factors (VEGF) are the major regulators of angiogenesis and regulate endothelial cells with cell proliferation, migration, and sprouting in the early stages of angiogenesis [105]. A novel pathway for VEGF signaling transduction had been reported, such that VEGF receptor activation led to NAADP and TPC2-dependent Ca2+ release from acidic Ca2+ stores, which in turn controlled angiogenic response. Pharmacologically using the NAADP antagonist Ned-19 or genetically using TPCN2−/− mice, it was found to dramatically reduce angiogenic responses to VEGF in vitro and in vivo [106]. The same mechanism was later confirmed by a study using the natural product Naringenin to inhibit the VEGF-induced angiogenesis [107]. An implication of these studies is the possibility to target TPC2 to develop anti-angiogenics as a strategy for cancer treatment.
Uncontrolled cell proliferation is another basic feature of cancers. A recent study has discovered that NAADP-induced Ca2+ release was blocked by genetic silencing of TPC1, and a pharmacological and genetic blockade of TPC1 dramatically reduced fetal bovine serum (FBS) and induced Ca2+ release and proliferation of metastatic colorectal cancer (mCRC) cells established from liver metastasis of human patients, thereby hinting at TPC1 being a novel therapeutic target in mCRC patients [108]. Metastatic invasion is the major cause of cancer-related deaths. A study has unveiled that TPCs played a crucial role in the formation of metastasis, as silencing TPC1 and TPC2 reduced the adhesion and migration of invasive tumor cells in vitro, and pharmacological TPC inhibition and siRNA silencing of TPC2 reduced the formation of lung metastasis in vivo [109]. However, in this study, the activation of TPCs by NAADP was not directly proved, suggesting only an involvement of trafficking of β1-integrin, a protein that is prominently involved in tumor migration. Take into account the important role of Ca2+ homeostasis in β1-integrin trafficking and the molecular mechanisms of NAADP-induced TPC activation, the TPC-mediated Ca2+ signaling in the metastasis process should be independently resolved.
Besides, there are some studies on the functions of TPCs in other physiological processes related to tumorigenesis. For example, NAADP-evoked Ca2+ signals through TPC1 and TPC2 sustained glutamate-induced autophagy in SHSY5Y neuroblastoma cells [110]. Silencing of TPC2 attenuated epidermal growth factor-induced vimentin expression in MDA-MB-468 breast cancer cells [111]. TPC2 overexpression in 4T1 mouse breast cancer cell lines and human HeLa cervical cancer cell lines inhibited the fusion of autophagosomes and lysosomes, causing the accumulation of autophagosomes [112]. Besides, TPC2 overexpression led to the evocation of the defects of pigmentation that is closely related to the development of melanoma, and its interactivity with Rab GTPases underpinned NAADP-evoked Ca2+ signals [113]. Taking all of the abovementioned together, TPCs are druggable targets that can interfere with tumorigenesis, angiogenesis, and metastasis.
4. Ryanodine Receptors (RyRs)
The RyRs represent another class of calcium channel with the regulation by NAD+ and its metabolites. RyRs are located on the sarcoplasmic and endoplasmic reticulum (SR/ER) forming a series of intracellular Ca2+ channels. There are three major structurally similar RyRs mammalian isoforms: RyR1, RyR2, and RyR3. RyR1 and RyR2 are the major RyR isoforms in skeletal and cardiac muscle, respectively, and RyR3 is expressed in various tissues along with the other two isoforms [114]. Dating back to 1994 and 1995, studies have found that NAD+ could increase the open probability of RyR1 and RyR2 [115,116]. It was then reported that RyR1 contained several dehydrogenase and NAD+/NADH oxidoreductase domains, and some residues that participate in NADP+ binding in isocitrate dehydrogenase were conserved in RyR1, suggesting that the channel may be capable of binding to NAD+ metabolites [8,117]. Later, single channel recordings from RyRs incorporated into lipid bilayers revealed that NADH (2 mM) inhibited the activity of RyR channels by 84% in permeabilized rat ventricular myocytes [118]. These results all suggest that NAD+/NADH is the direct modulator of RyRs.
Later on, more studies have focused on the activation of RyRs by cADPR. It was first proposed that cADPR could activate RyRs in 1991 [3]. The abovementioned studies in 1994 and 1995 also suggested similar conclusions [115,116]. All three RyR isoforms have been shown to mediate cADPR-induced Ca2+ release [119,120]. One of our own pieces of research, using a caged cADPR analogue, also confirmed its activation on RyR2 and RyR3 in Jurkat T cells [121]. However, evidence regarding whether cADPR acts directly on the receptors is lacking. RyR was isolated from the cellular environment and incorporated into artificial membranes under voltage-clamp conditions, which could avoid confounding cellular factors and decide the direct interaction of cADPR on the RyR channel. Numerous studies have found that cADPR had no effect on the gating of all the three types of RyRs [122]. These results suggest that cADPR may not act directly on RyRs, but via some accessory proteins to activate RyRs. Two cADPR-binding proteins—140- and 100-kDa proteins—have been identified in sea urchin egg homogenates by 8-N3-cADPR, an analog of cADPR, as a photoaffinity probe [123]. Moreover, calmodulin-dependent protein kinase II (CaMKII), calmodulin, and FK506-binding protein, FKBP12.6 have been shown to be required for cADPR action [124,125,126]. A recent study by synthesizing a novel photoaffinity labeling cADPR agonist identified glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as one of the bridging proteins between cADPR and RyRs [127].
Besides cADPR, involvement of RyRs for NAADP-activated Ca2+ mobilization has also been evidenced. For instance, NAADP induced Ca2+ release from rat heart microsomes, and RyR2 activated by NAADP from dog heart incorporated into bilayer lipid membranes were observed [128]. In addition, nanomolar concentrations of NAADP triggered Ca2+ release from skeletal muscle SR, which was due to a direct action on RyR1, since NAADP increased the open probability of the purified RyR1 channel by using a single-channel recording [129]. Another series of studies showed that co-injection of the RyRs antagonists ruthenium red with NAADP abolished the Ca2+ signal from NAADP, and Jurkat T cells with largely reduced expression of RyRs did not respond to microinjections of NAADP, suggesting that the Ca2+ release channel sensitive to NAADP in T-lymphocytes is the RyRs [130,131]. However, none of these results had demonstrated the direct activation of RyRs by NAADP. A more recent study using combinatorial knockouts and antibodies against RyRs and TPCs compared their relative contribution to NAADP-induced Ca2+ release from permeabilized pancreatic acinar cells. It was observed that, with a sequence of RyR1 > TPC2 > RyR3 > TPC1 >> RyR2, and the primary, but very small, NAADP-elicited Ca2+, release via TPCs triggered the detectable Ca2+-induced Ca2+ release (CICR) via RyRs occurring from the granules and the ER [132].
Although further investigations are required to prove the molecular mechanisms of direct/indirect activation of RyRs by NAD+ and its metabolites mentioned above, their regulations on RyRs that involved in the physiological and pathological processes such as cancer development are well established. For example, in Namalwa B lymphoma cells, ryanodine stimulation of Ca2+ release decreased both CD38 protein abundance and cyclase activity, indicating a negative feedback mechanism between the RyRs channel and CD38, which could directly affect the signaling pathways of NAD+ metabolites catalyzed by CD38 [133]. Oxygen and glucose deprivation (OGD) due to insufficient blood circulation can decrease cancer cell survival and proliferation of solid tumors with the activation of adenosine 5′-monophosphate-activated protein kinase (AMPK). One study found that NAD(P)H: quinone oxidoreductase 1 (NQOD1) played a key role in the AMPK-induced cancer cell death in OGD through the CD38/cADPR/RyR/Ca2+/CaMKII signaling pathway [134]. Besides, there are also some reports that showed a high diversity of the RyRs expression in tumors. For example, in a research including 57 ductal, human breast cancer specimens, moderate-to-high expression of RyRs by immunostaining was found in 82% of the specimens, and there was a direct correlation between RyRs levels and tumor grades [135]. RyR2 was over-expressed in melanoma tissues [136], and RyR3 over-expression was detected in breast cancer [137]. However, in comparison with normal thyroid tissues, tissues derived from thyroid carcinoma exhibited decreased expression of RyR2, which was tightly associated with lymphatic metastasis, extracapsular extension, and the TNM stage [138]. Many studies have also reported the differential regulations of RyRs in the proliferation or migration of different types of cancer cells [139,140,141,142], but various functions of RyRs in respectively characterized malignant diseases are still needed to be clarified, especially the roles of NAD+ and its metabolites in the Ca2+ signaling pathway regulated by RyRs.
5. Transient Receptor Potential Channel Subtype Mucolipin 1 (TRPML1)
The transient receptor potential channel subtype mucolipin 1 (TRPML1) is an integral part of the acidic vesicles in the endolysosomal system. Similar to all TRP channels, each TRPML1 is composed of 4 subunits which possess 6 transmembrane spanning domains with cytosolic N and C terminals. It is widely distributed within the later vesicles of the endocytic pathway [143,144]. Research that fused the TRPML1 into lipid planar bilayers observed that NAADP activated Ca2+ release at concentrations of 1–1000 nM, and TRPML1 gene silencing reduced the extent of this NAADP-sensitive Ca2+ release. In addition, the blockade of TRPML1 by anti-TRPML1 antibodies almost abolished NAADP-induced activation of lysosomal Ca2+ channels, which provided the direct evidence to show that a NAADP-sensitive Ca2+ release is characteristic of the TRPML1 channel [145,146]. Research from the same group further claimed that NAADP-induced activation of the TRPML1 channel could not be observed in lysosomes from TRPML1−/− cells, but was restored by re-expressed TRPML1 into these cells. This work has also proved that NAADP regulated TRPML1 activation via promoting the interaction of endosomes and lysosomes, and thereby regulated lipid transport to lysosomes [147]. However, some studies put forward a contrary view that TRPML1 was not the target for NAADP, because neither overexpression of TRPML1 nor the dominant negative TRPML1 mutant D471K affected the NAADP-mediated Ca2+ signals [148]. However, there was a comment on the above research that proposed that a direct recording of lysosomal TRPML1 currents or measurements of Ca2+ release from lysosomes are needed for solid evidence to specify the TRPML1 channel as a NAADP-sensitive lysosome sensor [149].
Up to now, no evidence has been presented on the involvement of TRPML1 activated by NAADP in malignant transformation. There are only limited studies on the function of TRPML1 in cancer cell proliferation. For example, the increase of TRPML1 expression attenuated MAPK and mTORC1 signaling to sustain macropinocytosis, and avoid proteotoxic stress among melanoma cells [150]. TRPML1 maintains oncogenic mutations in the RAS family by mediating cholesterol de-esterification and transport, and reducing the proliferation of cancer cells that express oncogenic mutations by TRPML1 inhibition [151,152]. TRPML1 was specifically upregulated in triple-negative breast cancer (TNCB), which regulates TNBC development through controlling mTORC1 activity and the release of lysosomal ATP, while genetic downregulation or pharmacological inhibition of TRPML1 suppressed the growth of TNBC [153]. The functions of TRPML1 channels in tumorigenesis, especially the roles of NAD+ metabolites, still require further elucidation.
6. Conclusions and Expectation
In classical biochemistry, NAD+ and its metabolites are most frequently viewed as soluble electron carriers. Recent research suggests that these metabolites can also regulate cell signaling by acting as the modulator of ion channels, just like the calcium channels reviewed above (Figure 4). It should be noted that these ion channels often have multiple activators/regulators, for instance, RyRs can be activated by a variety of NAD+ metabolites, even the non-NAD+ metabolites ryanodine and caffeine. On the other hand, one NAD+ metabolite can also regulate various calcium channels, the NAADP for example, which may regulate all the four ion channels mentioned above. Moreover, since NAD+ metabolites are a mutual conversion and the concentrations of NAD+ metabolites change dynamically in vivo, it is important for us to comprehensively investigate how the contributions from different activators/regulators of these ion channels mediate intracellular Ca2+ signaling in the complicated tumor pathogenesis in future.
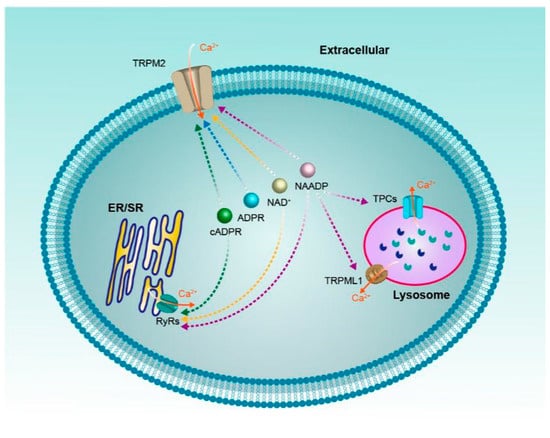
Figure 4.
NAD+ and its metabolites (ADPR, cADPR, NAADP) regulate calcium channels.
Professor Potter B.V.L. has made brilliant contributions to the identification of these NAD+ metabolites as these ion channels activators/regulators, which have been cited above. More importantly, based on the fact that these ion channels have been regarded as new cancer therapeutic targets, Professor Potter has designed and synthesized a series of ligands/inhibitors of these calcium channels [72,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168], which certainly play critical roles in the development of anti-cancer drugs targeting these ion channels. In addition, some potassium and sodium channels can also be activated by NAD+ and its metabolites, and there are also some calcium channels whose activity can be indirectly affected by NAD+ and its metabolites. For example, the P2 × 7 channel is activated by ADP-ribosyltransferase 2.2-dependent ADP ribosylation in the presence of extracellular NAD+ [169]. Moreover, the CD38-cADPR-RyRs signaling pathway modulates store-operated calcium entry through transient receptor potential ion channels (TRPCs) [170], which was also evidenced by co-immunoprecipitation of RyRs and TRPC3 [171], and gating of the TRPC under activation of RyRs [172]. All of these NAD+ metabolite-regulated ion channel-mediated signaling pathways have been increasingly demonstrated to play important roles in tumorigenesis, metastasis, and therapy. This field is still maturing, and is surely going to open doors to more exciting studies in the future.
Author Contributions
P.Y. drafted the review paper, X.C., Y.L., and M.W. made substantial contributions to the conception or design of the review, W.Y. did the final approval of the version. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of China (31872796 and 81371302 to W.Y., 32071102 to P.Y.), and Zhejiang Provincial Natural Science Foundation (LY19B020013 to P.Y.).
Acknowledgments
The authors would like to thank Yongjuan Zhao (Peking University Shenzhen Graduate School), Xiafei Yu (Zhejiang University), and Han Zhang (Peking University) for reading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, Y.; Liu, J.; Park, J.; Rai, P.; Zhai, R. Subcellular compartmentalization of NAD and its role in cancer: A sereNADe of metabolic melodies. Pharmacol. Ther. 2019, 200, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M. Interplay between compartmentalized NAD synthesis and consumption: A focus on the PARP family. Genes Dev. 2020, 34, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; Lee, H.; Busa, W. Ca2+-induced Ca2+ release in sea urchin egg homogenates: Modulation by cyclic ADP-ribose. Science 1991, 253, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santosargumedo, L.; Parkhouse, R.M.E.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Xie, X.J.; Li, W.H.; Liu, J.; Chen, Z.; Zhang, B.; Li, T.; Li, S.L.; Lu, J.G.; Zhang, L. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 2019, 15, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Bhatnagar, A.; Sadoshima, J. Overview of pyridine nucleotides review series. Circul. Res. 2012, 111, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Kilfoil, P.; Tipparaju, S.; Barski, O.; Bhatnagar, A. Regulation of ion channels by pyridine nucleotides. Circul. Res. 2013, 112, 721–741. [Google Scholar] [CrossRef]
- Ussher, J.; Jaswal, J.; Lopaschuk, G. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circul. Res. 2012, 111, 628–641. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Heikal, A. Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobiol. B Biol. 2009, 95, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Milisav, I. NAD+ as the Link Between Oxidative Stress, Inflammation, Caloric Restriction, Exercise, DNA Repair, Longevity, and Health Span. Rejuvenation Res. 2016, 19, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.; Canto, C.; Wanders, R.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Choupani, J.; Mansoori Derakhshan, S.; Bayat, S.; Alivand, M.; Shekari Khaniani, M. Narrower insight to SIRT1 role in cancer: A potential therapeutic target to control epithelial-mesenchymal transition in cancer cells. J. Cell. Physiol. 2018, 233, 4443–4457. [Google Scholar] [CrossRef]
- Chini, E.; Chini, C.; Espindola Netto, J.; de Oliveira, G.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef]
- Patra, K.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Tedeschi, P.; Bansal, N.; Kerrigan, J.; Abali, E.; Scotto, K.; Bertino, J. NAD+ Kinase as a Therapeutic Target in Cancer. Clin. Cancer Res. 2016, 22, 5189–5195. [Google Scholar] [CrossRef]
- Demarest, T.G.; Babbar, M.; Okur, M.N.; Dan, X.; Croteau, D.L.; Fakouri, N.B.; Mattson, M.P.; Bohr1, V.A. NAD+ Metabolism in Aging and Cancer. Annu. Rev. Cancer Biol. 2019, 3, 105–130. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yang, W.; Zou, J.; Beech, D. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, H.; Kamm, G.; Pohle, J.; Reis, F.; Heppenstall, P.; Wende, H.; Siemens, J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 2016, 353, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; McNaughton, P. TRPM2 and warmth sensation. Pflug. Arch. 2018, 470, 787–798. [Google Scholar] [CrossRef]
- Yang, W.; Zou, J.; Xia, R.; Vaal, M.; Seymour, V.; Luo, J.; Beech, D.; Jiang, L. State-dependent inhibition of TRPM2 channel by acidic pH. J. Biol. Chem. 2010, 285, 30411–30418. [Google Scholar] [CrossRef]
- Yang, W.; Manna, P.; Zou, J.; Luo, J.; Beech, D.; Sivaprasadarao, A.; Jiang, L. Zinc inactivates melastatin transient receptor potential 2 channels via the outer pore. J. Biol. Chem. 2011, 286, 23789–23798. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Beck, A.; Kolisek, M.; Bagley, L.; Fleig, A.; Penner, R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006, 20, 962–964. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- Lange, I.; Penner, R.; Fleig, A.; Beck, A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium 2008, 44, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.; Fleig, A.; Dunn, C.; Bagley, L.; Launay, P.; Schmitz, C.; Stokes, A.; Zhu, Q.; Bessman, M.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Toth, B.; Szollosi, A.; Chen, J.; Csanady, L. Structure of a TRPM2 channel in complex with Ca explains unique gating regulation. eLife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, 6421. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Winkler, P.; Sun, W.; Lv, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018, 562, 145–149. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Hsu, A.; Borschel, W.; Borgnia, M.; Lander, G.; Lee, S. Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat. Commun. 2019, 10, 3740. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Roth, B.; Lv, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 2019, 8, e50175. [Google Scholar] [CrossRef]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef]
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jvngling, E.; Zitt, C.; Lvckhoff, A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 2002, 277, 23150–23156. [Google Scholar] [CrossRef]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695. [Google Scholar] [CrossRef]
- Heiner, I.; Eisfeld, J.; Lvckhoff, A. Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium 2003, 33, 533–540. [Google Scholar] [CrossRef]
- Perraud, A.; Shen, B.; Dunn, C.; Rippe, K.; Smith, M.; Bessman, M.; Stoddard, B.; Scharenberg, A. NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. [Google Scholar] [CrossRef]
- Toth, B.; Iordanov, I.; Csanady, L. Putative chanzyme activity of TRPM2 cation channel is unrelated to pore gating. Proc. Natl. Acad. Sci. USA 2014, 111, 16949–16954. [Google Scholar] [CrossRef]
- Iordanov, I.; Mihalyi, C.; Toth, B.; Csanady, L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife 2016, 5, e17600. [Google Scholar] [CrossRef]
- Kuhn, F.; Luckhoff, A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.; Takanishi, C.; Shen, B.; Kang, S.; Smith, M.; Schmitz, C.; Knowles, H.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef]
- Yu, P.; Xue, X.; Zhang, J.; Hu, X.; Wu, Y.; Jiang, L.; Jin, H.; Luo, J.; Zhang, L.; Liu, Z.; et al. Identification of the ADPR binding pocket in the NUDT9 homology domain of TRPM2. J. Gen. Physiol. 2017, 149, 219–235. [Google Scholar] [CrossRef]
- Morrissette, J.; Heisermann, G.; Cleary, J.; Ruoho, A.; Coronado, R. Cyclic ADP-ribose induced Ca2+ release in rabbit skeletal muscle sarcoplasmic reticulum. FEBS Lett. 1993, 330, 270–274. [Google Scholar] [CrossRef]
- Toth, B.; Csanady, L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J. Biol. Chem. 2010, 285, 30091–30102. [Google Scholar] [CrossRef] [PubMed]
- Toth, B.; Iordanov, I.; Csanady, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J. Gen. Physiol. 2015, 145, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Miller, B. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- DeBerardinis, R.; Chandel, N. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Yoon, B.; Yang, E.; Kim, S. The ADP-ribose reactive NUDIX hydrolase isoforms can modulate HIF-1α in cancer cells. Biochem. Biophys. Res. Commun. 2018, 504, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Grozio, A.; Lasiglie, D.; Basile, G.; Sturla, L.; Magnone, M.; Sociali, G.; Soncini, D.; Caffa, I.; Poggi, A.; et al. The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J. Biol. Chem. 2012, 287, 40924–40937. [Google Scholar] [CrossRef]
- Gao, G.; Wang, W.; Tadagavadi, R.; Briley, N.; Love, M.; Miller, B.; Reeves, W. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, L. Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 2018, 9, 195. [Google Scholar] [CrossRef]
- Ye, M.; Yang, W.; Ainscough, J.; Hu, X.; Li, X.; Sedo, A.; Zhang, X.; Zhang, X.; Chen, Z.; Li, X.; et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014, 5, e1541. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, W.; Tong, Q.; Conrad, K.; Hirschler-Laszkiewicz, I.; Bayerl, M.; Kim, J.; Cheung, J.; Miller, B. Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am. J. Physiol. Cell Physiol. 2013, 304, C548–C560. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hoffman, N.; Shanmughapriya, S.; Bao, L.; Keefer, K.; Conrad, K.; Merali, S.; Takahashi, Y.; Abraham, T.; Hirschler-Laszkiewicz, I.; et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2α. J. Biol. Chem. 2014, 289, 36284–36302. [Google Scholar] [CrossRef]
- Bao, L.; Chen, S.; Conrad, K.; Keefer, K.; Abraham, T.; Lee, J.; Wang, J.; Zhang, X.; Hirschler-Laszkiewicz, I.; Wang, H.; et al. Depletion of the Human Ion Channel TRPM2 in Neuroblastoma Demonstrates Its Key Role in Cell Survival through Modulation of Mitochondrial Reactive Oxygen Species and Bioenergetics. J. Biol. Chem. 2016, 291, 24449–24464. [Google Scholar] [CrossRef] [PubMed]
- Oz, A.; Celik, O. Curcumin inhibits oxidative stress-induced TRPM2 channel activation, calcium ion entry and apoptosis values in SH-SY5Y neuroblastoma cells: Involvement of transfection procedure. Mol. Membr. Biol. 2016, 33, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Hirschler-Laszkiewicz, I.; Chen, S.; Bao, L.; Wang, J.; Zhang, X.; Shanmughapriya, S.; Keefer, K.; Madesh, M.; Cheung, J.; Miller, B. The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am. J. Physiol. Cell Physiol. 2018, 315, C571–C586. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Fu, Z.; Mai, C.; Wang, W.; Wei, L.; Li, D.; Li, C.; Jiang, L. Increasing the TRPM2 Channel Expression in Human Neuroblastoma SH-SY5Y Cells Augments the Susceptibility to ROS-Induced Cell Death. Cells 2019, 8, 28. [Google Scholar] [CrossRef]
- Akpinar, O.; Ozsimsek, A.; Gvzel, M.; NazirogŸlu, M. Clostridium botulinum neurotoxin A induces apoptosis and mitochondrial oxidative stress via activation of TRPM2 channel signaling pathway in neuroblastoma and glioblastoma tumor cells. J. Recept. Signal Transduct. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.; MacEwan, D.; Bowles, K.; Rushworth, S. Oxidative stress responses and Nrf2 in human leukaemia. Oxid. Med. Cell. Longev. 2015, 2015, 454659. [Google Scholar] [CrossRef]
- Jaramillo, M.; Zhang, D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Kim, J.; Xu, E.; Sacks, D.; Lee, J.; Shu, L.; Xia, B.; Kong, A. Identification and functional studies of a new Nrf2 partner IQGAP1: A critical role in the stability and transactivation of Nrf2. Antioxid. Redox Signal. 2013, 19, 89–101. [Google Scholar] [CrossRef]
- Bao, L.; Festa, F.; Freet, C.; Lee, J.; Hirschler-Laszkiewicz, I.; Chen, S.; Keefer, K.; Wang, H.; Patterson, A.; Cheung, J.; et al. The Human Transient Receptor Potential Melastatin 2 Ion Channel Modulates ROS Through Nrf2. Sci. Rep. 2019, 9, 14132. [Google Scholar] [CrossRef]
- Hopkins, M.; Feng, X.; Liu, M.; Parker, L.; Koh, D. Inhibition of the transient receptor potential melastatin-2 channel causes increased DNA damage and decreased proliferation in breast adenocarcinoma cells. Int. J. Oncol. 2015, 46, 2267–2276. [Google Scholar] [CrossRef]
- Koh, D.; Powell, D.; Blake, S.; Hoffman, J.; Hopkins, M.; Feng, X. Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep. 2015, 34, 1589–1598. [Google Scholar] [CrossRef]
- Lin, R.; Wang, Y.; Chen, Q.; Liu, Z.; Xiao, S.; Wang, B.; Shi, B. TRPM2 promotes the proliferation and invasion of pancreatic ductal adenocarcinoma. Mol. Med. Rep. 2018, 17, 7537–7544. [Google Scholar] [CrossRef]
- Kuhn, F.; Watt, J.; Potter, B.; Luckhoff, A. Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci. Rep. 2019, 9, 4985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xu, W.; Xu, Z.; Qi, C.; Li, Y.; Cheng, J.; Liu, L.; Wu, Y.; Gao, J.; Ye, J. The overexpressed functional transient receptor potential channel TRPM2 in oral squamous cell carcinoma. Sci. Rep. 2016, 6, 38471. [Google Scholar] [CrossRef]
- Zeng, X.; Sikka, S.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.; Pottle, J.; Taylor, J.; Li, M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Almasi, S.; Long, C.; Sterea, A.; Clements, D.; Gujar, S.; El Hiani, Y. TRPM2 Silencing Causes G2/M Arrest and Apoptosis in Lung Cancer Cells via Increasing Intracellular ROS and RNS Levels and Activating the JNK Pathway. Cell Physiol. Biochem. 2019, 52, 742–757. [Google Scholar] [PubMed]
- Almasi, S.; Kennedy, B.; El-Aghil, M.; Sterea, A.; Gujar, S.; Partida-Sanchez, S.; El Hiani, Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.; Misovic, M.; Szteyn, K.; Shumilina, E.; Rudner, J.; Huber, S. Targeting TRPM2 Channels Impairs Radiation-Induced Cell Cycle Arrest and Fosters Cell Death of T Cell Leukemia Cells in a Bcl-2-Dependent Manner. Oxid. Med. Cell. Longev. 2016, 2016, 8026702. [Google Scholar] [CrossRef]
- Lavorgna, G.; Chiacchiera, F.; Briganti, A.; Montorsi, F.; Pasini, D.; Salonia, A. Expression-profiling of apoptosis induced by ablation of the long ncRNA TRPM2-AS in prostate cancer cell. Genom. Data 2015, 3, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, Q.; Zhang, C.; Xu, W.; Xu, G.; Zhao, X.; Liu, X.; Du, Y. Long non-coding RNA TRPM2-AS as a potential biomarker for hepatocellular carcinoma. Ir. J. Med. Sci. 2018, 187, 621–628. [Google Scholar] [CrossRef]
- Ulareanu, R.; Chiritoiu, G.; Cojocaru, F.; Deftu, A.; Ristoiu, V.; Stanica, L.; Mihailescu, D.; Cucu, D. N-glycosylation of the transient receptor potential melastatin 8 channel is altered in pancreatic cancer cells. Tumor. Biol. 2017, 39, 1010428317720940. [Google Scholar] [CrossRef]
- Ma, L.; Xie, X.; Ma, L.; Pang, J.; Xiong, X.; Zheng, H.; Shen, X.; Wen, Z.; Wang, H. Downregulated long non-coding RNA TRPM2-AS inhibits cisplatin resistance of non-small cell lung cancer cells via activation of p53-p66shc pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2626–2634. [Google Scholar] [PubMed]
- Sun, T.; Song, Y.; Yu, H.; Luo, X. Identification of lncRNA TRPM2-AS/miR-140-3p/PYCR1 axis’s proliferates and anti-apoptotic effect on breast cancer using co-expression network analysis. Cancer Biol. Ther. 2019, 20, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chang, C.; Wang, B.; Li, H. ELK1-induced upregulation of lncRNA TRPM2-AS promotes tumor progression in gastric cancer by regulating miR-195/HMGA1 axis. J. Cell. Biochem. 2019, 120, 16921–16933. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Lv, Q.; Szeto, V.; Wong, R.; Zhu, S.; Zhang, Y.; Feng, Z.; Sun, H. TRPM2-AS inhibits the growth, migration, and invasion of gliomas through JNK, c-Jun, and RGS4. J. Cell. Physiol. 2020, 235, 4594–4604. [Google Scholar] [CrossRef]
- Pan, L.; Li, Y.; Jin, L.; Li, J.; Xu, A. TRPM2-AS promotes cancer cell proliferation through control of TAF15. Int. J. Biochem. Cell Biol. 2020, 120, 105683. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lin, L.; Luo, D.; Shi, L.; Chen, W.; Fan, H.; Li, Z.; Ma, X.; Ni, P.; Yang, L.; et al. Long noncoding RNA TRPM2-AS acts as a microRNA sponge of miR-612 to promote gastric cancer progression and radioresistance. Oncogenesis 2020, 9, 29. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, Y.; Xie, Y.; Leng, X.; He, M.; Song, F. Inhibition of gastric cancer cell apoptosis by long noncoding RNA TRPM2-AS via mitogen-activated protein kinase and activators of transduction-3. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Cao, Q.; Qian, S.; Wang, N.; Zhang, L.; Wang, W.; Shen, H. TRPM2 mediates histone deacetylase inhibition-induced apoptosis in bladder cancer cells. Cancer Biother. Radiopharm. 2015, 30, 87–93. [Google Scholar] [CrossRef]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.; Ulasli, M.; Demiryurek, A.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumor. Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef]
- Yu, F.; Yarov-Yarovoy, V.; Gutman, G.; Catterall, W. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef]
- Ruas, M.; Chuang, K.; Davis, L.; Al-Douri, A.; Tynan, P.; Tunn, R.; Teboul, L.; Galione, A.; Parrington, J. TPC1 has two variant isoforms, and their removal has different effects on endo-lysosomal functions compared to loss of TPC2. Mol. Cell. Biol. 2014, 34, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ma, J.; Parrington, J.; Calcraft, P.; Galione, A.; Evans, A. Calcium signaling via two-pore channels: Local or global, that is the question. Am. J. Physiol. Cell Physiol. 2010, 298, C430–C441. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.; Lam, A.; Rietdorf, K.; Galione, A.; Sitsapesan, R. Reconstituted human TPC1 is a proton-permeable ion channel and is activated by NAADP or Ca2+. Sci. Signal. 2014, 7, ra46. [Google Scholar] [CrossRef]
- Patel, S.; Churamani, D.; Brailoiu, E. NAADP-evoked Ca signals through two-pore channel-1 require arginine residues in the first S4-S5 linker. Cell Calcium 2017, 68, 1–4. [Google Scholar] [CrossRef]
- Guo, J.; Zeng, W.; Jiang, Y. Tuning the ion selectivity of two-pore channels. Proc. Natl. Acad. Sci. USA 2017, 114, 1009–1014. [Google Scholar] [CrossRef]
- She, J.; Guo, J.; Chen, Q.; Zeng, W.; Jiang, Y.; Bai, X. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556, 130–134. [Google Scholar] [CrossRef]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.; Jiang, Y. Structural mechanisms of phospholipid activation of the human TPC2 channel. eLife 2019, 8, e45222. [Google Scholar] [CrossRef]
- Guse, A. Linking NAADP to ion channel activity: A unifying hypothesis. Sci. Signal. 2012, 5, pe18. [Google Scholar] [CrossRef]
- Morgan, A.; Davis, L.; Wagner, S.; Lewis, A.; Parrington, J.; Churchill, G.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, Y.; Alharbi, A.; Hanbashi, A.; Alhoshani, A.; Parrington, J. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol. Sci. 2020, 41, 582–594. [Google Scholar] [CrossRef]
- Alharbi, A.; Parrington, J. Endolysosomal Ca Signaling in Cancer: The Role of TPC2, From Tumorigenesis to Metastasis. Front. Cell Dev. Biol. 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, M.; Lee, Y.; Bang, L.; Garg, T.; Sohn, K.; Kim, D. Identification of epigenetic interactions between miRNA and DNA methylation associated with gene expression as potential prognostic markers in bladder cancer. BMC Med. Genom. 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ji, J.; Xu, Y.; Liu, R. Identification a novel set of 6 differential expressed genes in prostate cancer that can potentially predict biochemical recurrence after curative surgery. Clin. Transl. Oncol. 2019, 21, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity and Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 5121. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef]
- Nguyen, O.; Grimm, C.; Schneider, L.; Chao, Y.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef]
- Pereira, G.; Antonioli, M.; Hirata, H.; Ureshino, R.; Nascimento, A.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.; Smaili, S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef]
- Jahidin, A.; Stewart, T.; Thompson, E.; Roberts-Thomson, S.; Monteith, G. Differential effects of two-pore channel protein 1 and 2 silencing in MDA-MB-468 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yue, J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancer cells. Exp. Cell Res. 2018, 370, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Lin-Moshier, Y.; Keebler, M.; Hooper, R.; Boulware, M.; Liu, X.; Churamani, D.; Abood, M.; Walseth, T.; Brailoiu, E.; Patel, S.; et al. The Two-pore channel (TPC) interactome unmasks isoform-specific roles for TPCs in endolysosomal morphology and cell pigmentation. Proc. Natl. Acad. Sci. USA 2014, 111, 13087–13092. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N. Molecular Insights into Calcium Dependent Regulation of Ryanodine Receptor Calcium Release Channels. Adv. Exp. Med. Biol. 2020, 1131, 321–336. [Google Scholar]
- Sitsapesan, R.; McGarry, S.; Williams, A. Cyclic ADP-ribose competes with ATP for the adenine nucleotide binding site on the cardiac ryanodine receptor Ca2+-release channel. Circul. Res. 1994, 75, 596–600. [Google Scholar] [CrossRef]
- Sitsapesan, R.; Williams, A. Cyclic ADP-ribose and related compounds activate sheep skeletal sarcoplasmic reticulum Ca2+ release channel. Am. J. Physiol. 1995, 268, C1235–C1240. [Google Scholar] [CrossRef]
- Baker, M.; Serysheva, I.; Sencer, S.; Wu, Y.; Ludtke, S.; Jiang, W.; Hamilton, S.; Chiu, W. The skeletal muscle Ca2+ release channel has an oxidoreductase-like domain. Proc. Natl. Acad. Sci. USA 2002, 99, 12155–12160. [Google Scholar] [CrossRef]
- Zima, A.; Copello, J.; Blatter, L. Effects of cytosolic NADH/NAD+ levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J. Physiol. 2004, 555, 727–741. [Google Scholar] [CrossRef]
- Lee, H. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef]
- Guse, A. Biochemistry, biology, and pharmacology of cyclic adenosine diphosphoribose (cADPR). Curr. Med. Chem. 2004, 11, 847–855. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, Z.; Hao, B.; Zhao, Y.; Zhang, L.; Lee, H.; Zhang, L.; Yue, J. A novel fluorescent cell membrane-permeable caged cyclic ADP-ribose analogue. J. Biol. Chem. 2012, 287, 24774–24783. [Google Scholar] [CrossRef] [PubMed]
- Venturi, E.; Pitt, S.; Galfre, E.; Sitsapesan, R. From eggs to hearts: What is the link between cyclic ADP-ribose and ryanodine receptors? Cardiovasc. Ther. 2012, 30, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Walseth, T.; Aarhus, R.; Kerr, J.; Lee, H. Identification of cyclic ADP-ribose-binding proteins by photoaffinity labeling. J. Biol. Chem. 1993, 268, 26686–26691. [Google Scholar]
- Takasawa, S.; Ishida, A.; Nata, K.; Nakagawa, K.; Noguchi, N.; Tohgo, A.; Kato, I.; Yonekura, H.; Fujisawa, H.; Okamoto, H. Requirement of calmodulin-dependent protein kinase II in cyclic ADP-ribose-mediated intracellular Ca2+ mobilization. J. Biol. Chem. 1995, 270, 30257–30259. [Google Scholar] [CrossRef]
- Thomas, J.; Summerhill, R.; Fruen, B.; Churchill, G.; Galione, A. Calmodulin dissociation mediates desensitization of the cADPR-induced Ca2+ release mechanism. Curr. Biol. 2002, 12, 2018–2022. [Google Scholar] [CrossRef]
- Zhang, X.; Tallini, Y.; Chen, Z.; Gan, L.; Wei, B.; Doran, R.; Miao, L.; Xin, H.; Kotlikoff, M.; Ji, G. Dissociation of FKBP12.6 from ryanodine receptor type 2 is regulated by cyclic ADP-ribose but not beta-adrenergic stimulation in mouse cardiomyocytes. Cardiovasc. Res. 2009, 84, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sun, W.; Huang, L.; Zhu, K.; Pei, F.; Zhu, L.; Wang, Q.; Lu, Y.; Zhang, H.; Jin, H.; et al. Identifying Glyceraldehyde 3-Phosphate Dehydrogenase as a Cyclic Adenosine Diphosphoribose Binding Protein by Photoaffinity Protein-Ligand Labeling Approach. J. Am. Chem. Soc. 2017, 139, 156–170. [Google Scholar] [CrossRef]
- Mojzisova, A.; Krizanova, O.; Zacikova, L.; Kominkova, V.; Ondrias, K. Effect of nicotinic acid adenine dinucleotide phosphate on ryanodine calcium release channel in heart. Pflug. Arch. 2001, 441, 674–677. [Google Scholar] [CrossRef]
- Hohenegger, M.; Suko, J.; Gscheidlinger, R.; Drobny, H.; Zidar, A. Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem. J. 2002, 367, 423–431. [Google Scholar] [CrossRef]
- Langhorst, M.; Schwarzmann, N.; Guse, A. Ca2+ release via ryanodine receptors and Ca2+ entry: Major mechanisms in NAADP-mediated Ca2+ signaling in T-lymphocytes. Cell Signal. 2004, 16, 1283–1289. [Google Scholar] [CrossRef]
- Dammermann, W.; Guse, A. Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J. Biol. Chem. 2005, 280, 21394–21399. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, J.; Charlesworth, R.; Sherwood, M.; Ferdek, P.; Mikoshiba, K.; Parrington, J.; Petersen, O.; Gerasimenko, O. Both RyRs and TPCs are required for NAADP-induced intracellular Ca2+ release. Cell Calcium 2015, 58, 237–245. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.; Datar, S.; Mackrill, J. Activation of ryanodine receptor/Ca2+ release channels downregulates CD38 in the Namalwa B lymphoma. FEBS Lett. 2003, 554, 133–137. [Google Scholar] [CrossRef]
- Lee, H.; Oh, E.; Choi, B.; Park, M.; Lee, J.; Lee, J.; Park, H. NQO1-induced activation of AMPK contributes to cancer cell death by oxygen-glucose deprivation. Sci. Rep. 2015, 5, 7769. [Google Scholar] [CrossRef] [PubMed]
- Abdul, M.; Ramlal, S.; Hoosein, N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol. Oncol. Res. 2008, 14, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Deli, T.; Varga, N.; Adam, A.; Kenessey, I.; Raso, E.; Puskas, L.; Tovari, J.; Fodor, J.; Feher, M.; Szigeti, G.; et al. Functional genomics of calcium channels in human melanoma cells. Int. J. Cancer 2007, 121, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3′UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, D.; Chen, J.; He, G.; Gao, L. Low expression of ryanodine receptor 2 is associated with poor prognosis in thyroid carcinoma. Oncol. Lett. 2019, 18, 3605–3612. [Google Scholar] [CrossRef]
- Hakamata, Y.; Nishimura, S.; Nakai, J.; Nakashima, Y.; Kita, T.; Imoto, K. Involvement of the brain type of ryanodine receptor in T-cell proliferation. FEBS Lett. 1994, 352, 206–210. [Google Scholar] [CrossRef]
- Kang, H.; Kim, N.; Lee, C.; Lee, J.; Kang, W. Expression and function of ryanodine receptors in human melanocytes. J. Cell. Physiol. 2000, 185, 200–206. [Google Scholar] [CrossRef]
- Matyash, M.; Matyash, V.; Nolte, C.; Sorrentino, V.; Kettenmann, H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 2002, 16, 84–86. [Google Scholar] [CrossRef]
- Shin, D.; Leem, D.; Shin, J.; Kim, J.; Kim, K.; Choi, S.; Lee, M.; Choi, J.; Lee, K. Compound K induced apoptosis via endoplasmic reticulum Ca release through ryanodine receptor in human lung cancer cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef]
- Jaslan, D.; Bock, J.; Krogsaeter, E.; Grimm, C. Evolutionary Aspects of TRPMLs and TPCs. Int. J. Mol. Sci. 2020, 21, 4181. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, P. Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J. Biol. Chem. 2007, 282, 25259–25269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jin, S.; Yi, F.; Li, P. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J. Cell. Mol. Med. 2009, 13, 3174–3185. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, M.; Han, W.; Li, P. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(-/-) cells. Am. J. Physiol. Cell Physiol. 2011, 301, C421–C430. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Jha, A.; Li, Q.; Soyombo, A.; Dickinson, G.; Churamani, D.; Brailoiu, E.; Patel, S.; Muallem, S. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J. Biol. Chem. 2011, 286, 22934–22942. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, Y.; Abais, J.; Ritter, J.; Zhang, F. Cyclic ADP-Ribose and NAADP in Vascular Regulation and Diseases. Messenger 2013, 2, 63–85. [Google Scholar] [CrossRef] [PubMed]
- Kasitinon, S.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.; Morrison, S. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell Rep. 2019, 28, 2293–2305.e9. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.; Naji, A.; Clemons, K.; Wong, C.; Villanueva, M.; Gregory, S.; Karagas, N.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Venkatachalam, K. TRPML1 and RAS-driven cancers-exploring a link with great therapeutic potential. Channels 2019, 13, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.; Rizvi Syeda, A.; Shen, B.; Richard Derek, C.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Baszczynski, O.; Watt, J.; Rozewitz, M.; Fliegert, R.; Guse, A.; Potter, B. Synthesis of phosphonoacetate analogues of the second messenger adenosine 5′-diphosphate ribose (ADPR). RSC Adv. 2020, 10, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Baszczynski, O.; Watt, J.; Rozewitz, M.; Guse, A.; Fliegert, R.; Potter, B. Synthesis of Terminal Ribose Analogues of Adenosine 5′-Diphosphate Ribose as Probes for the Transient Receptor Potential Cation Channel TRPM2. J. Org. Chem. 2019, 84, 6143–6157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Watt, J.; Cordiglieri, C.; Dammermann, W.; Mahon, M.; Flugel, A.; Guse, A.; Potter, B. Small Molecule Antagonists of NAADP-Induced Ca Release in T-Lymphocytes Suggest Potential Therapeutic Agents for Autoimmune Disease. Sci. Rep. 2018, 8, 16775. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Bauche, A.; Wolf Perez, A.; Watt, J.; Rozewitz, M.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef]
- Fliegert, R.; Watt, J.; Schobel, A.; Rozewitz, M.; Moreau, C.; Kirchberger, T.; Thomas, M.; Sick, W.; Araujo, A.; Harneit, A.; et al. Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349. Biochem. J. 2017, 474, 2159–2175. [Google Scholar] [CrossRef]
- Swarbrick, J.; Graeff, R.; Zhang, H.; Thomas, M.; Hao, Q.; Potter, B. Cyclic adenosine 5′-diphosphate ribose analogs without a “southern” ribose inhibit ADP-ribosyl cyclase-hydrolase CD38. J. Med. Chem. 2014, 57, 8517–8529. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Takano, S.; Sakaguchi, N.; Kudoh, T.; Murayama, T.; Sakurai, T.; Hashii, M.; Higashida, H.; Weber, K.; Guse, A.; et al. Design, Synthesis, and Chemical and Biological Properties of Cyclic ADP-4-Thioribose as a Stable Equivalent of Cyclic ADP-Ribose. Messenger 2014, 3, 35–51. [Google Scholar] [CrossRef][Green Version]
- Swarbrick, J.; Graeff, R.; Garnham, C.; Thomas, M.; Galione, A.; Potter, B. ‘Click cyclic ADP-ribose’: A neutral second messenger mimic. Chem. Commun. 2014, 50, 2458–2461. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Kirchberger, T.; Swarbrick, J.; Bartlett, S.; Fliegert, R.; Yorgan, T.; Bauche, A.; Harneit, A.; Guse, A.; Potter, B. Structure-activity relationship of adenosine 5′-diphosphoribose at the transient receptor potential melastatin 2 (TRPM2) channel: Rational design of antagonists. J. Med. Chem. 2013, 56, 10079–10102. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Liu, Q.; Graeff, R.; Wagner, G.; Thomas, M.; Swarbrick, J.; Shuto, S.; Lee, H.; Hao, Q.; Potter, B. CD38 Structure-Based Inhibitor Design Using the N1-Cyclic Inosine 5′-Diphosphate Ribose Template. PLoS ONE 2013, 8, e66247. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, J.; Potter, B. Total synthesis of a cyclic adenosine 5′-diphosphate ribose receptor agonist. J. Org. Chem. 2012, 77, 4191–4197. [Google Scholar] [CrossRef]
- Moreau, C.; Kirchberger, T.; Zhang, B.; Thomas, M.; Weber, K.; Guse, A.; Potter, B. Aberrant cyclization affords a C-6 modified cyclic adenosine 5′-diphosphoribose analogue with biological activity in Jurkat T cells. J. Med. Chem. 2012, 55, 1478–1489. [Google Scholar] [CrossRef]
- Moreau, C.; Ashamu, G.; Bailey, V.; Galione, A.; Guse, A.; Potter, B. Synthesis of cyclic adenosine 5′-diphosphate ribose analogues: A C2′endo/syn “southern” ribose conformation underlies activity at the sea urchin cADPR receptor. Org. Biomol. Chem. 2011, 9, 278–290. [Google Scholar] [CrossRef]
- Dammermann, W.; Zhang, B.; Nebel, M.; Cordiglieri, C.; Odoardi, F.; Kirchberger, T.; Kawakami, N.; Dowden, J.; Schmid, F.; Dornmair, K.; et al. NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10678–10683. [Google Scholar] [CrossRef]
- Zhang, B.; Wagner, G.; Weber, K.; Garnham, C.; Morgan, A.; Galione, A.; Guse, A.; Potter, B. 2′-deoxy cyclic adenosine 5′-diphosphate ribose derivatives: Importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [Google Scholar] [CrossRef]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. ADP-ribosylation of P2 × 7: A matter of life and death for regulatory T cells and natural killer T cells. Curr. Top. Microbiol. Immunol. 2015, 384, 107–126. [Google Scholar]
- Thompson, M.; White, T.; Chini, E.N. Modulation of store-operated Ca2+ entry by cyclic-ADP-ribose. Braz. J. Med. Biol. Res. 2006, 39, 739–748. [Google Scholar] [CrossRef]
- Kiselyov, K.; Shin, D.; Wang, Y.; Pessah, I.; Allen, P.; Muallem, S. Gating of store-operated channels by conformational coupling to ryanodine receptors. Mol. Cell 2000, 6, 421–431. [Google Scholar] [CrossRef]
- Kiselyov, K.; Shin, D.; Shcheynikov, N.; Kurosaki, N.; Muallem, S. Regulation of Ca2+-release-activated Ca2+ current (Icrac) by ryanodine receptors in inositol 1,4,5-trisphosphate-receptor-deficient DT40 cells. Biochem. J. 2001, 360, 17–22. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).