
The Pictet-Spengler Reaction Updates Its Habits

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
My chameleon soulKeeps turning me around(A. Alexopoulos)
1. Introduction
2. Variations on the Classic Pictet-Spengler Reaction
3. Update (2011–2015) of Pictet-Spengler Reactions
I think it’s timeTo do an update(Roger Turner)
3.1. Tetrahydroisoquinolines and Tetrahydro-β-Carbolines
3.2. Polyheterocycles
3.3. Total Synthesis of Complex Alkaloid Natural Products
4. Dressing with Fashionable Clothes a Classic Reaction
Not to be old fashionedI’ll put something red(Emily Dickinson)
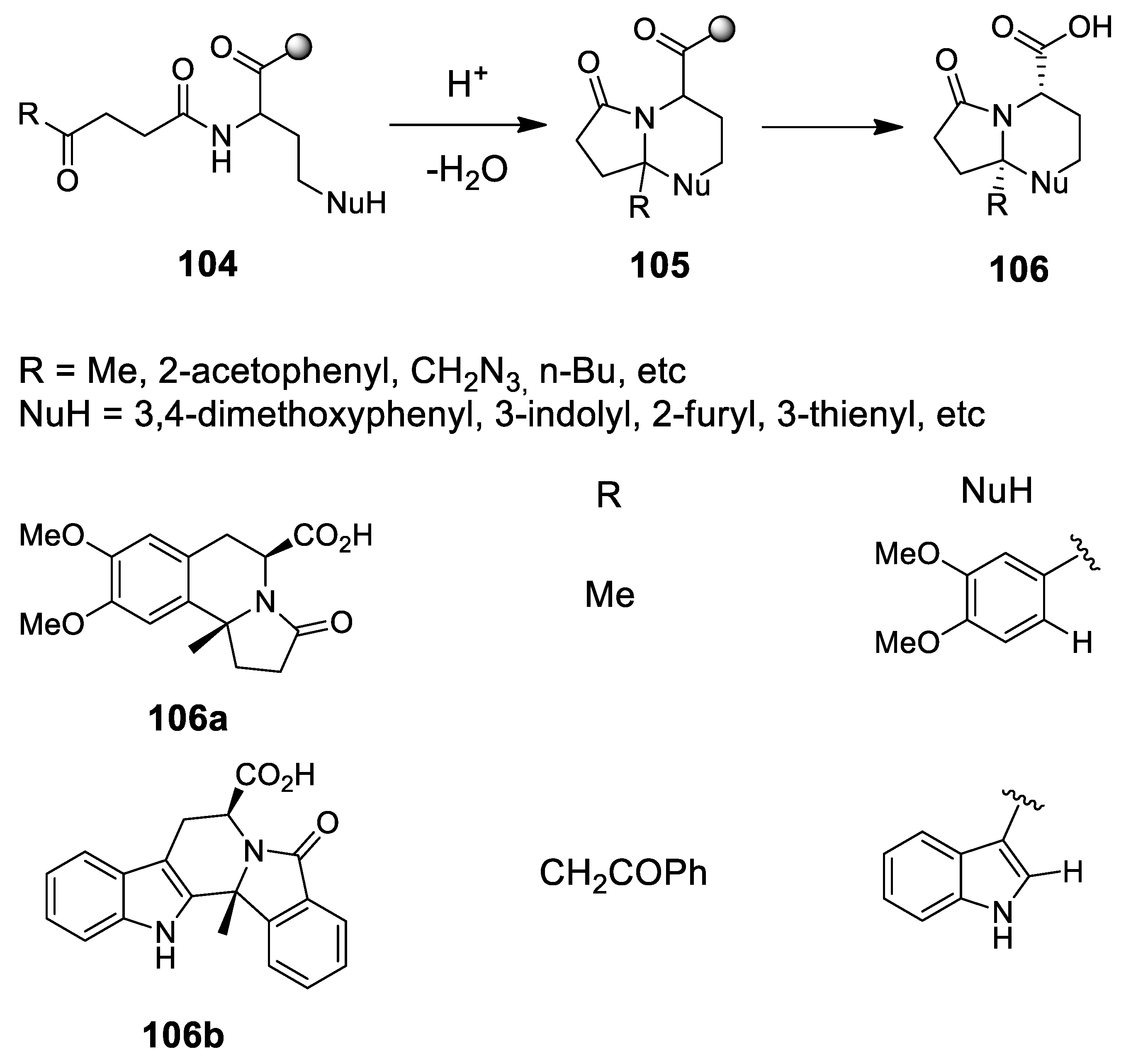
4.1. A New Scenography: Updating the Solid Phase Strategy
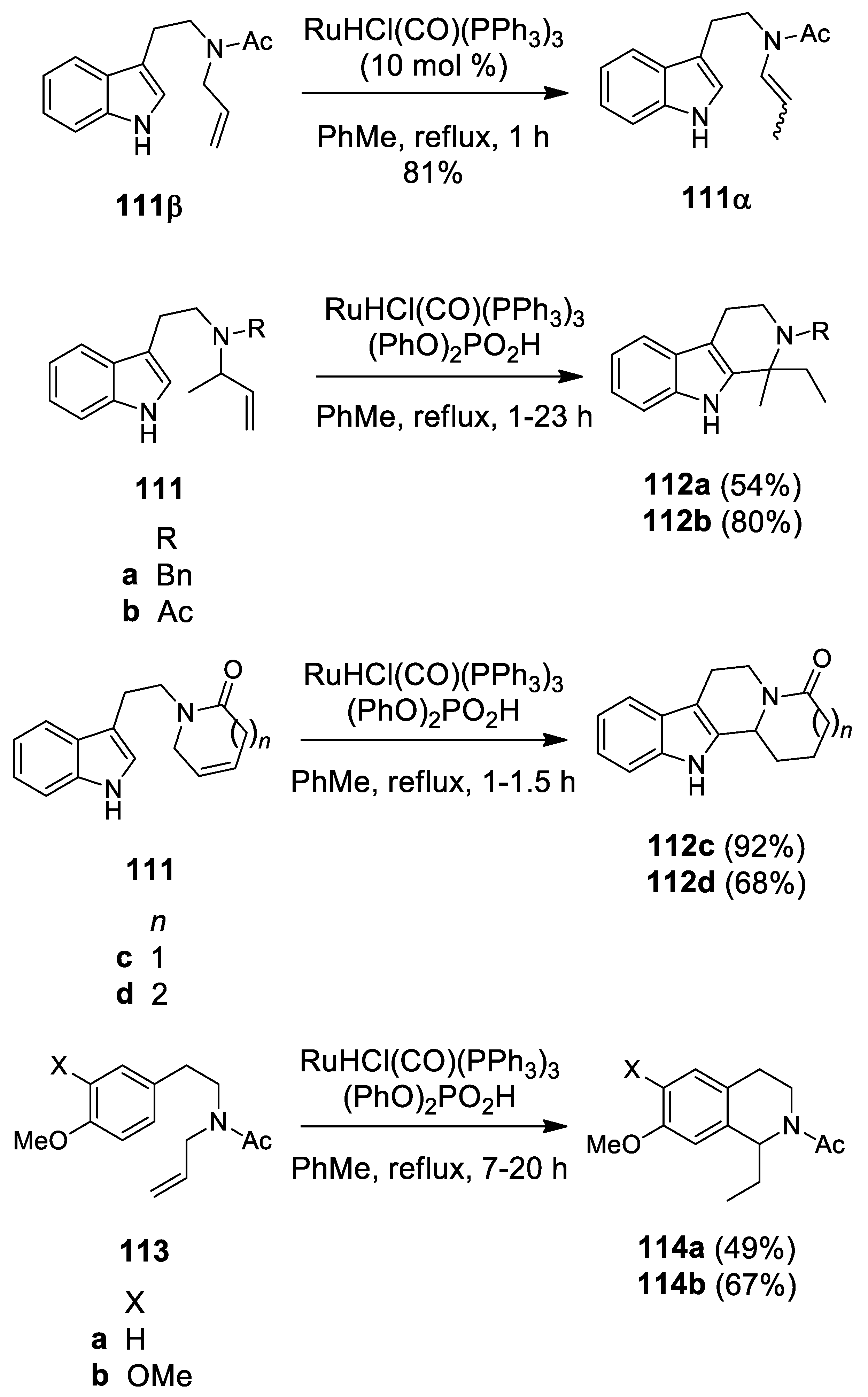
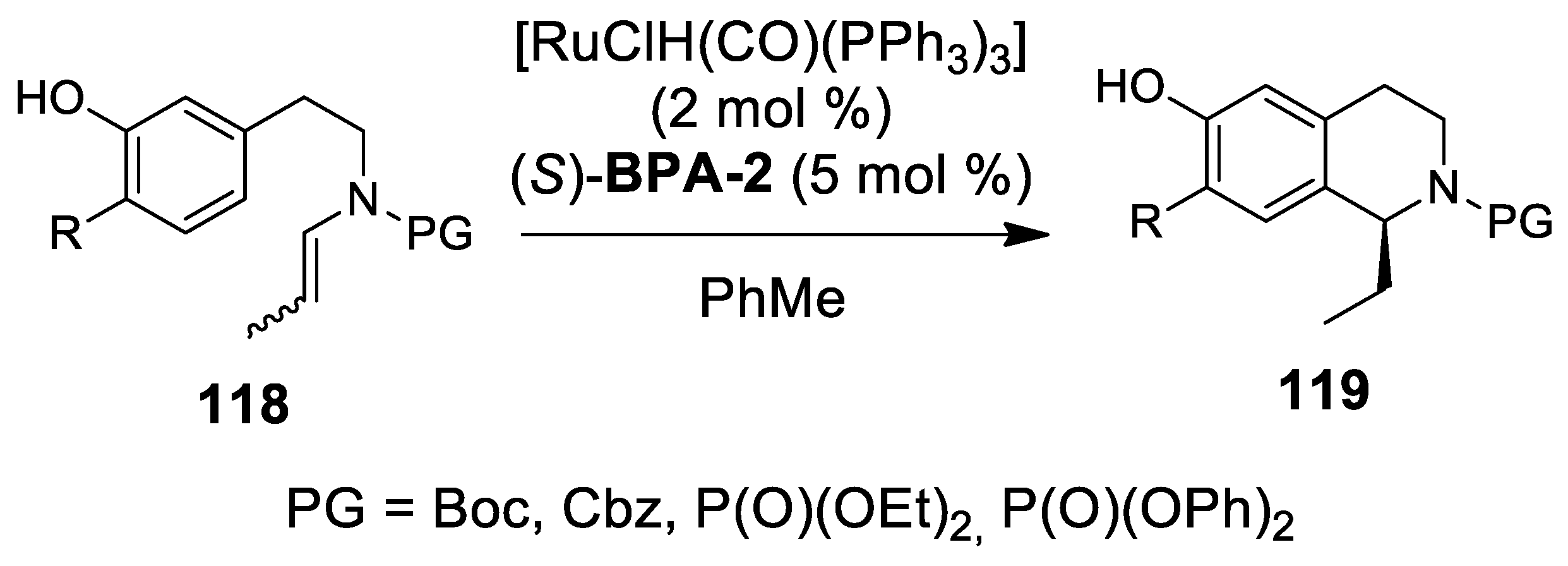
4.2. Ruthenium-Catalyzed N-Acyliminium Cyclization
4.3. All in One Pot
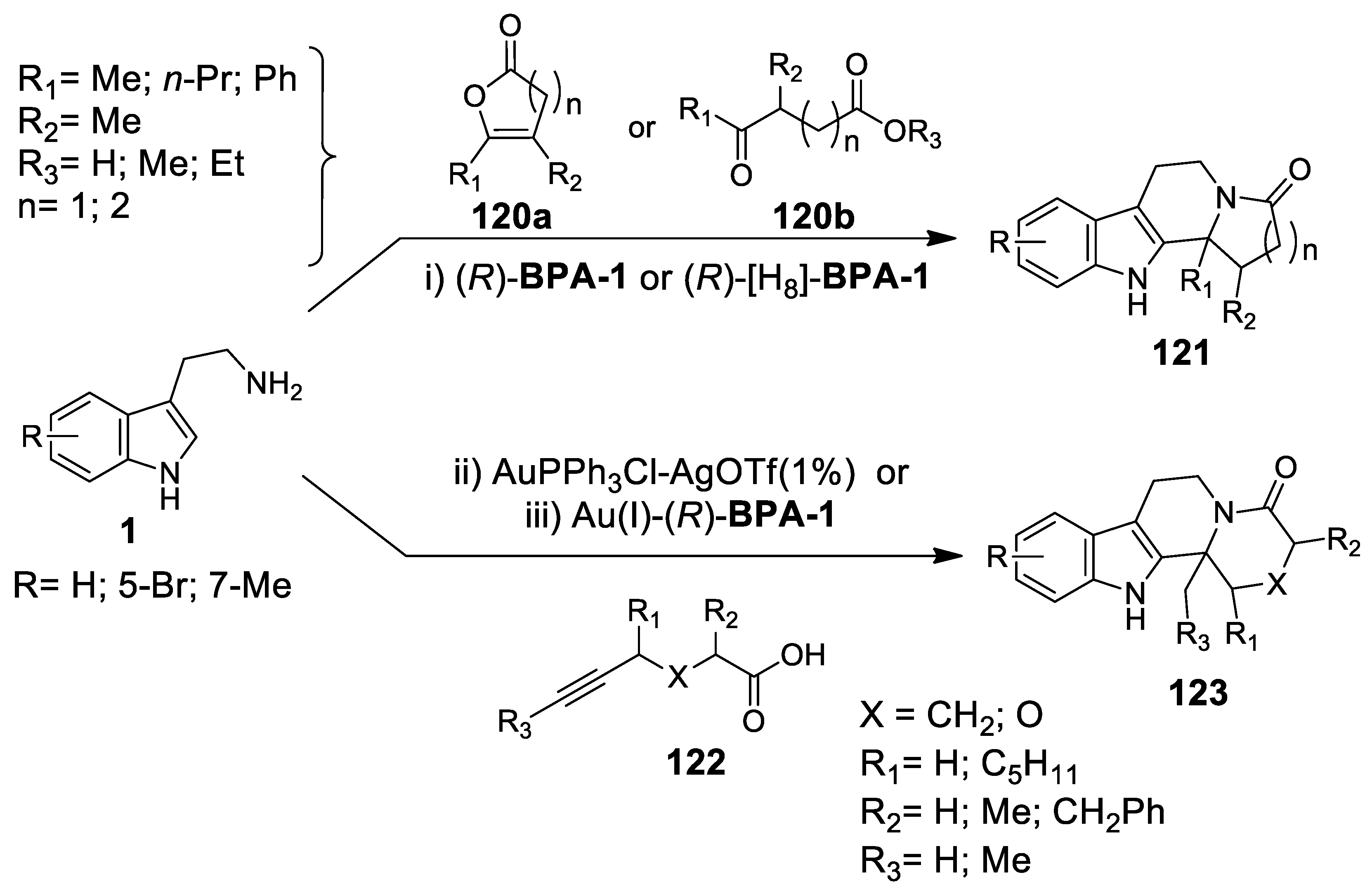
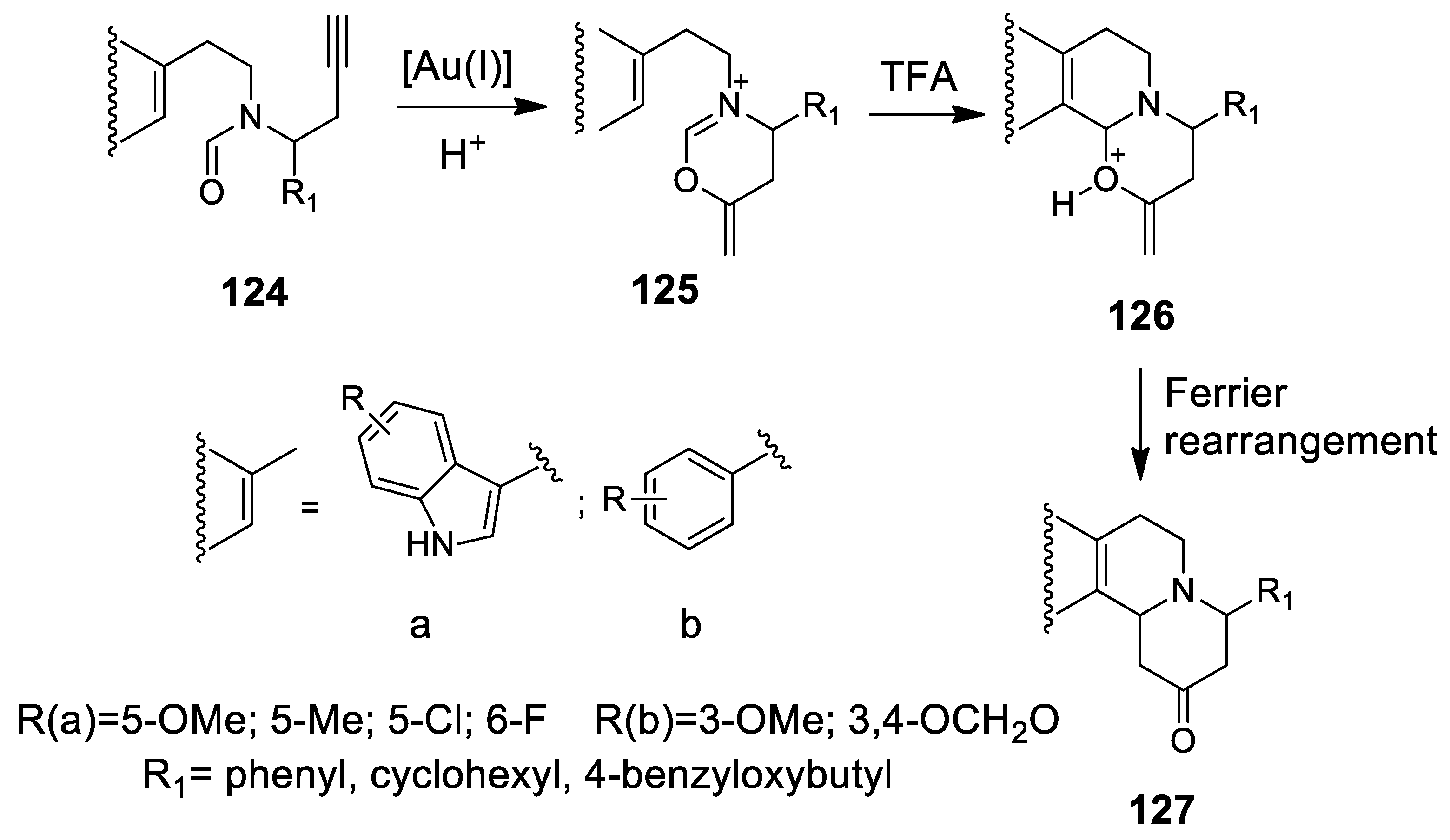
4.3.1. Au(I)-Catalyzed N-Acyliminium Cyclization Cascade
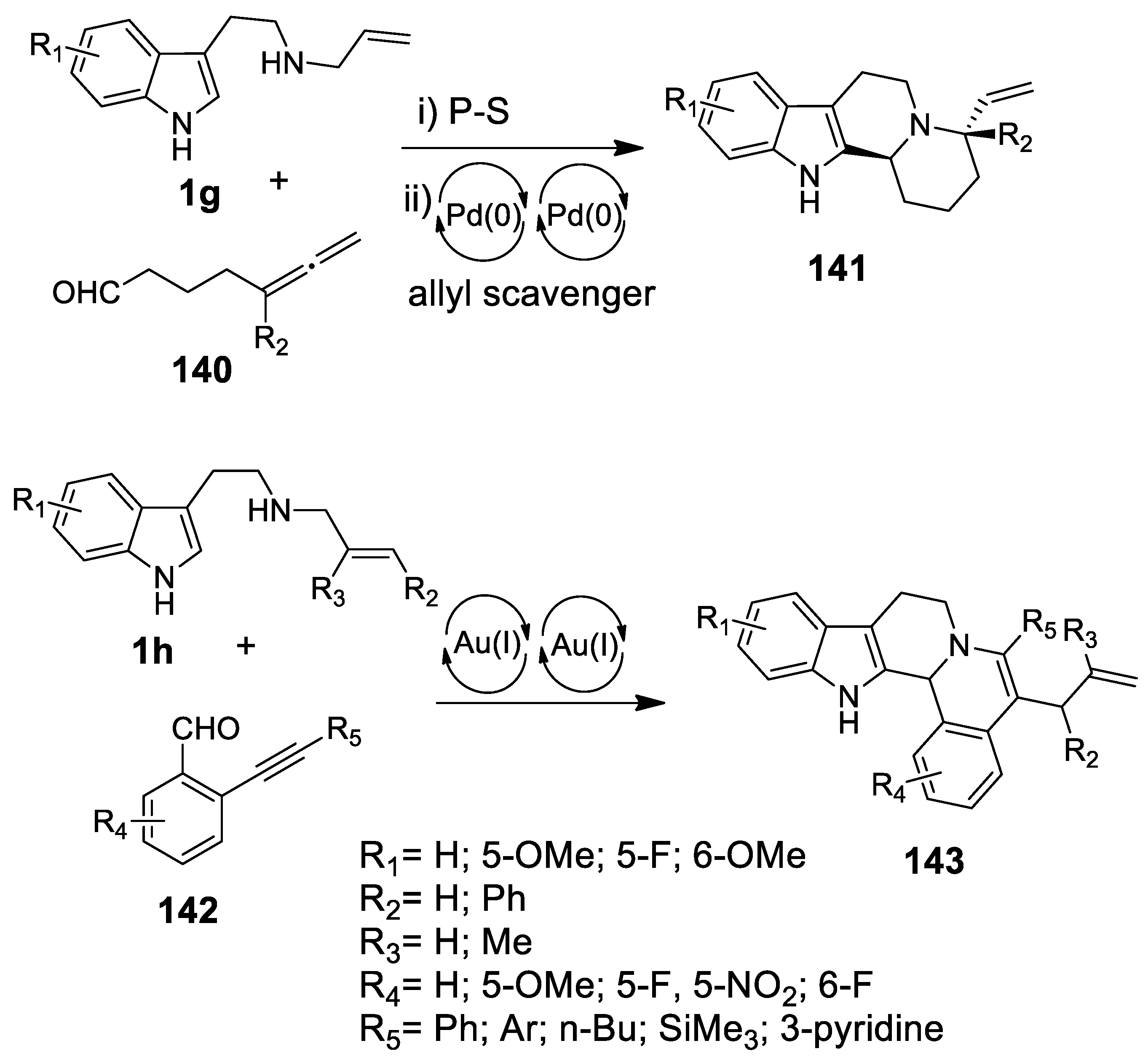
4.3.2. Michael Addition/Pictet-Spengler Reaction Sequences: Update 2011–2015
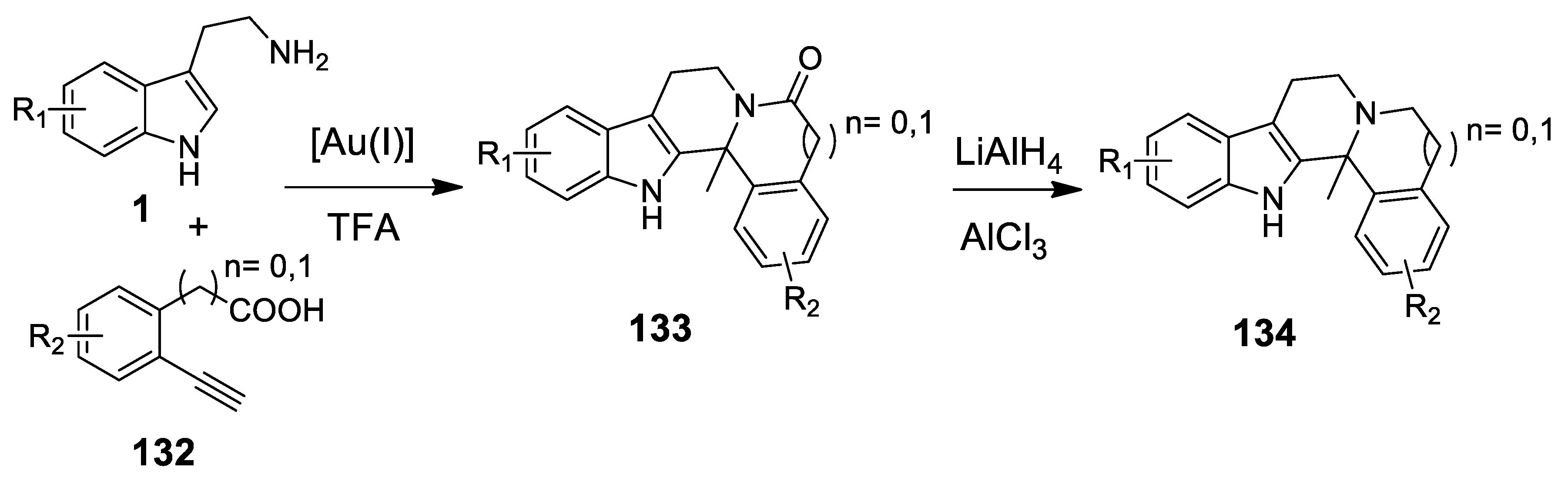
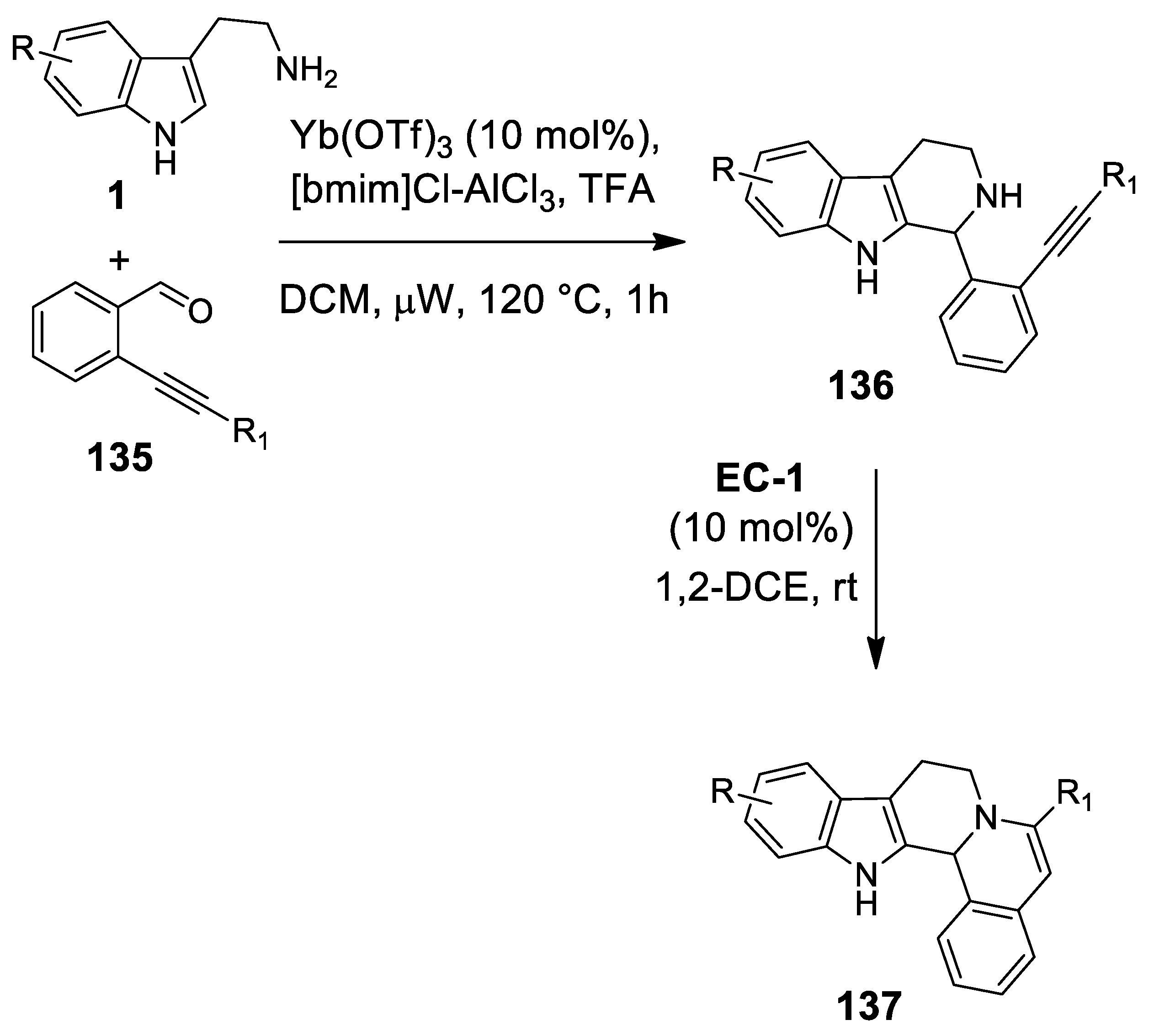
4.3.3. Other Pictet-Spengler Reactions in Cascade Sequences: Update (2011–2015)
5. The Ring-a Ring of Multi-Component Reactions
Awake–awake!Now come and makeA ring–a ring of roses(A.S. Stephens)
5.1. Ugi Meets Pictet-Spengler
5.2. Update of Ugi/Pictet-Spengler Combinations
6. The Enzymatic Pictet-Spengler: Briefing and Update 2011–2015
We are star-crossed;cursed to walkdivergent paths(Grace, crossroad poems)
6.1. Pictet-Spenglerases
6.1.1. Biogenetic Studies
6.1.2. Biosynthesis: Update 2011–2015
6.1.3. Biocatalysis
6.1.4. Biotransformations: Update 2011–2015
6.2. Chemistry and biology of Pictet-Spengler Reaction
6.2.1. Iso-Pictet-Spengler
6.2.2. Orthogonal Bioconjugation
6.2.3. Unnatural Compounds from Fungal Pictet-Spengler Biosynthesis
7. Conclusions: Drawing a Veil over Act II
No encore.As the curtain falls,All that is waiting is silence.(Spike Harper)
Author Contributions
Funding
Conflicts of Interest
References
- Ingallina, C.; D’Acquarica, I.; Delle Monache, G.; Ghirga, F.; Quaglio, D.; Ghirga, P.; Berardozzi, S.; Markovic, V.; Botta, B. The Pictet Spengler reaction still on stage. Curr. Pharm. Des. 2016, 22, 1808–1850. [Google Scholar] [CrossRef]
- Marianoff, B.E.; Zhang, H.-C.; Cohen, J.H.; Turchi, I.J.; Marianoff, C.A. Cyclizations of N-acyliminium ions. Chem. Rev. 2004, 104, 1431–1629. [Google Scholar] [CrossRef]
- Yokoyama, A.; Ohwada, T.; Shudo, K. Prototype Pictet-Spengler reactions catalyzed by superacids. Involvement of dicationic superelectrophiles. J. Org. Chem. 1999, 64, 611–617. [Google Scholar] [CrossRef]
- Barley, P.D.; Moore, M.H.; Smith, D.I.; Vernon, J.M. Enhancing the yield and the diastereoselectivity of the Pictet-Spengler reaction: A highly efficient route to cis-1,3-disubstituted tetrahydro-β-carbolines. Tetrahedron Lett. 1994, 35, 3587–3588. [Google Scholar]
- Pulka, K.; Misicka, A. Influence of reaction conditions on products of the Pictet-Spengler condensation. Tetrahedron 2011, 67, 1955–1959. [Google Scholar] [CrossRef]
- Larghi, E.L.; Amongero, M.; Bracca, A.B.J.; Kaufman, T. The intermolecular Pictet-Spengler condensation with chiral carbonyl derivatives in the stereoselective syntheses of optically-active isoquinoline and indole alkaloid. ARKIVOK 2005, 2015, 98–153. [Google Scholar]
- Schmidt, G.; Waldmann, H.; Henke, H.; Burkard, M. Asymmetric Pictet-Spengler Reactions Employing N,N-Phthaloyl Amino Acids as Chiral Auxiliary Groups. Angew. Chem. Int. Ed. 1995, 34, 2402–2403. [Google Scholar]
- Schmidt, G.; Waldmann, H.; Henke, H.; Burkard, M. Asymmetric Control in the Pictet-Spengler Reaction by means of N-protected Amino Acids as Chiral Auxiliary Groups. Chem. Eur. J. 1996, 2, 1566–1571. [Google Scholar] [CrossRef]
- Yamada, H.; Kawate, T.; Matsumizu, M.; Nishida, A.; Yamaguchi, K.; Nakagawa, M. Chiral Lewis Acid-mediated Enantioselective Pictet-Spengler Reaction of Nb-Hydroxy-tryptamine with Aldehydes. J. Org. Chem. 1998, 63, 6348–6354. [Google Scholar] [CrossRef]
- Kawate, T.; Yamanaka, M.; Nagakawa, M. Chiral Auxiliary Approach to the Asymmetric Pictet-Spengler Reaction of Tryptamines. Heterocycles 1999, 50, 1033–1039. [Google Scholar]
- Ascic, E.; Hansen, C.L.; Le Quement, S.T.; Nielsen, T.E. Synthesis of tetrahydro-β-carboline via isomerization of N-allyltryptamines: A metal-catalyzed variation on the Pictet-Spengler theme. Chem. Commun. 2012, 48, 3345–3347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gholamzadeh, P. The Pictet-Spengler Reaction: A Powerful Strategy for the Synthesis of Heterocycles. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 127, pp. 153–226. [Google Scholar]
- Sharma, S.; Joshi, G.; Kalra, S.; Singh, S.; Kumar, R. Synthetic versus Enzymatic Pictet-Spengler Reaction: An Overview. Curr. Org. Synth. 2018, 15, 924–939. [Google Scholar] [CrossRef]
- Magnus, N.A.; Ley, C.P.; Pollock, P.M.; Wepsiec, J.P. Pictet-Spengler based synthesis of a bisarylmaleimide Glicogen Synthase Kinase 3 inhibitor. Org. Lett. 2010, 12, 3700–3703. [Google Scholar] [CrossRef] [PubMed]
- Neelamegam, R.; Hellenbrand, T.; Schroeder, F.A.; Wang, C.; Hooker, J.M. Imaging evaluation of 5HT2C agonists, [11C]WAY-163909 and [11C]vabicaserin, formed by Pictet –Spengler Cyclization. J. Med. Chem. 2014, 57, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Raheem, I.T.; Thiara, P.S.; Petersen, E.A.; Jacobsen, E.N. Enantioselective Pictet-Spengler type cyclizations of hydroxylactams: H-bond donor catalysis by anion binding. J. Am. Chem. Soc. 2007, 129, 13404–13405. [Google Scholar] [CrossRef] [PubMed]
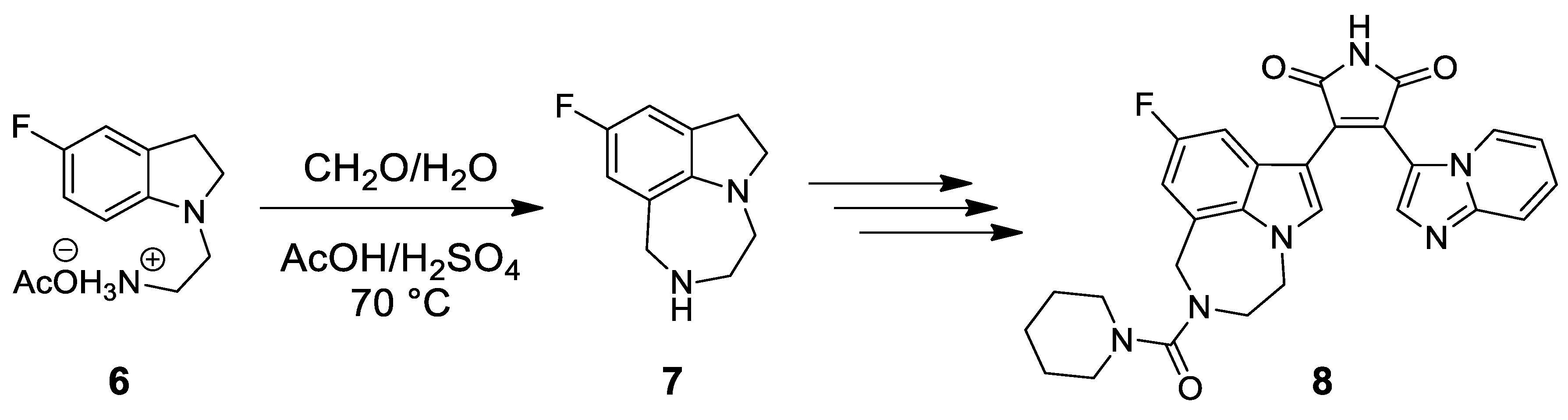
- Owen, R.T.; Castaner, R.; Bolos, J.; Estivill, C. Dual orexin OX1/OX2 antagonist. Treatment of sleep disorders. Drugs Fut. 2009, 34, 5–10. [Google Scholar] [CrossRef]
- Chaudary, S.; Harding, W.W. Synthesis of C-Homoaporphines via microwave-assisted direct acylation. Tetrahedron 2011, 67, 569–575. [Google Scholar] [CrossRef]
- Berhal, F.; Wu, Z.; Zhang, Z.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective synthesis of 1-aryltetrahydroisoquinoline through iridium catalyzed asymmetric hydrogenation. Org. Lett. 2012, 14, 3308–3311. [Google Scholar] [CrossRef]
- Chang, M.; Li, W.; Zhang, X. A highly efficient and enantio-selective access to tetrahydroisoquinoline alkaloids: Asymmetric hydrogenation with an iridium catalyst. Angew. Chem. Int. Ed. 2011, 50, 10679–10681. [Google Scholar] [CrossRef]
- Fañanás-Mistral, M.; Teichert, J.; Fernández-Salas, J.A.; Heijnen, D.; Feringa, B.L. Enantio-selective synthesis of almorexant via iridium catalysed intramolecular allylic amidation. Org. Biomol. Chem. 2013, 11, 4521–4525. [Google Scholar] [CrossRef][Green Version]
- Teichert, J.; Fañanás-Mistral, M.; Feringa, B.L. Iridium-cata-lyzed asymmetric intramolecular allylic amidation: Enantioselective synthesis of chiral tetrahydro-isoquinolines and saturated nitrogen heterocycles. Angew. Chem. Int. Ed. 2011, 50, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Seidel, D. The azomethine ylide route to amine C-H functionalization: Redox version of classic reactions and a pathway to new transformations. Acc. Chem. Res. 2015, 48, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Das, D.; Seidel, D. Azomethine ylide annulation: Facile access to polycyclic ring systems. Chem. Sci. 2011, 2, 233–236. [Google Scholar] [CrossRef]
- Wang, S.; Chai, Z.; Zhou, S.; Wang, S.; Zhu, X.; Wei, Y. A novel Lewis acid catalyzed [3 + 3] annulation strategy for the syntheses of tetrahydro-β-carbolines and tetrahydroisoquinolines. Org. Lett. 2013, 15, 2628–2631. [Google Scholar] [CrossRef]
- Schlosser, M.; Simig, G.; Geneste, H. Three complementary methods offering access to 5-substituted 1,2,3,4-tetrahydroisoquinolines. Tetrahedron 1998, 54, 9023–9032. [Google Scholar] [CrossRef]
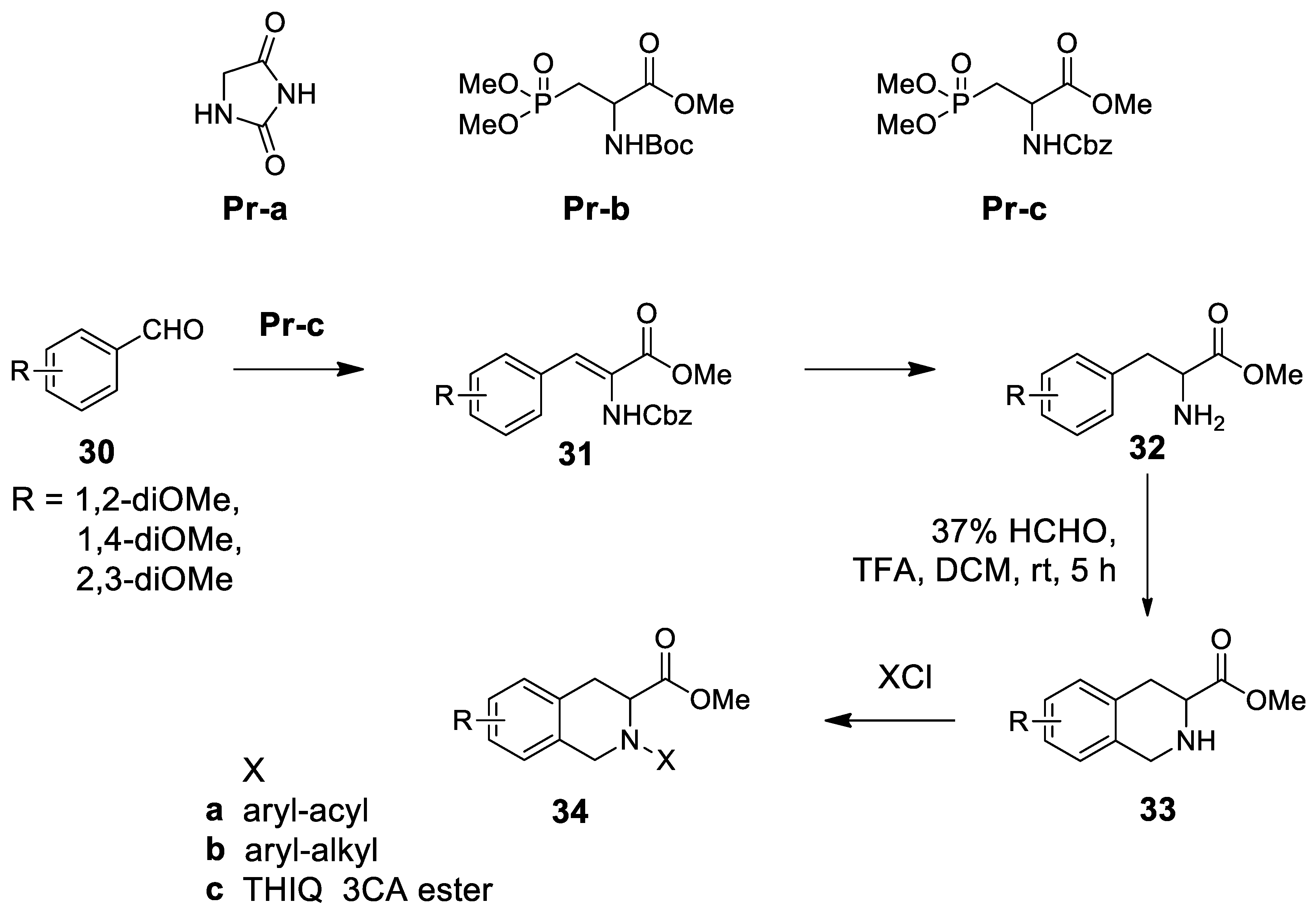
- Zhang, X.; Zhang, J.; Zhang, L.; Feng, J.; Xu, Y.; Yuan, Y.; Fang, H.; Xu, W. Design, synthesis and biological evaluation of novel 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as aminopeptidase N/CD13 inhibitors. Bioorg. Med. Chem. 2011, 19, 6015–6025. [Google Scholar] [CrossRef]
- Zou, Z.H.; Lan, X.B.; Qian, H.; Huang, W.L.; Li, Y.M. Synthesis and evaluation of furoxan based nitric oxide-releasing derivatives of tetrahydroisoquinoline as anticancer and multidrug resistance reversal agents. Bioorg. Med. Chem. Lett. 2011, 21, 5934–5938. [Google Scholar] [CrossRef]
- Zou, Z.H.; Lan, X.B.; Tang, C.L.; Zhu, X.Y.; Liu, B.M.; Qian, H.; Huang, W.L.; Li, Y.M. Synthesis and biological evaluation of a series of 6,7-dimethoxy-1-(3,4-dimethoxybenzyl)-2-substituted tetrahydro-isoquinoline derivatives. Med. Chem. 2012, 8, 711–716. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. Phosphate mediated biomimetic synthesis of tetrahydroisoquinoline alkaloids. Chem. Commun. 2011, 47, 3242–3244. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Liang, A.; Desai, U.R. Designing non-saccharide, allosteric activators of antithrombin for accelerated inhibition of factor Xa. J. Med. Chem. 2011, 54, 6125–6138. [Google Scholar] [CrossRef]
- Al-Horani Desai, U.R. Electronically rich N-substituted tetrahydoisoquinoline 3-carboxilic acid esters: Concise synthesis and conformationl studies. Tetrahedron 2012, 68, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Leese, M.P.; Jourdan, F.L.; Dohle, W.; Kimberley, M.R.; Thomas, M.P.; Bai, R.; Hamel, E.; Ferrandis, E.; Potter, B.V.L. Steroidomimetic tetrahydroisoquinolines for the design of new microtubule disruptors. ACS Med. Chem. Lett. 2012, 3, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Leese, M.P.; Jourdan, F.L.; Major, M.R.; Dohle, W.; Hamel, E.; Ferrandis, E.; Fiore, A.; Kasprzyk, P.G.; Potter, B.V.L. Tetrahydro-isoquinolinone-based steroidomimetic and chimeric microtubule disruptors. ChemMedChem 2014, 9. [Google Scholar] [CrossRef]
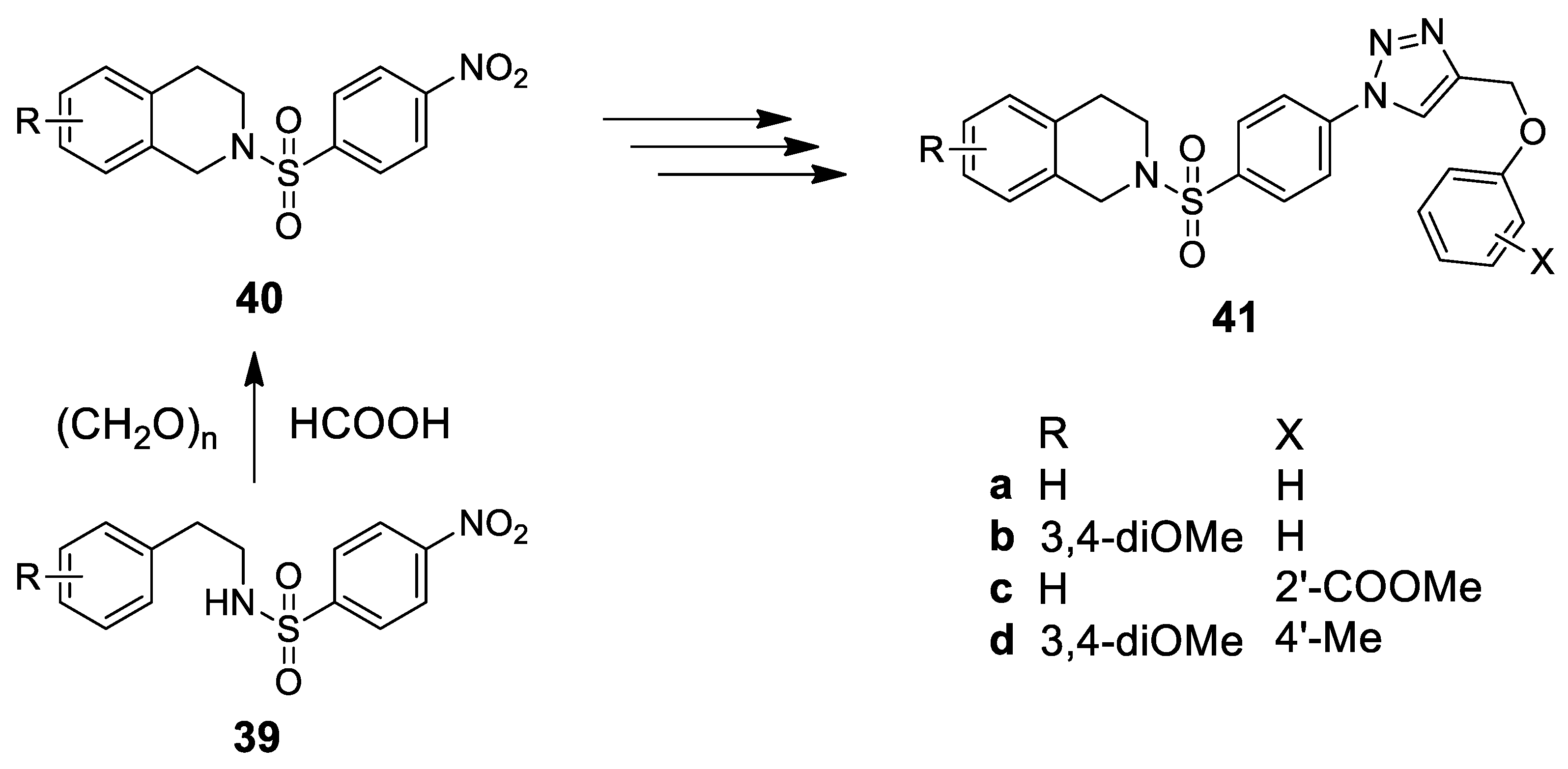
- Pingaew, R.; Worachartcheewan, A.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, cytotoxicity and QSAR study of N-tosyl-1,2,3,4-tetrahydro-isoquinoline derivatives. Arch. Pharm. Res. 2013, 36, 1066–1077. [Google Scholar] [CrossRef]
- Pingaew, R.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Design, synthesis and molecular docking studies of novel N-benzenesulfonyl-1,2,3,4-tetrahydroisoquinoline-based triazole with potential anticancer activity. Eur. J. Med. Chem. 2014, 81, 192–203. [Google Scholar] [CrossRef]
- Xu, G.; Guo, J.; Yan, Z.; Wang, N.; Liu, Z.-Z. Synthesis and citotoxicity of dinuclear platinum (II) complexes of (1S,3S)-1,2,3,4-tetrahydroisoquinolines. Chin. Chem. Lett. 2013, 24, 186–188. [Google Scholar] [CrossRef]
- Mons, E.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Organocatalytic enantioselective Pictet-Spengler reactions for the syntheses of 1-substituted 1,2,3,4-tetrahydro-isoquinolines. J. Org. Chem. 2014, 79, 7380–7390. [Google Scholar] [CrossRef]
- Guzman, J.D.; Pesnot, T.; Barrera, D.A.; Davies, H.M.; McMahon, E.; Evangelopoulos, D.; Mortazavi, P.N.; Munshi, T.; Maitra, A.; Lamming, E.D.; et al. Tetrahydroisoquinolines affect the whole-cell phenotype of Mycobacterium tuberculosis by inhibiting the ATP-dependent MurE ligase. J. Antimicrob. Chemoter. 2015, 70, 1691–1703. [Google Scholar] [CrossRef]
- Ruiz-Olalla, A.; Würdemann, M.A.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Organocatalytic enantioselective Pictet-Spengler approach to biologically relevant 1-benzyl-1,2,3,4-tetrahydroisoquinoline alkaloids. J. Org. Chem. 2015, 80, 5125–5132. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Chang, Y.-T.; Lin, C.-L.; Liaw, C.-C.; Kuo, Y.H.; Tu, L.-C.; Yeh, S.F.; Chern, J.-W. Synthesis of 1-substituted carbazolyl-1,2,3,4-tetrahydro- and carbazolyl-3,4-dihydro-β-carboline analogs as potential antitumor agents. Mar. Drugs 2011, 9, 256–277. [Google Scholar] [CrossRef]
- Painter, T.O.; Wang, L.; Majumder, S.; Xie, X.-Q.; Brummond, K.M. Diverging DOS strategy using an allene-containing tryptophan scaffold and a library design that maximize biologically relevant chemical space while minimizing the number of compounds. ACS Comb. Sci. 2011, 13, 166–174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arai, T.; Wasai, M.; Yokoyama, N. Easy access to fully functionalized chiral tetrahydro-β-carboline alkaloids. J. Org. Chem. 2011, 76, 2909–2916. [Google Scholar] [CrossRef] [PubMed]
- Duce, S.; Pesciaioli, F.; Gramigna, L.; Bernardi, L.; Mazzanti, A.; Ricci, A.; Bartoli, G.; Bencivenni, G. An easy entry to optically active spiroindolinones: Chiral Brønsted acid-catalysed Pictet-Spengler reactions of isatins. Adv. Synth. Catal. 2011, 353, 860–864. [Google Scholar] [CrossRef]
- Badillo, J.J.; Silva-Garcia, A.; Shupe, B.H.; Fettinger, J.C.; Franz, A.K. Enantioselective Pictet-Spengler reactions of isatins for the synthesis of spiroindolones. Tetrahedron Lett. 2011, 52, 5550–5553. [Google Scholar] [CrossRef]
- Zou, B.; Yap, P.; Sonntag, L.-S.; Leong, S.Y.; Yeung, B.K.S.; Keller, T.H. Mechanistic study of the spiroindolones: A new class of antimalarial. Molecules 2012, 17, 10131–10141. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Hayano, M.; Shimada, K.; Stockwell, B.R. Design and synthesis of Pictet-Spengler condensation products that exhibit oncogenic-RAS synthetic lethality and induce non-apoptotic cell death. Bioorg. Med. Chem. Lett. 2012, 22, 5707–5713. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, D.; Gödecke, T.; Chen, S.-N.; White, J.; Lankin, D.C.; Pauli, G.F.; van Breemen, R.B. Mass spectrometric dereplication of nitrogen-containing constituents of black cohosh (Cimifuga racemosa L.). Fitoterapia 2012, 83, 441–460. [Google Scholar] [CrossRef]
- Huang, D.; Xu, F.; Lin, X.; Wang, Y. High enantioselective Pictet-Spengler reaction catalyzed by SPINOL phosphoric acids. Chem. Eur. J. 2012, 18, 3148–3152. [Google Scholar] [CrossRef]
- Rashid, N.; Alam, S.; Hasan, M.; Khan, K.M.; Duddeck, H.; Pescitelli, G.; Kenéz, A.; Antus, S.; Kurtán, T. Cis-diastereoselectivity in Pictet-Spengler reaction of L-tryptophan and Electronic Circular Dicroism Studies. Chirality 2012, 24, 789–795. [Google Scholar] [CrossRef]
- Brummond, K.M.; Goodell, J.R.; LaPorte, M.G.; Wang, L.; Xie, X.-Q. Synthesis and in silico screening of a library of β-carboline-containing compounds. Beilstein J. Org. Chem. 2012, 8, 1048–1058. [Google Scholar] [CrossRef]
- Edwankar, R.V.; Edwankar, C.R.; Namioshi, O.A.; Deschamps, J.R.; Cook, J.M. Brönsted acid mediated cyclization of enaminones. Rapid and efficient access to the tetracyclic framework of the Strychnos alkaloids. J. Nat. Prod. 2012, 75, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fan, Z.; Dong, J.; Shi, X.-X.; Lu, X. Novel asymmetric total syntheses of (R)-pyridindolol, (R)-pyrindolol K1, and (R)-pyrindolol K2 via a mild one-pot aromatization of N-tosyl-tetrahydro-β-carboline with (S)-2,3-isopropylidene-L-glyceraldehyde as the source of chirality. Tetrahedron Asymmetry 2013, 24, 633–637. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, M.; Nand Rai, N.; Savant, D. Mild and efficient cyanuric chloride catalyzed Pictet-Spengler reaction. Beilstein J. Org. Chem. 2013, 9, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-R.; Wu, H.; Xiang, B.; Yu, W.-B.; Han, L.; Jia, Y.-X. Highly enantioselective construction of trifluoromethylated carbon quaternary stereocenters via nickel-catalyzed Friedel-Crafts alkylation reaction. J. Am. Chem. Soc. 2013, 135, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Lesma, G.; Cecchi, R.; Cagnotto, A.; Gobbi, M.; Mereghetti, F.; Musolino, M.; Sacchetti, A.; Silvani, A. Tetrahydro-β-carboline-basedspyrocyclic lactam as type II’ β-turn: Application to the synthesis and biological evaluation of somatostatine mimetics. J. Org. Chem. 2013, 78, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Pulka, K.; Slupska, M.; Puszko, M.; Wilczek, M.; Kozminski, W.; Misicka, A. peptides and peptidoaldehydes as substrates for the Pictet-Spengler reaction. J. Pept. Sci. 2013, 19, 433–440. [Google Scholar] [CrossRef] [PubMed]
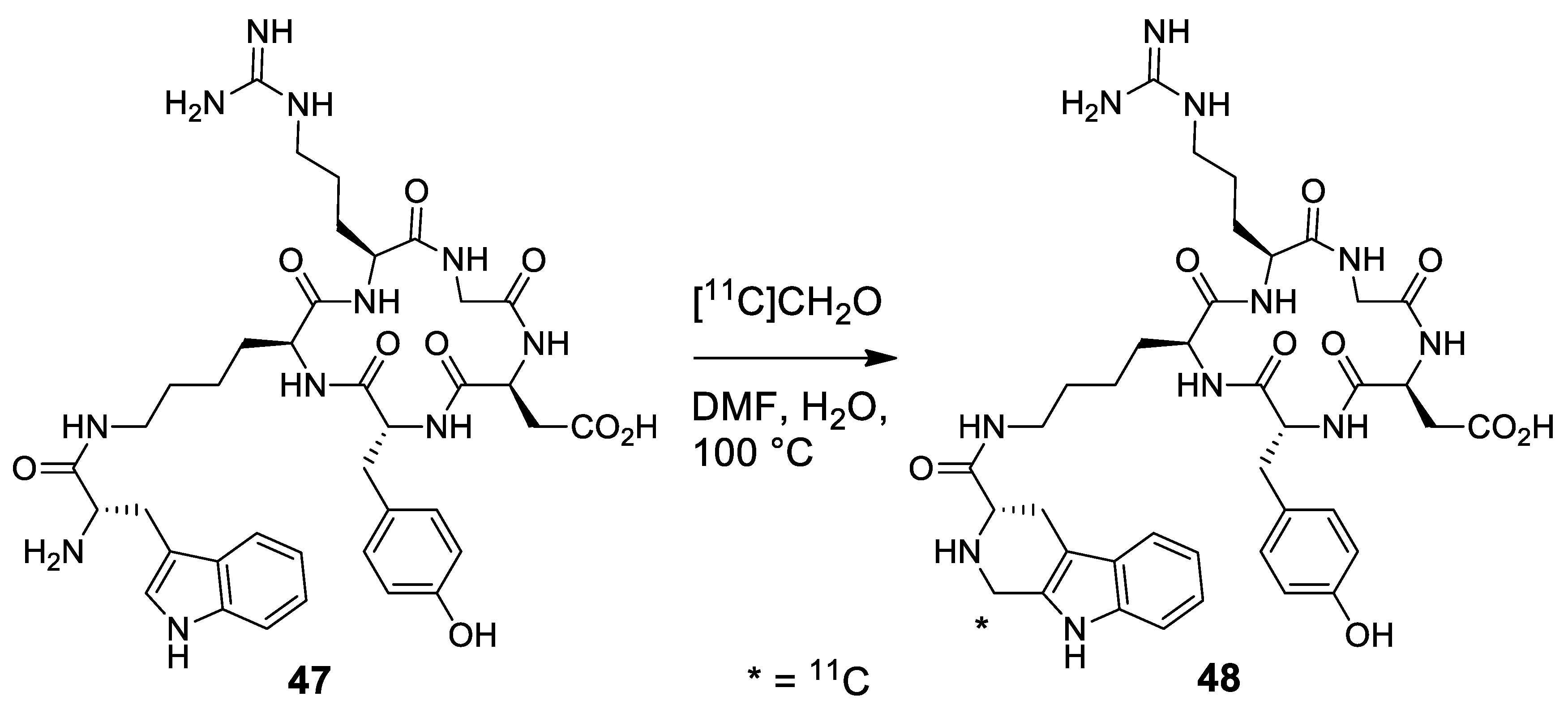
- Hanyu, M.; Takada, Y.; Hashimoto, H.; Kawamura, K.; Zhang, M.-R.; Fukumura, T. Carbon-11 radiolabeling of an oligopeptide containing tryptophan hydrochloride via a Pictet-Spengler reaction using carbon-11 formaldehyde. J. Pept. Sci. 2013, 19, 663–668. [Google Scholar] [CrossRef]
- Wang, L.-N.; Shen, S.-L.; Qu, J. Simple and efficient synthesis of tetrahydro-β-carboline via the Pictet-Spengler reaction in1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). RSC Adv. 2014, 4, 30733–30741. [Google Scholar] [CrossRef]
- Brokamp, R.; Bergmann, B.; Müller, I.B.; Bienz, S. Stereoselective preparation of pyridoxal 1,2,3,4-tetrahydro-β-carboline derivatives and the influence of their absolute and relative configuration on the proliferation of the malaria parasite Plasmodium falciparum. Bioorg Med. Chem. 2014, 22, 1832–1837. [Google Scholar] [CrossRef]
- Kabeshov, M.A.; Musio, B.; Murray, P.R.D.; Browne, D.L.; Ley, S.V. Expedient preparation of nazlinine and a small library of indole alkaloid using flow electrochemistry as an enabling technology. Org. Lett. 2014, 16, 4618–4621. [Google Scholar] [CrossRef]
- Mittal, N.; Sun, D.X.; Seidel, D. Conjugate-base-stabilized Brønsted acids: Catalytic enantioselective Pictet-Spengler reactions with unmodified tryptamine. Org. Lett. 2014, 16, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Slupska, M.; Pulka-Ziach, K.; Deluga, E.; Sosnowski, P.; Wilenska, B.; Kozminski, W.; Misicka, A. Synthesis of rigid tryptophan mimetics by the diastereoselective Pictet-Spengler reaction of β3-homo-tryptophan derivatives with chiral α-amino aldehydes. J. Pept. Sci. 2015, 21, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Vavsari, V.F.; Dianati, V.; Ramezanpour, S.; Balalaie, S. Stereoselective synthesis of functionalized tetrahydro-β-carbolines via Pictet-Spengler reaction. Synlett 2015, 26, 1955–1960. [Google Scholar] [CrossRef]
- Todd, M.H. Catalytic, Asymmetric Pictet-Spengler Reaction. OpenWetWare. 2016. Available online: https://openwetware.org/wiki/Todd:Catalytic,_Asymmetric_Pictet-Spengler_Reaction (accessed on 10 January 2020).
- Dalpozzo, R. The chiral pool in the Pictet-Spengler reaction for the synthesis of β-carbolines. Molecules 2016, 21, 699. [Google Scholar] [CrossRef]
- Mastranzo, V.M.; Yuste, F.; Ortiz, B.; Sánchez-Obregón, R.; García Ruano, J.L. Asymmetric synthesis of (S)-(−)-xylopinine.Use of the sulfinyl group as an ipso director in aromatic SE. J. Org. Chem. 2011, 76, 5036–5041. [Google Scholar] [CrossRef]
- Mastranzo, V.M.; Olivares Romero, J.L.; Yuste, F.; Ortiz, B.; Sánchez-Obregón, R.; García Ruano, J.L. Asymmetric synthesis of (S)-(−)-tetrahydropalmatine and (S)-(−)-canadine via sulfinyl directed Pictet-Spengler cyclization. Tetrahedron 2012, 68, 1266–1271. [Google Scholar] [CrossRef]
- Gómez-SanJuan, A.G.; Sotomayor, S.; Lete, E. Enantioselective intramolecular α-amidoalkylation reaction in the synthesis of pyrrolo[2,1-a]isoquinolines. Tetrahedron Lett. 2012, 53, 2157–2159. [Google Scholar] [CrossRef]
- Petersen, R.; Cohrt, A.E.; Petersen, M.Å.; Wu, P.; Clausen, M.H.; Nielsen, T.E. Synthesis of hexahydropyrrolo[2,1-a]isoquinoline compound libraries through a Pictet-Spengler cyclization/metal catalyzed cross coupling/amidation sequence. Bioorg. Med. Chem. 2015, 23, 2466–2469. [Google Scholar] [CrossRef]
- Lin, Y.D.; Cho, C.-L.; Ko, C.-W.; Pultte, A.; Wu, Y.-T. Palladium catalyzed annulation of 2,2’-diiodobiphenyls with alkynes: Synthesis and application of phenanthrenes. J. Org. Chem. 2012, 77, 9979–9988. [Google Scholar] [CrossRef]
- Su, B.; Cai, C.; Wang, Q. Enantioselective approach to 13a-methylphenanthroindozilidine A. J. Org. Chem. 2012, 77, 7981–7987. [Google Scholar] [CrossRef]
- Larsson, R.; Blanco, N.; Johansson, M.; Sterner, O. Synthesis of C-1 indol-3-yl substituted tetrahydroisoquinoline derivatives via a Pictet-Spengler approach. Tetrahedron Lett. 2012, 53, 4966–4970. [Google Scholar] [CrossRef]
- Jida, M.; Soueidan, O.-M.; Deprez, B.; Laconde, G.; Deprez-Poulain, R. Racemic and diastereoselective construction of indole alkaloid under solvent- and catalyst-free microwave-assisted Pictet-Spengler condensation. Green Chem. 2012, 14, 909–911. [Google Scholar] [CrossRef]
- Arumugam, N.; Raghunatan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno[4,3-b]pyrroles and indolizino[6,7-b]indoles as potent antimicrobial and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- El-Gamil, D.S.; Ahmed, N.S.; Gary, B.D.; Piazza, G.A.; Engel, M.; Hartmann, R.W.; Abadi, A.H. Design of novel β-carboline derivatives with pendant 5-bromothienyl and their evaluation as phosphodiesterase-5 inhibitors. Arch. Pharm. 2013, 346, 23–33. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, H.; Zhang, J.; Li, Y. Enantioselective synthesis and antimicrobial activities of tetrahydro-β-carboline diketopiperazines. Chirality 2013, 10, 656–662. [Google Scholar] [CrossRef]
- Airaghi, F.; Fiorati, A.; Lesma, G.; Musolino, M.; Sacchetti, A.; Silvani, A. The diketopiperazine-fused tetrahydro-β-carboline scaffold as a model peptidomimetic with an unusual α-turn secondary structure. Beilstein J. Org. Chem. 2013, 9, 147–154. [Google Scholar] [CrossRef]
- Lood, C.S.; Koskinen, A.M.P. Synthesis of (S)- and (R)-harmicine from proline: An approach toward tetrahydro-β-carbolines. Eur. J. Org. Chem. 2014, 2014, 2357–2364. [Google Scholar] [CrossRef]
- Lood, C.S.; Nieger, M.; Koskinen, A.M.P. Enantiospecific gram scale synthesis of (S)-eleagnine. Tetrahedron 2015, 71, 5019–5024. [Google Scholar] [CrossRef]
- Gurram, M.; Gyimóthy, B.; Wang, R.; Lam, S.Q.; Ahmed, F.; Herr, R.J. Concise enantiospecific, stereoselective syntheses of (±)-crispine A and its (−)-antipode. J. Org. Chem. 2011, 76, 1605–1613. [Google Scholar] [CrossRef]
- Yokoya, M.; Ito, H.; Saito, N. Synthesis of renieramycins: Construction of the core ring system of cribrostatin 4 through modified Pictet-Spengler cyclization of 3,6-bisarylpiperazine-2,5-dione with diethoxyethyl benzoate. Chem. Pharm. Bull. 2011, 59, 787–792. [Google Scholar] [CrossRef]
- Yokoya, M.; Ito, H.; Saito, N. Chemistry of renieramycins. Part 11: Total synthesis of (±)-cribrostatin 4. Tetrahedron 2011, 67, 9185–9192. [Google Scholar] [CrossRef]
- Yokoya, M.; Shinada-Fujino, K.; Saito, N. Chemistry of renieramycins. Part 9: Stereocontrolled total synthesis of (±)-renieramycin. Tetrahedron Lett. 2011, 52, 2446–2449. [Google Scholar] [CrossRef]
- Yokoya, M.; Shinada-Fujino, K.; Yoshida, S.; Mimura, M.; Takada, H.; Saito, N. Chemistry of renieramycins. Part 12: An improved total synthesis of (±)-renieramycin G. Tetrahedron 2012, 68, 4166–4181. [Google Scholar] [CrossRef]
- Petit, L.; Banwell, M.G.; Willis, A.C. The total synthesis of the crinine alkaloid hamayne via a Pd(0)-catalyzed intramolecular Alder-ene reaction. Org. Lett. 2011, 13, 5800–5803. [Google Scholar] [CrossRef]
- Huntley, R.J.; Funk, R.L. Total synthesis of (±)-γ-lycorane via the electrocyclic ring closure of a divinylpyrroline. Tetrahedron Lett. 2011, 52, 6671–6674. [Google Scholar] [CrossRef][Green Version]
- Dong, W.; Liu, W.; Liao, X.; Guan, B.; Chen, S.; Liu, Z. Asymmetric total synthesis of (−)-saframycin A from L-tyrosine. J. Org. Chem. 2011, 76, 5363–5368. [Google Scholar] [CrossRef]
- Kerschgens, I.P.; Claveau, E.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Total syntheses of mitragynine, paynantheine and specogynine via an enantioselective thiourea-catalysed Pictet-Spengler reaction. Chem. Commun. 2012, 48, 12243–12245. [Google Scholar] [CrossRef]
- Flink, H.; Jokela, R. Microwave-assisted total synthesis of tangutorine. Tetrahedron 2012, 68, 33811–33814. [Google Scholar] [CrossRef]
- Yoshida, A.; Akaiwa, M.; Asakawa, T.; Hamashima, Y.; Yokoshima, S.; Fukuyama, T.; Kan, T. Total synthesis of (−)-lemonomycin. Chem. Eur. J. 2012, 18, 11192–11195. [Google Scholar] [CrossRef]
- Jiménez-Somarriba, A.; Williams, R.M. Synthetic studies on lemonomycin: Construction of the tetracyclic core. Tetrahedron 2013, 69, 7505–7512. [Google Scholar] [CrossRef][Green Version]
- Lebold, T.P.; Wood, J.L.; Deitch, J.; Lodewik, M.W.; Tantillo, D.J.; Sarpong, R. A divergent approach to the synthesis of the yohimbinoid alkaloids venenetine and alstovenine. Nat. Chem. 2013, 5, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Dong, W.; Liu, W.; Yan, Z.; Wang, N.; Liu, Z. Synthesis and cytotoxicity of 3-acrylic amide derivatives of the simplified saframycin-ecteinascidin skeleton prepared from L-DOPA. Eur. J. Med. Chem. 2013, 62, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Z.; Wang, Y.; Tang, Y.-F.; Chen, S.-Z.; Chen, X.-G.; Li, H.-Y. Synthesis and antitumor activity of simplified ecteinascidin–saframycin analogs. Bioorg. Med. Chem. Lett. 2006, 16, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Edwankart, C.R.; Edwankart, R.V.; Namjashi, O.A.; Liao, X.; Cook, J.M. Stereospecific approach to the synthesis of ring-A oxygenated Sarpagine indole alkaloids. Total synthesis of the dimeric P-(+)-dispegatrine and six other monomeric indole alkaloids. J. Org. Chem. 2013, 78, 6471–6478. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, H.; Chen, X. Asymmetric total synthesis of (−)-jorunnamycins A and C and (−)-jorumycin from L-tyrosine. J. Nat. Prod. 2013, 76, 1789–1795. [Google Scholar] [CrossRef]
- Liu, H.; Chen, R.; Chen, X. A rapid and efficient access to renieramycin-type alkaloids featuring a temperature–dependent stereoselective cyclization. Org. Biomol. Chem. 2014, 12, 1633–1640. [Google Scholar]
- Craig, D.; Goldberg, F.W.; Pett, R.V.; Tholen, N.T.H.; White, A.J.P. Aziridine-based concise synthesis of (±)-alstonerine. Chem.Commun. 2013, 49, 9275–9277. [Google Scholar] [CrossRef]
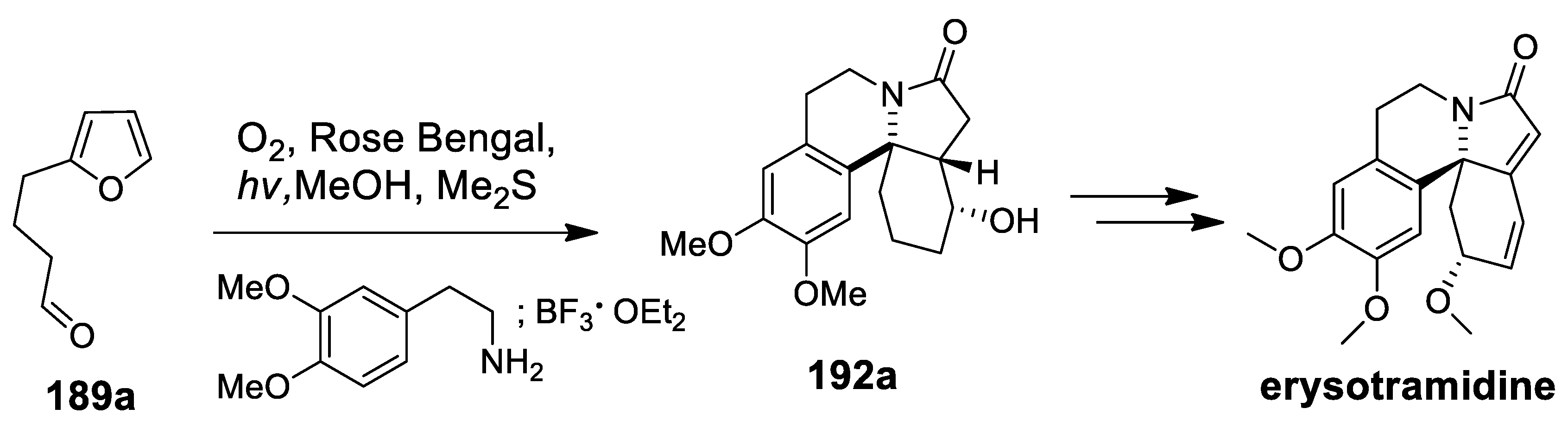
- L’Homme, C.; Menard, M.A.; Canesi, S. Synthesis of the Erythrina Alkaloid Erysotramidine. J. Org. Chem. 2014, 79, 8481–8485. [Google Scholar] [CrossRef]
- Maertens, G.; L’Homme, C.; Canesi, S. Total synthesis of natural products using hypervalent iodine reagents. Front. Chem. 2015, 2, 1–16. [Google Scholar] [CrossRef]
- Granger, B.A.; Jewett, I.T.; Butler, J.D.; Martin, S.F. Concise total synthesis of (±)-actynophyllic acid. Tetrahedron 2014, 70, 4094–4104. [Google Scholar] [CrossRef]
- Nakai, K.; Kubo, K.; Yokoya, M.; Saito, N. Preparation of renieramycin left-half model compounds. Tetrahedron 2014, 70, 6529–6545. [Google Scholar] [CrossRef]
- Yokoya, M.; Kobayashi, K.; Sato, M.; Saito, N. Chemistry of renieramycins. Part 14: Total synthesis of renieramycin I and practical synthesis of cribrostatin 4 (renieramycin H). Mar. Drugs 2015, 13, 4915–4933. [Google Scholar] [CrossRef] [PubMed]
- Herlè, B.; Wanner, M.J.; van Maarseveen, J.H.; Hiemstra, H. Total synthesis of (+)-yohimbine via an enantioselective organocatalytic Pictet-Spengler reaction. J. Org. Chem. 2011, 76, 8907–8912. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Oonishi, Y.; Takimoto, M.; Sato, Y. Total synthesis of (−)-corynantheidine by nickel-catalyzed carboxylative cyclization of enynes. Eur. J. Org. Chem. 2011, 2011, 2606–2609. [Google Scholar] [CrossRef]
- Wanner, M.J.; Claveau, E.; van Maarseveen, J.H.; Hiemstra, H. Enantioselective syntheses of Corinanthe alkaloids by chiral Brønsted acid and palladium catalysis. Chem. Eur. J. 2011, 17, 13680–13683. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, G.O.; Wang, Z.-J.; Namjoshi, O.A.; Deschamps, J.R.; Cook, J.M. First stereospecific total synthesis of (−)-affinisine oxindole as well as facile entry into the C(7)-diastereomeric chitosenine stereochemistry. Tetrahedron Lett. 2015, 56, 3052–3056. [Google Scholar] [CrossRef]
- Casciaro, B.; Calcaterra, A.; Cappiello, F.; Mori, M.; Loffredo, M.R.; Ghirga, F.; Mangoni, M.L.; Botta, B.; Quaglio, D. Nigritanine as a New Potential Antimicrobial Alkaloid for the Treatment of Staphylococcus aureus-Induced Infections. Toxins 2019, 11, 511. [Google Scholar] [CrossRef]
- Bonnet, D.; Ganesan, A. Solid-phase synthesis of tetrahydro-β-carbolinehydantoins via the N-acyliminium Pictet-Spengler reaction and cyclative cleavage. J. Comb. Chem. 2002, 4, 546–548. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Sanderson, S.J.; Mottram, J.C.; Coombs, G.H.; Meldal, M. Combinatorial library of peptidotriazoles: Identification of [1,2,3]-triazoles inhibitors against a recombinant Leishmania mexicana cysteine protease. J. Comb. Chem. 2004, 6, 312–324. [Google Scholar] [CrossRef]
- Zhu, Z.; Sun, Z.Y.; Ye, Y.; McKittrick, B.; Greenlee, W.; Czamiecki, M.; Fawzi, A.; Zhang, H.; Lackowicz, J.E. Design and discovery of 1,3-benzodiazepine as novel dopamine antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 5218–5221. [Google Scholar] [CrossRef]
- Hall, D.G.; Manku, S.; Wang, F. Solution- and solid-phase strategies for the design, synthesis, and screening of libraries based on natural product template: A comprehensive survey. J. Comb. Chem. 2001, 3, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Breibauer, R.; Vetter, I.R.; Waldmann, H. From Protein Domains to Drug Candidates—Natural Products as Guiding Principles in the Design and Synthesis of Compound Libraries. Angew. Chem. Int. Ed. 2002, 41, 2878–2890. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Diness, F.; Meldal, M. The Pictet-Spengler reaction in solid-phase combinatorial chemistry. Curr. Opin. Drug Discov. Dev. 2003, 6, 801–814. [Google Scholar] [CrossRef]
- Le Quement, S.T.; Petersen, R.; Meldal, M.; Nielsen, T.E. N-Acyliminium Intermediates in Solid-Phase Synthesis. Biopolymers 2009, 94, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.E.; Meldal, M. Solid–phase synthesis of pyrroloisoquinolines via the intra-molecular N-acyliminium Pictet-Spengler reaction. J. Comb. Chem. 2005, 7, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.E.; Le Quement, S.; Meldal, M. Solid–phase synthesis of bicyclic dipeptide mimetics by intramolecular cyclization of alcohols, thiols, amines, and amides with N-acyliminium intermediates. Org. Lett. 2005, 7, 3601–3604. [Google Scholar] [CrossRef]
- Diness, F.; Beyer, J.; Meldal, M. Solid–phase synthesis of tetrahydro-β-carbolines and tetrahydroisoquinolines by stereoselective intramolecular N-carbamyliminium Pictet-Spengler reactions. Chem. Eur. J. 2006, 12, 8056–8066. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Le Quement, S.; Meldal, M. Solid–phase synthesis of aryl-substituted thienoindolizines: Sequential Pictet-Spengler, bromination and Suzuki cross-coupling reactions of thiophenes. J. Comb. Chem. 2008, 10, 447–455. [Google Scholar]
- Chanda, K.; Chou, C.T.; Lai, J.J.; Lin, S.-F.; Yellol, G.S.; Sun, C.M. Traceless synthesis of diketopiperazine fused tetrahydro-β-carbolines on soluble polymer support. Mol. Divers. 2011, 15, 569–581. [Google Scholar] [CrossRef]
- Komnatnyy, V.V.; Givskov, M.; Nielsen, T.E. Solid-Phase Synthesis of Structurally Diverse Heterocycles by an Amide–Ketone Condensation/N-Acyliminium Pictet-Spengler Sequence. Chem. Eur. J. 2012, 18, 16793–16800. [Google Scholar] [CrossRef]
- Ascic, E.; Jensen, J.F.; Nielsen, T.E. Synthesis of Heterocycles through a Ruthenium–Catalyzed Tandem Ring-Closing Metathesis/Isomerization/N-Acyliminium Cyclization Sequence. Angew. Chem. Int. Ed. 2011, 50, 5188–5191. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.L.; Clausen, J.W.; Ohm, R.G.; Ascic, E.; Le Quement, S.T.; Tanner, D.; Nielsen, T.E. Ruthenium hydride/Brønsted Acid-catalyzed tandem isomerization/N-acyliminium cyclization sequence for the synthesis of tetrahydro-β-carbolines. J. Org. Chem. 2013, 78, 12545–12565. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Liang, X.W.; Wang, S.G.; Zhang, J.W.; Zhang, X.; You, S.L. Ring-closing metathesis/isomerization/Pictet-Spengler cascade via ruthenium/chiral phosphoric acid sequential catalysis. Org. Lett. 2012, 14, 5022–5025. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Liang, X.W.; Wang, S.G.; You, S.L. An olefin isomerization/asymmetric Pictet-Spengler cascade via sequential catalysis of ruthenium alkylidene and chiral phosphoric acid. Org. Biomol. Chem. 2013, 11, 1602–1605. [Google Scholar] [CrossRef] [PubMed]
- Toda, Y.; Terada, M. Relay Catalysis by a Ruthenium Complex-Chiral Brønsted Acid Binary System for Ternary Reaction Sequence involving enantioselective Pictet-Spengler-Type Cyclization as the key step. Synlett 2013, 24, 752–756. [Google Scholar]
- Tietze, L.F. Domino Reactions in Organic Synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Parsons, P.J.; Penkett, C.S.; Shell, A.J. Tandem Reactions in Organic Synthesis: Novel Strategies for Natural Product elaboration and the development of new synthetic methodology. Chem. Rev. 1996, 96, 195–206. [Google Scholar] [CrossRef]
- Nicolau, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade Reactions in Total Synthesis. Angew. Chem. Int. Ed 2006, 45, 7134–7186. [Google Scholar] [CrossRef]
- Enders, D.; Grondal, C.; Hütti, M.R. Asymmetric Organocatalytic Domino Reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef]
- Padwa, A.; Bur, S.K. The Domino Way to Heterocycles. Tetrahedron 2007, 63, 5341–5378. [Google Scholar] [CrossRef]
- Eschenbrenner-Lux, V.; Düchert, H.; Khedkar, V.; Bruss, H.; Waldmann, H.; Kumar, K. Cascade syntheses routes to the centrocountins. Chem. Eur. J. 2013, 19, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Bur, S.K.; Padwa, A. The Pummerer reaction: Methodology and strategy for the synthesis of heterocyclic compounds. Chem. Rev. 2004, 104, 2401–2432. [Google Scholar] [CrossRef] [PubMed]
- Padwa, A.; Danca, M.D.; Hardcastle, K.I.; McClure, M.S. A short diastereoselective synthesis of the putative alkaloid jamtine using a tandem Pummerer/Mannich cyclization sequence. J. Org. Chem. 2003, 68, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Padwa, A.; Heidelbaugh, T.M.; Kuethe, J.T.; McClure, M.S.; Wang, Q. Tandem Pummerer/Mannich cyclization cascade of α-sulfinylamides as a method to prepare aza-heterocycles. J. Org. Chem. 2002, 67, 5928–5937. [Google Scholar] [CrossRef]
- Padwa, A.; Danca, M.D. Total synthesis of (±)-jamtine using a thionium/N-acyliminium ion cascade. Org. Lett. 2002, 4, 715–717. [Google Scholar] [CrossRef]
- Padwa, A.; Waterson, A.G. Studies dealing with thionium ion promoted Mannich cyclization reactions. J. Org. Chem. 2000, 65, 235–244. [Google Scholar] [CrossRef]
- Yang, T.; Campbell, L.; Dixon, D.J. A Au(I)-Catalyzed N-Acyl Iminium Ion Cyclization Cascade. J. Am. Chem. Soc. 2007, 128, 12070–12071. [Google Scholar] [CrossRef]
- Muratore, M.E.; Holloway, C.A.; Pilling, A.W.; Storer, R.I.; Trevitt, G.; Dixon, D.J. Enantioselective Brønsted acid-catalyzed N-acyliminium cyclization cascades. J. Am. Chem. Soc. 2009, 131, 10796–10797. [Google Scholar] [CrossRef]
- Holloway, C.A.; Muratore, M.E.; Storer, R.I.; Dixon, D.J. Direct enantioselective Brønsted acid catalyzed N-acyliminium cyclization cascades of tryptamines and ketoacids. Org. Lett. 2010, 12, 4720–4723. [Google Scholar] [CrossRef]
- Wang, Y.M.; Lackner, A.D.; Toste, F.D. Developments of catalysts and ligands for enantioselective gold catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, T.; Yang, Z. Strategic innovation in the total synthesis of complex natural products using gold catalysis. Nat. Prod. Rep. 2014, 31, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, S.M.; Konala, A.; Patil, N.T. When gold meets chiral Brønsted acid catalysts: Extending the boundaries of enantioselective gold catalysis. Chem. Commun. 2014, 50, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Graham, T.H. Gold-catalyzed formation of heterocycles–an enabling new technology for medicinal chemistry. Drug Discov. Today Technol. 2013, 10, e3–e14. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Hashmi, A.S. Gold catalysis in total synthesis—An update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L. Access to electron rich arene–fused hexahydroquinolizinones via Gold catalysis-triggered cascade process. Angew. Chem. Int. Ed. 2012, 51, 7301–7304. [Google Scholar] [CrossRef]
- Gregory, A.W.; Jakubec, P.; Turner, P.; Dixon, D.J. Gold and BINOL-phosphoric acid catalyzed enantioselective hydroamination/N-sulfonyliminium cyclization cascade. Org. Lett. 2013, 15, 4330–4333. [Google Scholar] [CrossRef]
- Pérez-Galán, P.; Delpont, N.; Herrero-Gómez, E.; Maseras, F.; Echavarren, A.M. Metal-arene interactions in dialkylbiarylphosphane complexes of copper, silver, and gold. Chemistry 2010, 16, 5324–5332. [Google Scholar] [CrossRef]
- Nieto-Oberhuber, C.; López, S.; Muñoz, M.P.; Cárdenas, D.J.; Buñuel, E.; Nevado, C.; Echavarren, A.M. Divergent mechanisms for the skeletal rearrangement and [2+2] cycloaddition of enynes catalyzed by gold. Angew. Chem. Int. Ed. 2005, 44, 6146–6148. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Yang, N.; Chen, Y.; Zhou, Y.; Ji, X.; Zhang, L.; Wang, J.; Xie, X.; Liu, H. Gold(I)-catalyzed cascade approach for the synthesis of tryptamine-based polycyclic privileged as α1-adrenergic receptor antagonists. J. Org. Chem. 2013, 78, 10802–10811. [Google Scholar] [CrossRef]
- Iqbal, N.; Fiksdahl, A. Gold (I)-catalyzed benz[c]azepin-4-ol synthesis by intermolecular [5 + 2] cycloaddition. J. Org. Chem. 2013, 78, 7885–7895. [Google Scholar] [CrossRef]
- Danda, A.; Kumar, K.; Waldmann, H. A general catalytic reaction sequence to access alkaloid-inspired indole polycycles. Chem. Commun. 2015, 51, 7536–7539. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shi, M. Yb(OTf)3-or (Au(I)-catalyzed domino intramolecular hydroamination and ring-opening of sulfonamide-substituted 1,1-vinylidenecyclopropanediesters. Chem. Eur. J. 2011, 17, 13160–13165. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, R.L.; Sherry, B.D.; Kang, E.J.; Toste, F.D. Gold (I)-catalyzed enantioselective intramolecular hydroamination of allenes. J. Am. Chem. Soc. 2007, 129, 2452–2453. [Google Scholar] [CrossRef] [PubMed]
- Gobé, V.; Guinchard, X. Pd(0)-catalyzed tandem deprotection/cyclization of tetrahydro-β-carbolines on allenes: Application to the synthesis of indolo[2,3-a]quinolizidines. Org. Lett. 2014, 16, 1924–1927. [Google Scholar] [CrossRef]
- Gobé, V.; Guinchard, X. Stereoselective synthesis of chiral polycyclic indolic architectures trough Pd0-catalyzed tandem deprotection/cyclization of tetrahydro-β-carbolines on allenes. Chem. Eur. J. 2015, 21, 8511–8520. [Google Scholar] [CrossRef]
- Gobé, V.; Retailleau, P.; Guinchard, X. Self-relay gold(I)-catalyzed Pictet-Spengler/cyclization cascade reaction for the rapid elaboration of pentacyclic indole derivatives. Chem. Eur. J. 2015, 21, 17587–17590. [Google Scholar] [CrossRef]
- Wu, X.; Dai, X.; Nie, L.; Fang, H.; Chen, J.; Ren, Z.; Cao, W.; Zhao, G. Organocatalyzed enantioselective one-pot three-component access to indoloquinolizidines by a Michael addition-Pictet-Spengler sequence. Chem. Commun. 2010, 46, 2733–2735. [Google Scholar] [CrossRef]
- Fang, H.; Wu, X.; Nie, L.; Dai, X.; Chen, J.; Cao, W.; Zhao, G. Diastereoselective synthesis of indoloquinolizidines by a Pictet-Spengler lactamization cascade. Org. Lett. 2010, 12, 5366–5369. [Google Scholar] [CrossRef]
- Wu, X.; Dai, X.; Fang, H.; Nie, L.; Chen, J.; Cao, W.; Zhao, G. One Pot Three-Component Syntheses of Indoloquinolizidine Derivatives Using an Organocatalytic Michael Addition and Subsequent Pictet-Spengler Cyclization. Chem. Eur. J. 2011, 17, 10510–10514. [Google Scholar] [CrossRef]
- Dai, X.; Wu, X.; Fang, H.; Nie, L.; Chen, J.; Deng, H.; Cao, W.; Zhao, G. Enantioselective organocatalyzed cascade reactions to highly functionalized quinolizidines. Tetrahedron 2011, 67, 3034–3040. [Google Scholar] [CrossRef]
- Wu, X.; Fang, H.; Liu, Q.; Nie, L.; Chen, J.; Cao, W.; Zhao, G. Diastereoselective cascade reactions toward substituted diazaindeno[2,1-α]-phenanthrenes. Tetrahedron 2011, 67, 7251–7257. [Google Scholar] [CrossRef]
- Rueping, M.; Volla, C.M.R. Brønsted-acid catalyzed condensation-Michael reaction-Pictet-Spengler cyclization—Highly stereoselective synthesis of indoloquinolizidines. RSC Adv. 2011, 1, 79–82. [Google Scholar] [CrossRef]
- Rueping, M.; Volla, C.M.R.; Bolte, M.; Raabe, G. General and Efficient Organocatalytic Synthesis of Indoloquinolizidines, Pyridoquinazolines and Quinazolinones through a One-Pot Domino Michael Addition-Cyclization-Pictet-Spengler or 1,2-Amine Addition Reaction. Adv. Synth. Catal. 2011, 353, 2853–2859. [Google Scholar] [CrossRef]
- Albrecht, A.; Morana, F.; Fraile, A.; Jørgensen, K.A. Organophosphorus reagents in organocatalysis: Synthesis of optically active α-methylene-δ-lactones and δ-lactams. Chem. Eur. J. 2012, 18, 10348–10354. [Google Scholar] [CrossRef]
- Zhu, H.-L.; Ling, J.-B.; Xu, P.-F. α-Oxo-γ-butyrolactam, N-containing pronucleophile in organocatalytic one-pot assembly of butyrolactam-fused indoloquinolizidines. J. Org. Chem. 2012, 77, 7737–7743. [Google Scholar] [CrossRef]
- Mayer, S.F.; Kroutil, W.; Faber, K. Cooperative multi-catalyst system for one-pot organic transformation. Chem. Soc. Rev. 2004, 33, 301–312. [Google Scholar]
- Veum, L.; Hanefeld, U. Carrier enabled catalytic reaction cascade. Chem. Commun. 2006, 825–831. [Google Scholar] [CrossRef]
- Pilling, A.W.; Böhmer, J.; Dixon, D.J. Site-Isolated Base–and Acid-mediated Michael initiated cascades. Angew. Chem. Int. Ed. 2007, 46, 5428–5430. [Google Scholar] [CrossRef]
- Pilling, A.W.; Böhmer, J.; Dixon, D.J. Site isolated base and acid-catalyzed azaspyrocyclization cascades. Chem. Commun. 2008, 7, 832–834. [Google Scholar] [CrossRef]
- Muratore, M.E.; Shi, L.; Pilling, A.W.; Storer, R.I.; Dixon, D.J. Exploiting a novel size exclusion phenomenon for enantioselective acid/base cascade catalysis. Chem. Commun. 2012, 48, 6351–6353. [Google Scholar] [CrossRef]
- Hong, B.-C.; Liao, W.-K.; Dange, N.S.; Liao, J.-H. One-Pot Organocatalytic Enantioselective Domino Double-Michael Reaction and Pictet-Spengler–Lactamization Reaction. A Facile Entry to the “Inside Yohimbane” System with Five Contiguous Stereogenic Centers. Org. Lett. 2013, 15, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Aillaud, I.; Barber, D.M.; Thompson, A.L.; Dixon, D.J. Enantoselective Michael Addition/Iminium Ion Cyclization Cascades of Tryptamine-Derived Ureas. Org. Lett. 2013, 15, 2946–2949. [Google Scholar] [CrossRef]
- Sridharan, V.; Avendaño, C.; Menéndez, J.C. General, mild and efficient synthesis of β-enaminones catalyzed by ceric ammonium nitrate. Synlett 2007, 881–884. [Google Scholar] [CrossRef]
- Suryavanshi, P.A.; Sridharan, V.; Menéndez, J.C. A β-enaminone-initiated multicomponent domino reaction for the synthesis of indoloquinolizidines and benzoquinolines from acyclic precursors. Chem. Eur. J. 2013, 19, 13207–13215. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, L.-L.; Yan, C.-G. Facile construction of 1,2,6,7,12,12b-hexahydroindolo[2,3a]quinolizines via one-pot three component reactions of tryptamines, propiolate, and α-β-unsaturated aromatic aldehydes or ketones. Tetrahedron 2013, 69, 5451–5459. [Google Scholar] [CrossRef]
- Zhu, D.; Sun, J.; Yan, C.G. One-pot synthesis of 6,11-dihydro-5H-indolizino[8,7-b]indoles via sequential formation of β-enamino ester, Michael addition and Pictet-Spengler reactions. RSC Adv. 2014, 4, 62817–62826. [Google Scholar] [CrossRef]
- Dai, W.; Lu, H.; Li, X.; Shi, F.; Tu, S.-J. Diastereo- and enantioselective construction of bispiro oxindole scaffold containing a tetraydro-β-carboline moiety through an organocatalytic asymmetric cascade reaction. Chem. Eur. J. 2014, 20, 11382–11389. [Google Scholar] [CrossRef]
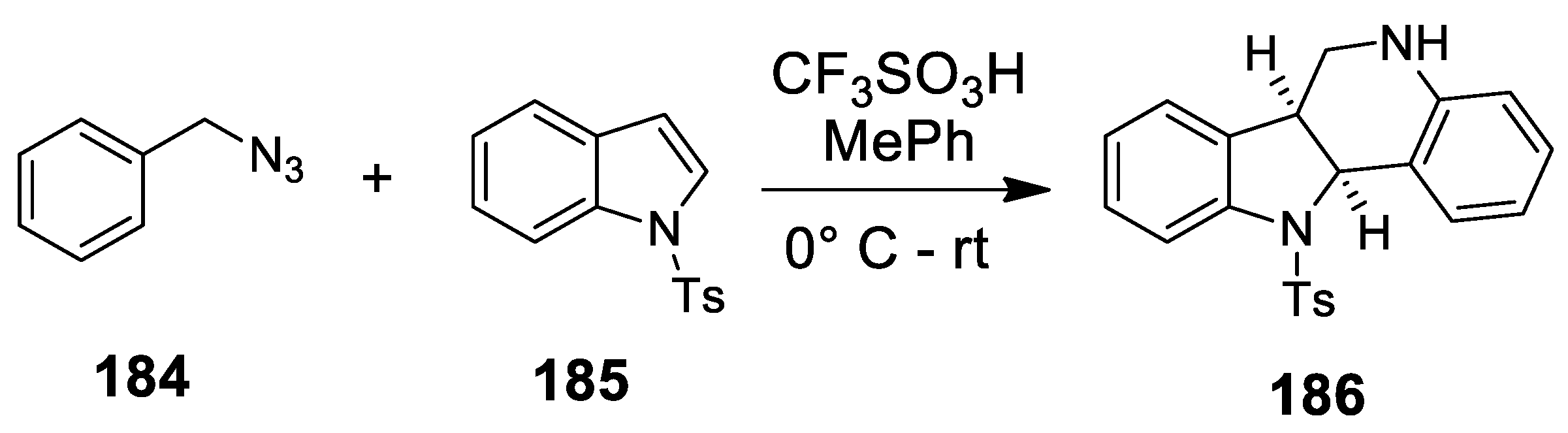
- Song, Z.; Zhao, Y.-M.; Zai, H. One step construction of tetrahydro-5H-indolo[3,2-c]quinolines from benzyl azides and indoles via a cascade reaction sequence. Org. Lett. 2011, 24, 6331–6333. [Google Scholar] [CrossRef]
- Zhang, W.; Bah, J.; Wohlfarth, A.; Franzén, J. A stereodivergent strategy for the preparation of corynantheine and ipecac alkaloids, their epimers, and analogues: Efficient total synthesis of (−)-di-hydrocorynantheol, (−)-protoemetinol, (−)-corinantheal, (−)-protoemetine, and related natural and nonnatural compounds chemistry. Chem. Eur. J. 2011, 17, 13814–13824. [Google Scholar]
- Yokosaka, T.; Nemoto, T.; Hamada, Y. An acid-promoted novel skeletal rearrangement initiated by intramolecular ipso-Friedel-Crafts-type addition to 3-alkylidene indolenium cations. Chem. Commun. 2012, 48, 5431–5433. [Google Scholar] [CrossRef]
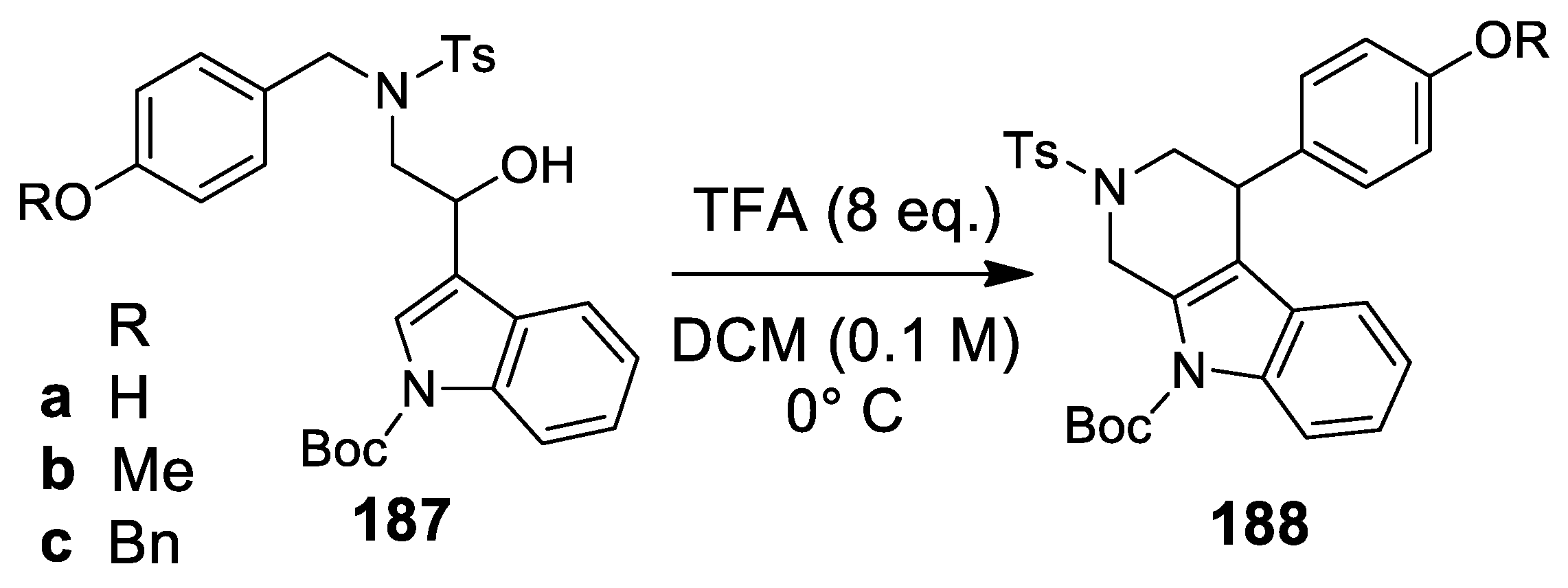
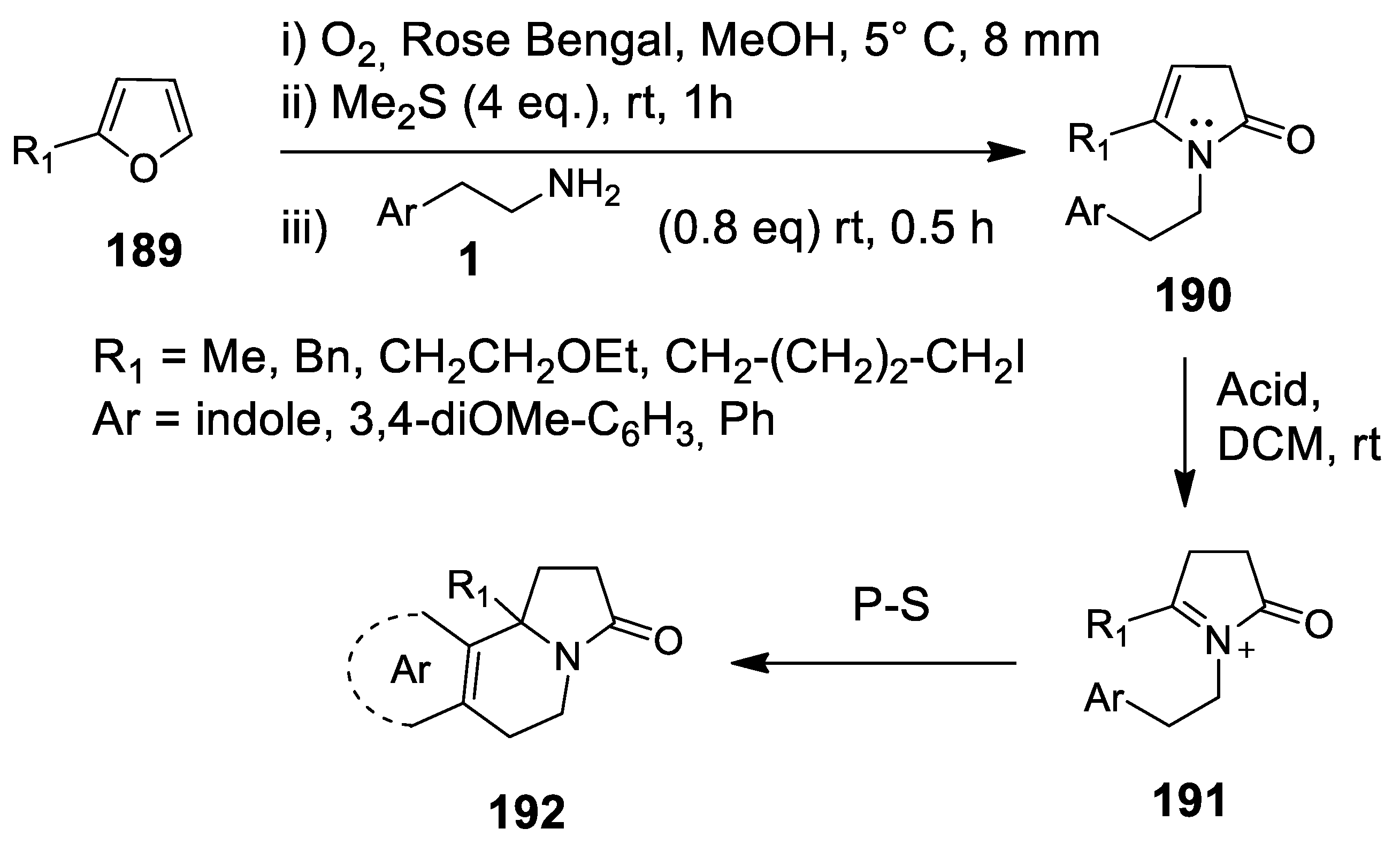
- Kalaitzakis, D.; Montagnon, T.; Antonatu, E.; Bardaji, N.; Vassilikogiannakis, G. From simple Furans to complex Nitrogen-bearing aromatic polycycles by means of a flexible and general reaction sequence initiated by singlet oxygen. Chem. Eur. J. 2013, 19, 10119–11021. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzakis, D.; Montagnon, T.; Antonatu, E.; Vassilikogiannakis, G. One-pot synthesis of the tetracyclic framework of the aromatic erythrina alkaloids from simple furans. Org. Lett. 2013, 15, 3714–3717. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, C.-H.; Yellol, G.S.; Tsai, C.H.; Dalvi, P.B.; Sun, C.-M. Diastereoselective synthesis of bridged polycyclic alkaloids via tandem acylation/intramolecular Diels–Alder reaction. J. Org. Chem. 2013, 78, 9738–9747. [Google Scholar] [CrossRef] [PubMed]
- Boomhoff, M.; Yadav, A.K.; Appun, J.; Schneider, C. Modular, flexible, and stereoselective synthesis of pyrroloquinolines: Rapid assembly of complex heterocyclic scaffolds. Org. Lett. 2014, 16, 6236–6239. [Google Scholar] [CrossRef] [PubMed]
- Jida, M.; Van der Poorten, O.; Guillemyn, K.; Urbanczyk-Lipkowska, Z.; Tourwé, D.; Ballet, S. T3P-promoted, mild, one-pot syntheses of constrained polycyclic lactam dipeptide analogues via stereoselective Pictet-Spengler and Meyers lactamization reactions. Org. Lett. 2015, 17, 4482–4485. [Google Scholar] [CrossRef]
- Prabhu, G.; Agarwal, S.; Sharma, V.; Madurkar, S.M.; Munshi, P.; Singh, S.; Sen, S. A natural product based DOS library of hybrid systems. Eur. J. Med. Chem. 2015, 95, 41–48. [Google Scholar] [CrossRef]
- Ugi, I. Recent progress in the chemistry of multicomponent reactions. Pure Appl. Chem. 2001, 73, 187–191. [Google Scholar] [CrossRef]
- Zhu, J.; Bienaymé, H. Multi-Component Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Sunderhaus, J.D.; Martin, S.F. Applications of Multicomponent Reactions to the synthesis of diverse heterocyclic scaffolds. Chem. Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef]
- Hulme, C.; Gore, V. “Multi-component Reactions: Emerging Chemistry in Drug Discovery” ‘From Xylocain to Crixivan’. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef]
- Ruijter, E.; Scheffelaar, R.; Orru, R.V.A. Multicomponent reaction Design in the quest for Molecular Complexity and Diversity. Angew Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef]
- Van der Heijden, G.; Ruijter, E.; Orru, R.V.A. Efficiency, Diversity, and Complexity with Multicomponent Reactions. Synlett 2013, 24, 666–687. [Google Scholar]
- Dömling, A.; Wang, R.; Wang, W. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Ganem, B. Strategies for the innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrücher, A.C. Ugi Reaktion. Angew. Chem. 1959, 71, 386. [Google Scholar]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanide. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Ugi, I.; Werner, B.; Dömling, A. The chemistry of Isocyanides, their MultiComponent Reactions and their Library. Molecules 2003, 8, 53–66. [Google Scholar] [CrossRef]
- Dömling, A. Recent Developments in Isocyanide Based MultiComponents Reactions in Applied Chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I. Isocyanide based multicomponent reaction in drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 324–331. [Google Scholar] [CrossRef]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reaction towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–596. [Google Scholar] [CrossRef]
- Mayer, J.P.; Bankaitis-Davis, D.; Zhang, J.; Beaton, G.; Bjergarde, K.; Andersen, C.M.; Goodman, B.A.; Herrera, C.J. Application of the Pictet-Spengler reaction in combinatorial chemistry. Tetrahedron Lett. 1996, 37, 5633–5636. [Google Scholar] [CrossRef]
- Lihu, Y.; Liangqin, G. Pictet-Spengler reaction on solid support. Tetrahedron Lett. 1996, 37, 5041–5044. [Google Scholar] [CrossRef]
- Hutchins, S.M.; Chapman, K.T. Solid phase synthesis of tetrahydroisoquinolines and tetrahydroimidazopyridine. Tetrahedron Lett. 1996, 37, 4865–4868. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. Concise synthesis of the cell cycle inhibitor demethoxy-fumitremorgin C. Tetrahedron Lett. 1997, 38, 4327–4328. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. The N-Acyliminium Pictet−Spengler Condensation as a Multicomponent Combinatorial Reaction on Solid Phase and Its Application to the Synthesis of Demethoxyfumitremorgin C Analogues. Org. Lett. 1999, 1, 1647–1649. [Google Scholar] [CrossRef]
- Van Loevezijin, A.; van Maarseeven, J.H.; Stegman, K.; Visser, G.M.; Koomen, G.-J. Solid phase synthesis of fumitremorgin, verruculogen and tryptostatin analogs based on a cyclization/cleavage strategy. Tetrahedron Lett. 1998, 39, 4737–4740. [Google Scholar] [CrossRef]
- El Kaim, L.; Gageat, M.; Gaultier, L.; Grimaud, L. New Ugi/Pictet-Spengler Multi-component Formation of Polycyclic Diketopiperazines from isocyanides and α-keto acids. Synlett 2007, 3, 502–503. [Google Scholar]
- Liu, H.; Dömling, A. Efficient and diverse synthesis of indole derivatives. J. Org. Chem. 2009, 74, 6895–6898. [Google Scholar] [CrossRef]
- Wang, W.; Herdtweck, E.; Dömling, A. Polycyclic indole alkaloid-type compounds by MCR. Chem. Commun. 2010, 46, 770–772. [Google Scholar] [CrossRef]
- Znabet, A.; Ruijter, E.; de Kanter, F.J.J.; Köhler, V.; Helliwell, M.; Turner, N.J.; Orru, R.V.A. Highly stereoselective synthesis of substituted prolyl peptides using a combination of bio-catalytic desymmetrization and multicomponent reactions. Angew. Chem. Int. Ed. 2010, 49, 5289–5292. [Google Scholar] [CrossRef]
- Znabet, A.; Zonneveld, J.; Janssen, E.; de Kanter, F.J.J.; Helliwell, M.; Turner, N.J.; Ruijter, E.; Orru, R.V.A. Asymmetric synthesis of synthetic alkaloids by a tandem biocatalysis/Ugi/Pictet-Spengler-type cyclization sequence. Chem. Commun. 2010, 46, 7706–7708. [Google Scholar] [CrossRef]
- Kaniskan, H.Ü.; Garner, P. An Efficient Synthetic Approach to Cyanocycline A and Bioxalomycin β2 via [C+NC+CC] Coupling. J. Am. Chem. Soc. 2007, 129, 15460–15461. [Google Scholar] [CrossRef]
- Neubauer, D.N. Almorexant, a dual orexin receptor antagonist for the treatment of insomnia. Curr. Opin. Investig. Drugs 2010, 11, 101–110. [Google Scholar]
- Hoever, P.; de Haas, S.; Winkler, J.; Schoemaker, R.C.; Chiossi, E.; van gerven, J.; Dingemanse, J. Orexin receptor antagonism, a new sleep pomoting paradigm: An ascending single-dose study with almorexant. Clin. Pharmacol. Ther. 2010, 87, 593–600. [Google Scholar] [CrossRef]
- Wang, W.; Ollio, S.; Herdtweck, H.; Dömling, A. Polycyclic compounds by Ugi-Pictet-Spengler sequence. J. Org. Chem. 2011, 76, 637–644. [Google Scholar] [CrossRef]
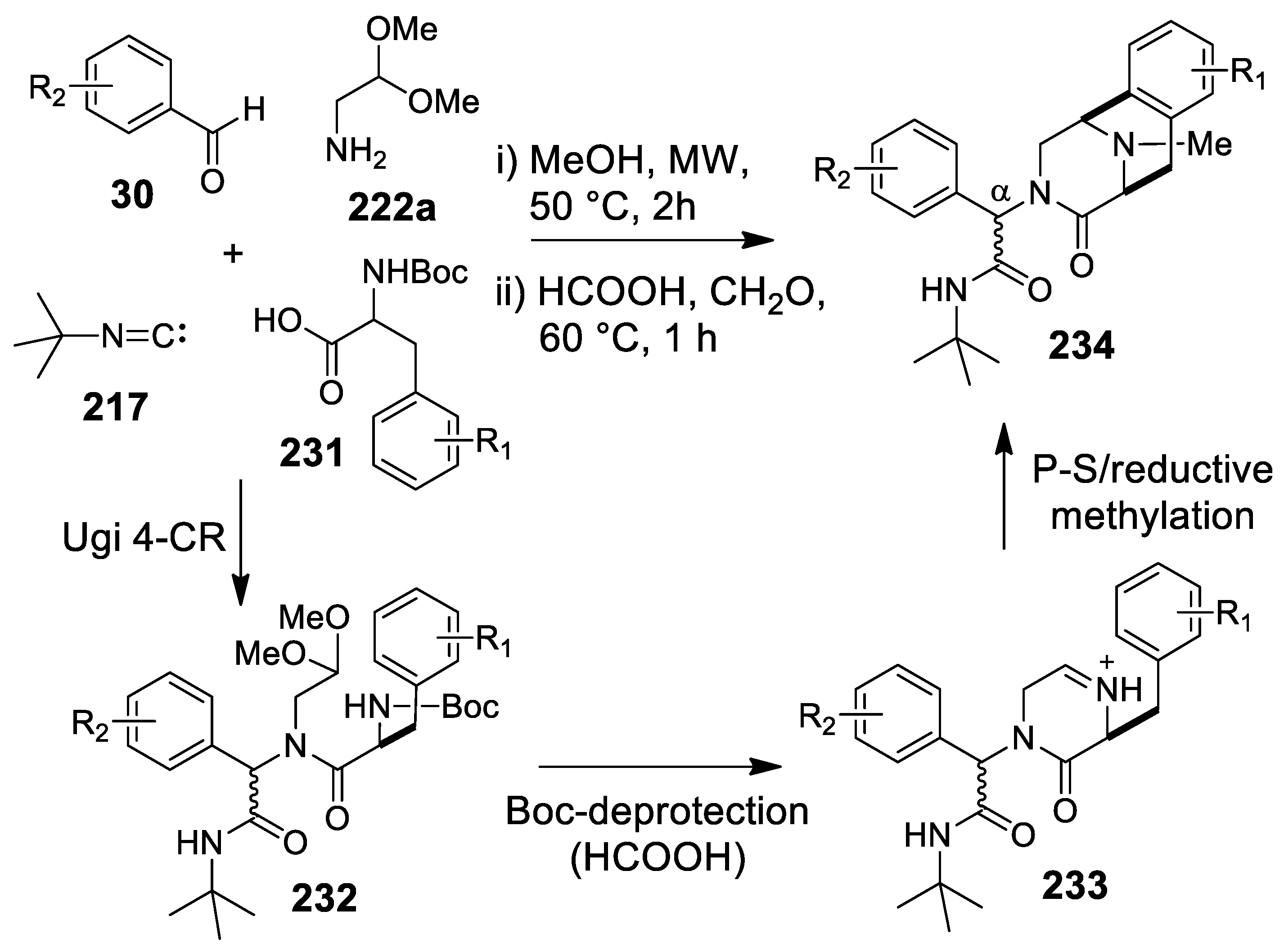
- Cano-Herrera, M.-A.; Miranda, L.D. Expedient entry to the piperazinohydroisoquinoline ring system using a sequential Ugi/Pictet-Spengler/reductive methylation reaction protocol. Chem. Commun. 2011, 47, 10770–10772. [Google Scholar] [CrossRef]
- Sacchetti, A.; Silvani, A.; Lesma, G.; Pilati, T. Phe-Ala-based diazaspirocyclic lactam as nucleator of type II’ β-turn. J. Org. Chem. 2011, 76, 833–839. [Google Scholar] [CrossRef]
- Lesma, G.; Cecchi, R.; Crippa, S.; Giovanelli, P.; Meneghetti, F.; Musolino, M.; Sacchetti, A.; Silvani, A. Ugi 4CR/Pictet-Spengler reaction as short route to tryptophan-derived peptidomimetics. Org. Biomol. Chem. 2012, 10, 9004–9012. [Google Scholar] [CrossRef]
- Pulka, K.; Feytens, D.; Misicka, A.; Tourwé, D. New tetracyclic tetrahydro-β-carbolines as tryptophan-derived peptidomimetics. Mol. Divers. 2010, 14, 97–108. [Google Scholar] [CrossRef]
- Liu, H.; William, S.; Herdtweck, E.; Botros, S.; Dömling, A. MCR synthesis of praziquantel derivatives. Chem. Biol. Drug Des. 2012, 79, 470–477. [Google Scholar] [CrossRef]
- Khoury, K.; Sinha, M.K.; Nagashima, T.; Herdtweck, E.; Dömling, A. Efficient assembly of iminodicarboxamides by a truly four component reaction. Angew. Chem. Int. Ed. 2012, 51, 10280–10283. [Google Scholar] [CrossRef]
- Sinha, M.K.; Khoury, K.; Herdtweck, E.; Dömling, A. Tricycles by a new Ugi Variation and Pictet-Spengler reaction in one-pot. Chem. Eur. J. 2013, 19, 8048–8052. [Google Scholar] [CrossRef]
- Sinha, M.K.; Khoury, K.; Herdtweck, E.; Dömling, A. Various cyclization scaffolds by a truly Ugi 4-CR. Org. Biomol. Chem. 2013, 11, 4792–4796. [Google Scholar] [CrossRef]
- Patil, P.; Khoury, K.; Herdtweck, E.; Dömling, A. MCR synthesis of tetracyclic tetrazole scaffold. Bioorg. Med. Chem. 2015, 23, 2699–2715. [Google Scholar] [CrossRef]
- Stöckigt, J.; Antonchik, A.P.; Wu, F.; Waldmann, H. The Pictet-Spengler Reaction in Nature and in Organic Chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef]
- Walsh, C.T.; Fischbach, M.A. Natural Products Version 2.0: Connecting Genes to Molecules. J. Am. Chem. Soc. 2010, 132, 2469–2473. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Resch, V. The role of biocatalysis in the asymmetric synthesis of alkaloids. RSC Adv. 2013, 3, 17602–17632. [Google Scholar] [CrossRef]
- Treimer, J.F.; Zenk, M.H. Purification and properties of strictosidine synthase, the key enzyme in indole alkaloid formation. Eur. J. Biochem. 1979, 101, 225–233. [Google Scholar] [CrossRef]
- Kutchan, T.M. Strictosidine: From alkaloid to enzyme to gene. Phytochemistry 1993, 32, 493–506. [Google Scholar] [CrossRef]
- O’Connor, S.E.; Maresh, J.J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar] [CrossRef]
- McCoy, E.; Galan, M.C.; O’Connor, S.E. Substrate specificity of strictosidine synthase. Bioorg. Chem. Med. Lett. 2006, 16, 2475–2478. [Google Scholar] [CrossRef] [PubMed]
- Maresh, J.J.; Giddings, L.A.; Friedrich, A.; Loris, E.A.; Panikar, S.; Trout, B.L.; Stöckigt, J.; Peters, B.; O’Connor, S.E. Strictosidine synthase: Mechanism of a Pictet-Spengler catalyzing enzyme. J. Am. Chem. Soc. 2008, 130, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Rueffer, M.; Nagakura, N.; Zenk, M.H. Strictosidine, the common precursor for monoterpenoid indole alkaloids with 3α and 3β configuration. Tetrahedron Lett. 1978, 19, 1593–1596. [Google Scholar] [CrossRef]
- Stöckigt, J.; Zenk, M.H. Isovincoside (strictosidine), the key intermediate in the enzymatic formation of indole alkaloids. FEBS Lett. 1977, 79, 233–237. [Google Scholar] [CrossRef]
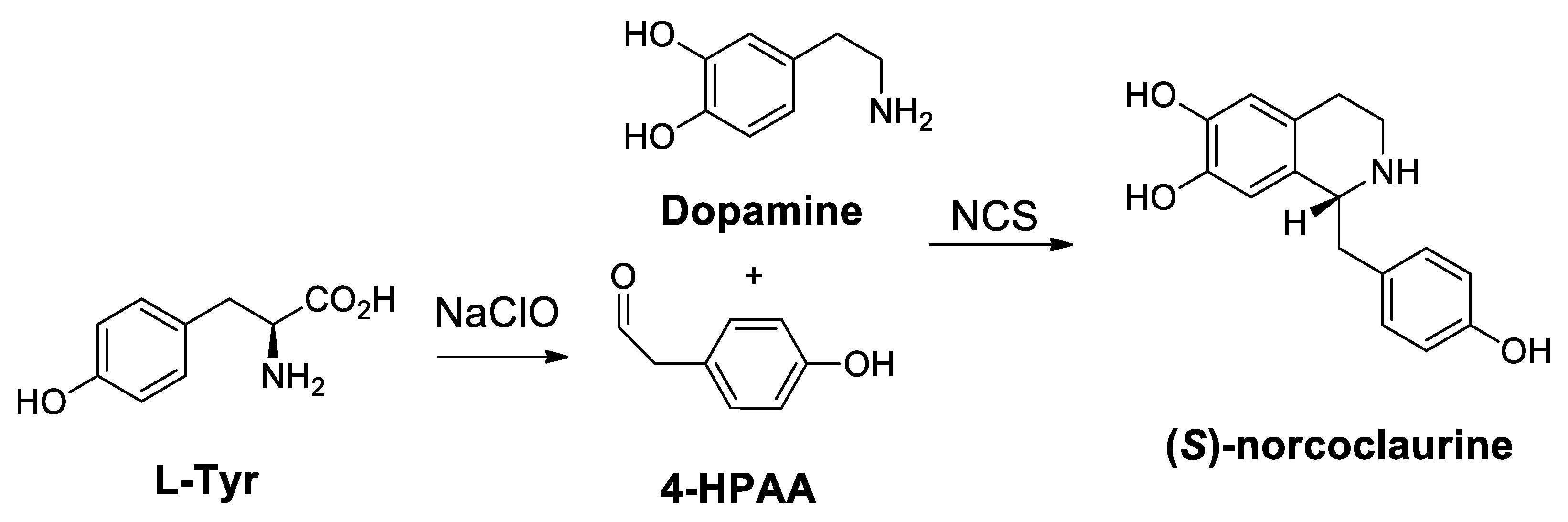
- Luk, L.Y.; Bunn, S.; Liscombe, D.K.; Facchini, P.J.; Tanner, M.E. Mechanistic studies on norcoclaurine synthase of benzylisoquinoline alkaloid biosynthesis: An enzymatic Pictet-Spengler reaction. Biochemistry 2007, 46, 10153–10161. [Google Scholar] [CrossRef]
- Samanani, N.; Facchini, P.J. Purification and characterization of norcoclaurine synthase. The first committed enzyme in benzylisoquinoline alkaloid biosynthesis in plants. J. Biol. Chem. 2002, 277, 33878–33883. [Google Scholar] [CrossRef]
- Mahmoudian, M.; Rahimi-Moghaddam, P. The anticancer activity of noscapine: A review. Recent Pat. Anticancer Drug Discov. 2009, 4, 92–97. [Google Scholar] [CrossRef]
- Ilari, A.; Franceschini, S.; Bonamore, A.; Arenghi, F.; Botta, B.; Macone, A.; Pasquo, A.; Bellucci, L.; Boffi, A. Structural basis of enzymatic (S)-norcoclaurine biosynthesis. J. Biol. Chem. 2009, 284, 897–904. [Google Scholar] [CrossRef]
- Bonamore, A.; Barba, M.; Botta, B.; Boffi, A.; Macone, A. Norcoclaurine synthase: Mechanism of an enantioselective Pictet-Spengler catalyzing enzyme. Molecules 2010, 15, 2070–2078. [Google Scholar] [CrossRef]
- Nagakura, N.; Höfle, G.; Zenk, M.H. Deacetylisopecoside: The key intermediate in the biosynthesis of the alkaloids cephaeline and emetine. J. Chem. Soc. Chem. Commun. 1978, 5, 896–898. [Google Scholar] [CrossRef]
- Nagakura, N.; Höfle, G.; Coggiola, D.; Zenk, M.H. The biosynthesis of Ipecac alkaloids andof ipecoside and alangiside. Planta Med. 1978, 34, 381–389. [Google Scholar] [CrossRef]
- Nomura, T.; Lara Quesada, A.; Kutchan, T.M. The new β-D-glucosidase in terpenoid-isoquinoline alkaloid biosynthesis in Psychotria ipecacuanha. J. Biol. Chem. 2008, 283, 34650–34659. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, K.; Watanabe, K.; Suda, H.; Oguri, H.; Oikawa, H. Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat. Chem. Biol. 2010, 6, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, K.; Minami, A.; Watanabe, K.; Oguri, H.; Oikawa, H. Pictet-Spenglerase involved in tetrahydroisoquinoline antibiotic biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 142–149. [Google Scholar] [CrossRef]
- Koketsu, K.; Minami, A.; Watanabe, K.; Oguri, H.; Oikawa, H. Chapter Five - The Pictet-Spengler mechanism involved in the biosynthesis of tetrahydroisoquinoline antitumor antibiotics: A novel function for a nonribososmal peptide synthetase. Methods Enzymol. 2012, 516, 79–98. [Google Scholar]
- Hiratsuka, T.; Koketsu, K.; Minami, A.; Kaneko, S.; Yamazaki, C.; Watanabe, K.; Oguri, H.; Oikawa, H. Core assembly mechanism of quinocarcin/SF-1739: Bimodular complex nonribosomal peptide synthetase for sequential Mannich-type reactions. Chem. Biol. 2013, 20, 1523–1535. [Google Scholar] [CrossRef]
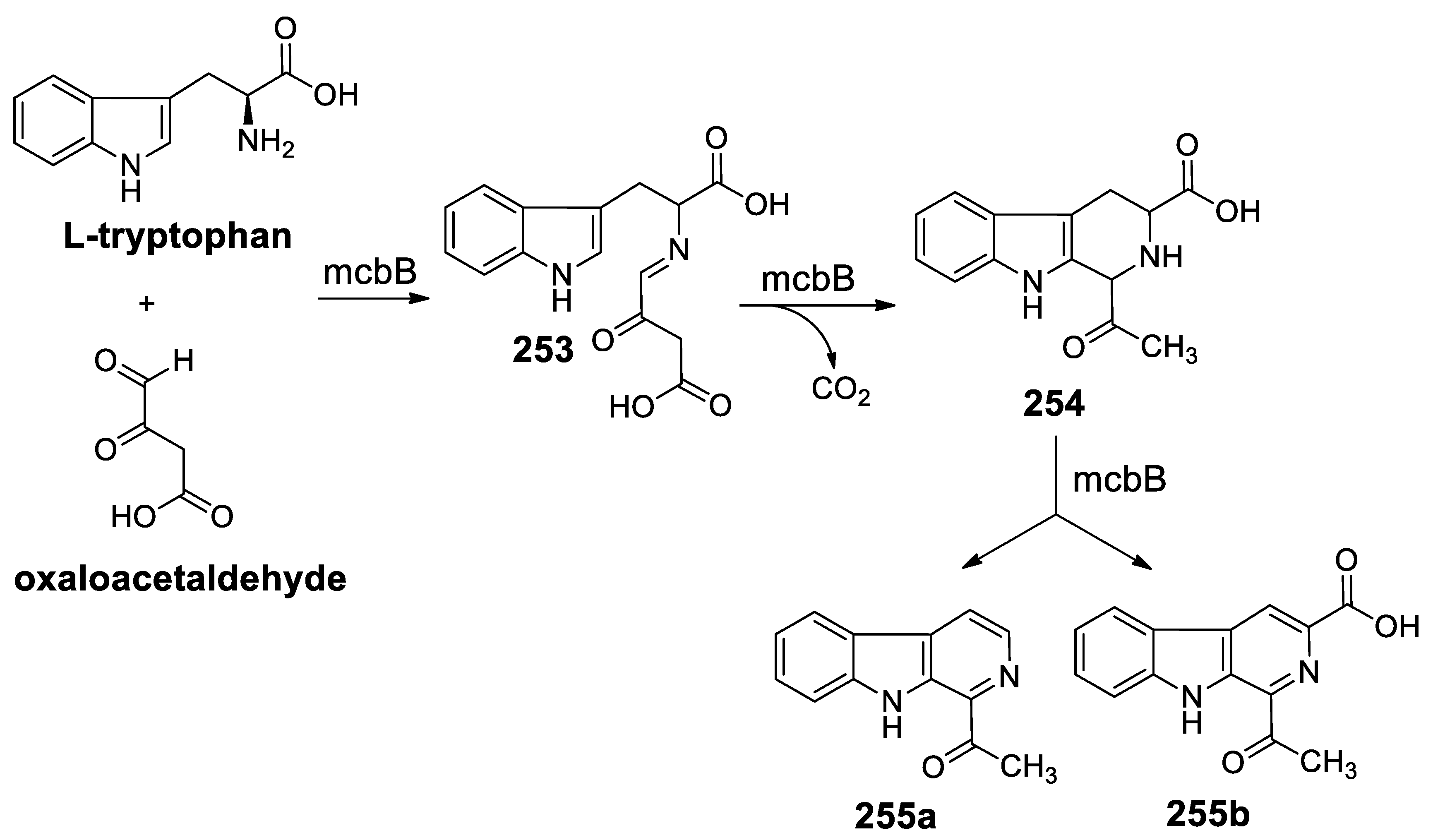
- Chen, Q.; Ji, C.; Song, Y.; Huang, H.; Ma, J.; Tian, X.; Ju, J. Discovery of McbB, an enzyme catalyzing the β-carboline skeleton construction in the marinacarboline biosynthetic pathway. Angew. Chem. Int. Ed. 2013, 52, 9980–9984. [Google Scholar] [CrossRef]
- Mori, T.; Hoshino, S.; Sahashi, S.; Wakimoto, T.; Matsui, T.; Morita, H.; Abe, I. Structural basis for β-carboline alkaloid production by the microbial homodimeric enzyme McbB. Chem. Biol. 2015, 22, 898–906. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Mi, Y.; Ju, J.; Chen, Q.; Zhang, H. Expression, crystallization and preliminary X-ray analysis of McbB, a multifunctional enzyme involved in β-carboline skeleton biosynthesis. Acta Crystallogr. Struct. Biol. Commun. 2014, 70, 1402–1405. [Google Scholar] [CrossRef]
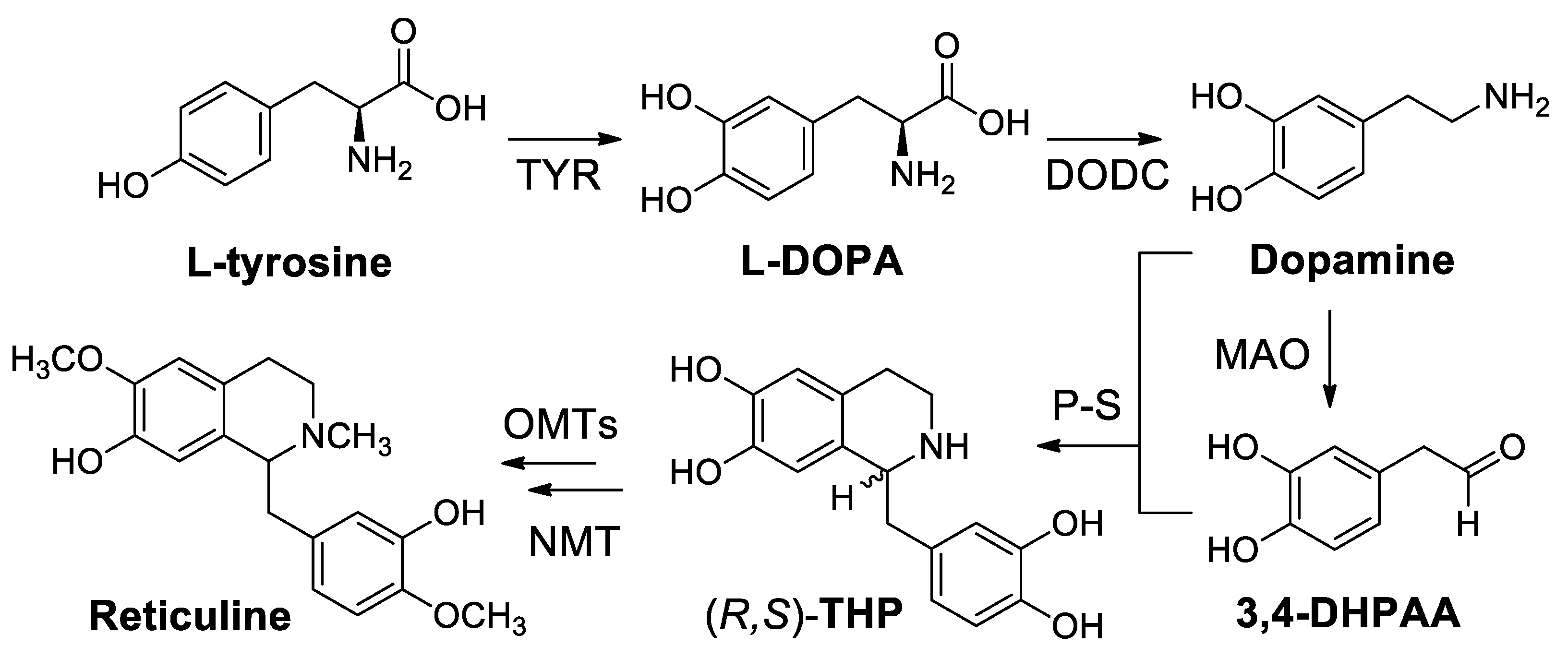
- Nakagawa, A.; Minami, H.; Kim, J.-S.; Koyanagi, T.; Katayama, T.; Sato, F.; Kumagai, H. A bacterial platform for fermentative production of plant alkaloids. Nature Commun. 2011, 2, 326. [Google Scholar] [CrossRef]
- Runguphan, W.; O’Connor, S.E. Metabolic reprogramming of periwinkle plant culture. Nat. Chem. Biol. 2009, 5, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Runguphan, W.; Maresh, J.J.; O’Connor, S.E. Silencing of tryptamine biosynthesis for production of nonnatural alkaloids in plant culture. Proc. Natl. Acad. Sci. USA 2009, 106, 13673–13678. [Google Scholar] [CrossRef]
- Faber, K. Biotransformation in Organic Synthesis, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Bornscheuer, U.T.; Kazlauskas, R.J. Hydrolases in Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Bernhardt, P.; Usera, A.R.; O’Connor, S.E. Biocatalityc asymmetric formation of tetrahydro-β-carbolines. Tetrahedron Lett. 2010, 51, 4400–4402. [Google Scholar] [CrossRef] [PubMed]
- Bonamore, A.; Rovardi, I.; Gasparrini, F.; Baiocco, P.; Barba, M.; Molinaro, C.; Botta, B.; Boffi, A.; Macone, A. An enzymatic, a steroselective synthesis of (S)-norcoclaurine. Green Chem. 2010, 12, 1623–1627. [Google Scholar] [CrossRef]
- Bonamore, A.; Calisti, L.; Calcaterra, A.; Ismail, O.H.; Gargano, M.; D’Acquarica, I.; Botta, B.; Boffi, A.; Macone, A. A Novel Enzymatic Strategy for the Synthesis of Substituted Tetrahydroisoquinolines. ChemistrySelect 2016, 1, 1525–1528. [Google Scholar] [CrossRef]
- Resch, V.; Schrittwieser, J.H.; Siirola, E.; Kroutil, W. Novel carbon-carbon formation for biocatalysis. Curr. Opin. Biotechnol. 2011, 22, 793–799. [Google Scholar] [CrossRef]
- Rath, C.M.; Janto, B.; Earl, J.; Ahmed, A.; Hu, F.Z.; Hiller, L.; Dahlgren, M.; Kreft, R.; Yu, F.; Wolff, J.J.; et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [Google Scholar] [CrossRef]
- Ruff, B.M.; Bräse, S.; O’Connor, S.E. Biocatalytic production of tetrahydroisoquinolines. Tetrahedron Lett. 2012, 53, 1071–1074. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. The catalytic potential of Coptis japonica NCS2 revealed–Development and utilisation of a fluorescamine-based assay. Adv. Synth. Catal. 2012, 354, 2997–3008. [Google Scholar] [CrossRef]
- Wu, F.; Zhu, H.; Sun, L.; Rajendran, C.; Wang, M.; Ren, X.; Panjikar, S.; Cherkasov, A.; Zou, H.; Stöckigt, J. Scaffold tailoring by a newly detected Pictet-Spenglerase activity of strictosidine synthase: From the common tryptoline skeleton to the rare piperazino-indole framework. J. Am. Chem. Soc. 2012, 134, 1498–1500. [Google Scholar] [CrossRef]
- Nagakawa, A.; Mattsuzaki, C.; Matsumura, E.; Koyanagi, T.; Katayama, T.; Yamamoto, K.; Sato, F.; Kumagai, H.; Minami, H. (R,S)-Tetrahydropapaveroline production by stepwise fermentation using engineered Escherichia coli. Sci. Rep. 2014, 4, 6695–6702. [Google Scholar] [CrossRef] [PubMed]
- Maresh, J.J.; Crowe, S.O.; Ralko, A.A.; Aparece, M.D.; Murphy, C.M.; Krzeskowiec, M.; Mullowney, M.W. Facile one-pot synthesis of tetrahydroisoquinolines from amino acids via hypochlorite-mediated decarboxylation and Pictet-Spengler condensation. Tetrahedron Lett. 2014, 55, 5047–5051. [Google Scholar] [CrossRef]
- Nishihachijo, M.; Hirai, Y.; Kawano, S.; Nishiyama, A.; Minami, H.; Katayama, T.; Yasohara, Y.; Sato, F.; Kumagai, H. Asymmetric synthesis of tetrahydroisoquinolines by enzymatic Pictet-Spengler reaction. Biosci. Biotechnol. Biochem. 2014, 78, 701–707. [Google Scholar] [CrossRef]
- Zhu, H.; Kerčmar, P.; Wu, F.; Rajendran, C.; Sun, L.; Wang, M.; Stöckigt, J. Using Strictosidine Synthase to prepare Novel Alkaloids. Curr. Med. Chem. 2015, 22, 1880–1888. [Google Scholar] [CrossRef]
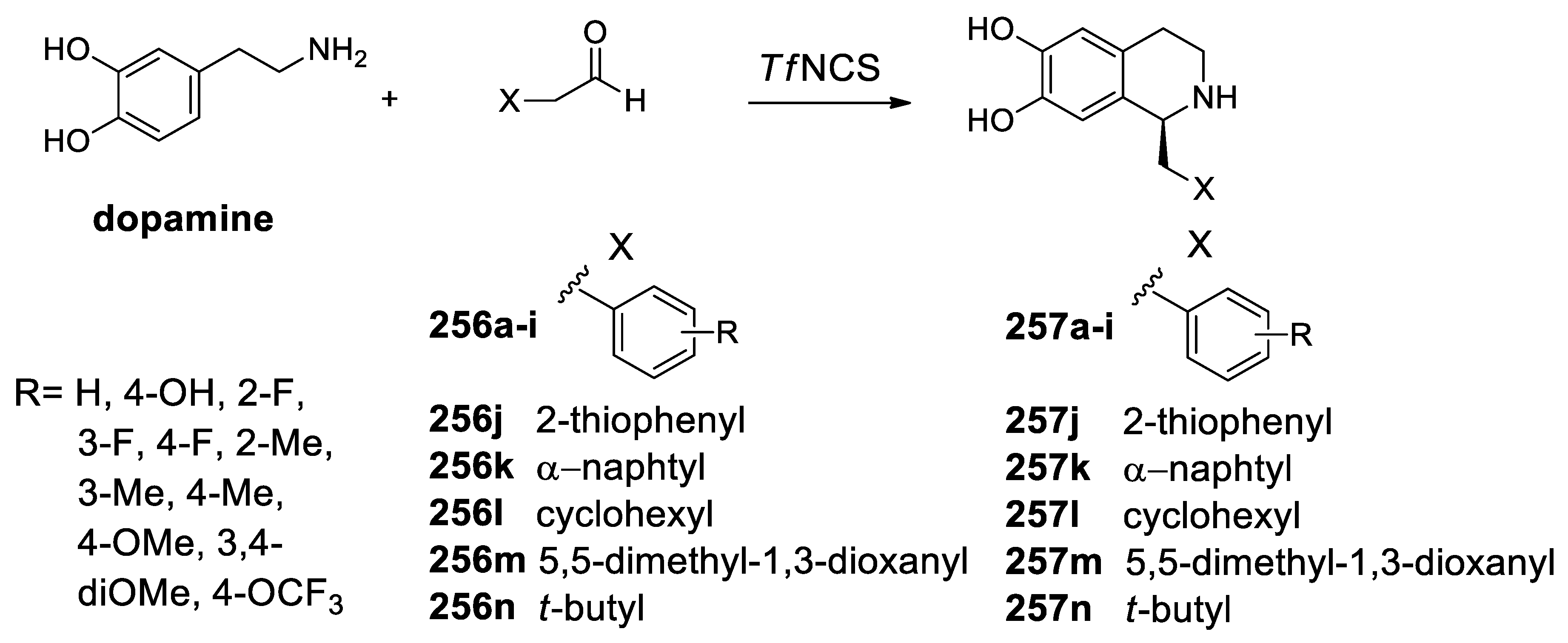
- Lichman, B.R.; Gershater, M.C.; Lamming, E.D.; Pesnot, T.; Sula, A.; Keep, N.H.; Hailes, H.C.; Ward, J.M. ‘Dopamine-first’ mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J. 2015, 282, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Pulka, K. Pictet-Spengler reactions for the synthesis of pharmaceutically relevant heterocycles. Curr. Opin. Drug Discov. Dev. 2010, 13, 669–684. [Google Scholar]
- Bridoux, A.; Millet, R.; Pommery, J.; Pommery, N.; Hemichart, J.-P. Synthesis and biological activity of N-aroyl tetrahydro-γ-carbolines. Bioorg. Med. Chem. 2010, 18, 3910–3924. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowsky, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted acid-thiourea cocatalysis: Enantioselective iso-Pictet-Spengler reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef]
- Subba Reddy, B.V.; Swain, M.; Madhusudana Reddy, S.; Yadar, J.S.; Sridhart, B. Gold-catalyzed domino cycloisomerization/Pictet-Spengler reaction of 2-(4-aminobut-1-yn-1-yl)anilines with aldehydes: Synthesis of tetrahydropyrido[4,3-b]indole scaffolds. J. Org. Chem. 2012, 14, 2610–2613. [Google Scholar] [CrossRef]
- Lee, Y.; Klausen, R.S.; Jacobsen, E.N. Thiourea-catalyzed enantioselective iso-Pictet-Spengler reactions. Org. Lett. 2011, 13, 5564–5567. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, H.; Leighton, J.L. Direct and highly enantioselective iso-Pictet-Spengler reactions with α-ketoamides: Access to underexplored indole core structures. Org. Lett. 2012, 14, 2610–2613. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, D.; Gu, H.; Lin, X. Enantioselective synthesis of benzazepinoindoles bearing trifluoromethylated quaternary stereocenters catalyzed by chiral phosphoric acids. Chem. Commun. 2014, 50, 7538–7541. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6988. [Google Scholar] [CrossRef] [PubMed]
- McKay, C.S.; Finn, M.G. Click chemistry in complex mixtures: Bioorthogonal bioconjugation. Chem. Biol. 2014, 21, 1075–1101. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as biocompatible method for the generation of stable protein conjugates. Bioconjug. Chem. 2013, 24, 840–851. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Agarwal, P.; Sletten, M. Pictet-Spengler Ligation for Protein Chemical Modification. Patent WO 2014078733 A1, 22 May 2014. [Google Scholar]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barnfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Albers, A.E.; Garofalo, A.W.; Drake, P.M.; Kudirka, R.; de Hart, G.W.; Barfield, R.M.; Baker, J.; Banas, S.; Rabuka, D. Exploring the effects of linker composition on site-specifically modified antibody-drug conjugates. Eur. J. Med. Chem. 2014, 88, 3–9. [Google Scholar] [CrossRef]
- Liu, J.; Hanne, J.; Britton, B.M.; Shoffner, M.; Albers, A.E.; Bennett, J.; Zatezalo, R.; Barfield, R.; Rabuka, D.; Lee, J.B.; et al. An efficient site-specific method for irreversible covalent labeling of proteins with a fluorophore. Sci. Rep. 2015, 5, 16883–16892. [Google Scholar] [CrossRef]
- Kudirka, R.; Rabuka, D. The Hydrazino-iso-Pictet-Spengler Ligation: A Versatile, Mild, and Efficient Aldehyde Conjugation Strategy To Generate Site-Specific, Positionally Programmable Antibody-Drug Conjugates. Am. Pharm. Rev. 2015. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/175076-The-Hydrazino-iso-Pictet-Spengler-Ligation-a-Versatile-Mild-and-Efficient-Aldehyde-Conjugation-Strategy-to-Generate-Site-specific-Positionally-Programable-Antibody-Drug-Conjugates/ (accessed on 10 January 2020).
- Li, L.; Deng, W.; Song, J.; Ding, W.; Zhao, Q.F.; Peng, C.; Song, W.W.; Tang, G.L.; Liu, W. Characterization of the Saframycin A Gene Cluster from Streptomyces lavendulae NRRL 11002 Revealing a Nonribosomal Peptide Synthetase System for Assembling the Unusual Tetrapeptidyl Skeleton in an Iterative Manner. J. Bacteriol. 2008, 190, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Imada, K.; Sakai, E.; Kato, H.; Kawabata, T.; Yoshinaga, S.; Nehira, T.; Terasawa, H.; Tsukamoto, S. Reticulins A and b and hyrtioreticulin F from the marine sponge Hyrtios reticulatus. Tetrahedron 2013, 69, 7051–7055. [Google Scholar] [CrossRef]
- Barker, S.A.; Harrison, R.E.; Monti, J.A.; Brown, G.B.; Christian, S.T. Identification and quantification of 1,2,3,4-tetrahydro-β-carboline, 2-methyl-1,2,3,4-tetrahydro-β-carboline, and 6-methoxy-1,2,3,4-tetrahydro-β-carboline as in vivo constituents of rat brain and adrenal gland. Biochem. Pharmacol. 1981, 30, 9–17. [Google Scholar] [CrossRef]
- Seigler, D.S. Plant Secondary Metabolism; Springer: Basel, Switzerland, 1998. [Google Scholar]
- Hanson, J.R. The Chemistry of Fungi; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Wiemann, P.; Keller, N.P. Strategies for mining fungal natural products. J. Ind. Microbiol. Biotechnol. 2014, 41, 301–313. [Google Scholar] [CrossRef]
- Yaegashi, J.; Oakley, B.R.; Wang, C.C. Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergyllus nidulans. J. Ind. Microbiol. Biotechnol. 2014, 41, 433–442. [Google Scholar] [CrossRef]
- Xu, W.; Gavia, D.J.; Tang, Y. Biosynthesis of fungal indole alkaloids. Nat. Prod. Rep. 2014, 31, 1474–1487. [Google Scholar] [CrossRef]
- Ding, G.; Song, Y.C.; Chen, J.R.; Xu, C.; Ge, H.M.; Wang, X.T.; Tan, R.X. Chaetoglobosin U, a cytochalasan alkaloid from endophitic Chaetonium globosum IFB-E019. J. Nat. Prod. 2006, 69, 302–304. [Google Scholar] [CrossRef]
- Jang, M.Y.; Feng, T.; Liu, J.K. N-Containing compounds of macromycetes. Nat. Prod. Res. 2011, 28, 783–808. [Google Scholar] [CrossRef]
- Ge, H.M.; Yan, W.; Guo, Z.K.; Luo, Q.; Feng, R.; Zang, L.Y.; Shen, Y.; Jao, R.; Xu, Q.; Tan, R.X. Precursor-directed fungal generation of novel halogenated chaetoglobosins with more preferable immunosuppressive action. Chem. Commun. 2011, 47, 2321–2323. [Google Scholar] [CrossRef]
- Yan, W.; Ge, H.M.; Wang, G.; Jiang, N.; Mei, Y.N.; Jiang, R.; Li, S.J.; Chen, C.J.; Jiao, R.H.; Xu, Q.; et al. Pictet-Spengler reaction-based biosynthetic machinery in fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 18138–18143. [Google Scholar] [CrossRef]
- Kasanah, N.; Farr, L.L.; Gholipour, A.; Wedge, D.E.; Hamann, M.T. Metabolism and resistance of Fusarium spp. To the manzamine alkaloids via a putative retro-pictet-spengler reaction and utility of the rational design of antimalarial and antifungal agents. Mar. Biotechnol. 2014, 16, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Gupta, A.K.; Cook, J.M. Reinvestigation of the mechanism of the acid-catalyzed epimerization of reserpine to isoreserpine. J. Org. Chem. 1989, 54, 4708–4712. [Google Scholar] [CrossRef]
- Van Linn, M.L.; Cook, J.M. Mechanistic studies of the cis to trans epimerization of trisubstituted 1,2,3,4-tetrahydro-β-carbolines. J. Org. Chem. 2010, 75, 3587–3599. [Google Scholar] [CrossRef] [PubMed]
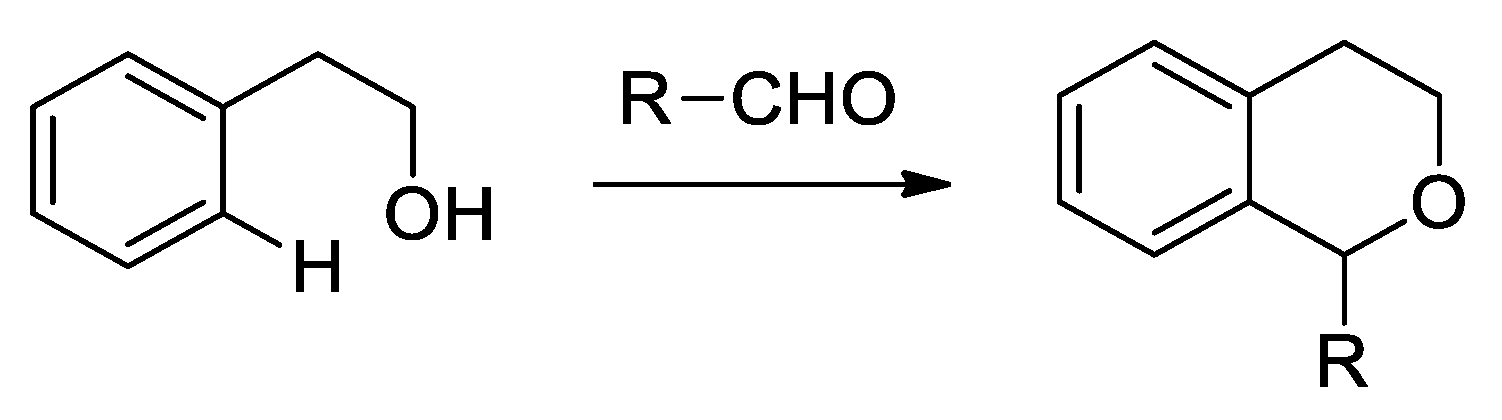
- Larghi, E.L.; Kaufman, T.S. Synthesis of Oxacycles Employing the Oxa-Pictet- Spengler Reaction: Recent Developments and New prospects. Eur. J. Org. Chem. 2011, 27, 5195–5231. [Google Scholar] [CrossRef]
- Larghi, E.L.; Kaufman, T.S. The Oxa-Pictet-Spengler Cyclization: Synthesis of Isochromans and Related Pyran Heterocycles. Synthesis 2006, 2, 187–220. [Google Scholar] [CrossRef]
- Zhu, Z.; Adili, A.; Zhao, C.; Seidel, D. Catalytic Enantioselective Approaches to the oxa-Pictet-Spengler Cyclization and Other 3,6-Dihydropyran-Forming Reactions. SynOpen 2019, 3, 77–90. [Google Scholar] [CrossRef]
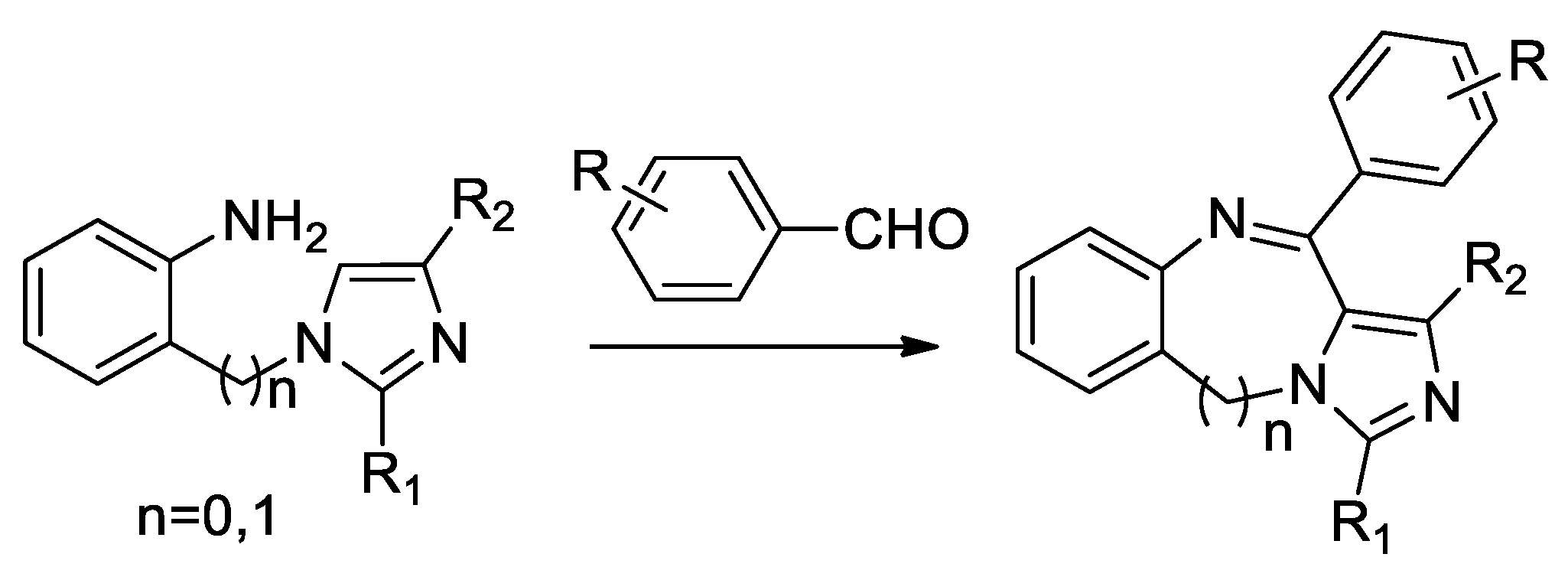
- Kundu, B.; Sawant, D.; Chhabra, R. A modified strategy for the Pictet-Spengler reaction leading to the synthesis of imidazoquinoxalines on solid phase. J. Comb. Chem. 2005, 7, 317–321. [Google Scholar] [CrossRef]
- Kundu, B.; Sawant, D.; Partani, P.; Kesarwani, A.P. New Application of Pictet-Spengler Reaction Leading to the Synthesis of an Unusual Seven-Membered Heterocyclic Ring System. J. Org. Chem. 2005, 70, 4889–4892. [Google Scholar] [CrossRef]
- Duggineni, S.; Sawant, D.; Saha, B.; Kundu, B. Application of the modified Pictet-Spengler reaction for the synthesis of thiazolo- and pyrazolo-quinolines. Tetrahedron 2006, 62, 3228–3241. [Google Scholar] [CrossRef]
- Xiang, J.; Zheng, L.; Chen, F.; Dang, Q.; Bai, X. A Cascade Reaction Consisting of a Pictet-Spengler-Type cyclization and Smiles Rearrangement: Application to the Synthesis of Novel Pyrrole-Fused Dihydropteridines. Org. Lett. 2007, 9, 765–767. [Google Scholar] [CrossRef]
- Sharma, S.; Saha, B.; Sawant, D.; Kundu, B. Synthesis of Novel N-Rich Polycyclic Skeletons Based on Azoles and Pyridines. J. Comb. Chem. 2007, 9, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zheng, X.F.; Liu, R.H.; Reiner, J.; Chang, J.B. Synthesis of novel substituted naphthoquino[b]-benzo[e][1,4]diazepines via Pictet-Spengler cyclization. Tetrahedron 2007, 63, 3389–3394. [Google Scholar] [CrossRef]
- Che, X.; Zheng, L.; Dang, Q.; Bai, X. Synthesis of Novel Pyrimidine Fused-8-Membered Heterocycles via Iminium Ion Cyclization Reactions. J. Org. Chem. 2008, 73, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nam, S.; Jove, R.; Yakushijin, K.; Horne, D.-H. Synthesis and anticancer activity of ageladine A and structural analogs. Bioorg. Med. Chem. Lett. 2010, 20, 83–86. [Google Scholar] [CrossRef] [PubMed]































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Scheme/Figure | P-S Carbonyl Partner and Catalyst | Note | [Ref] |
|---|---|---|---|---|
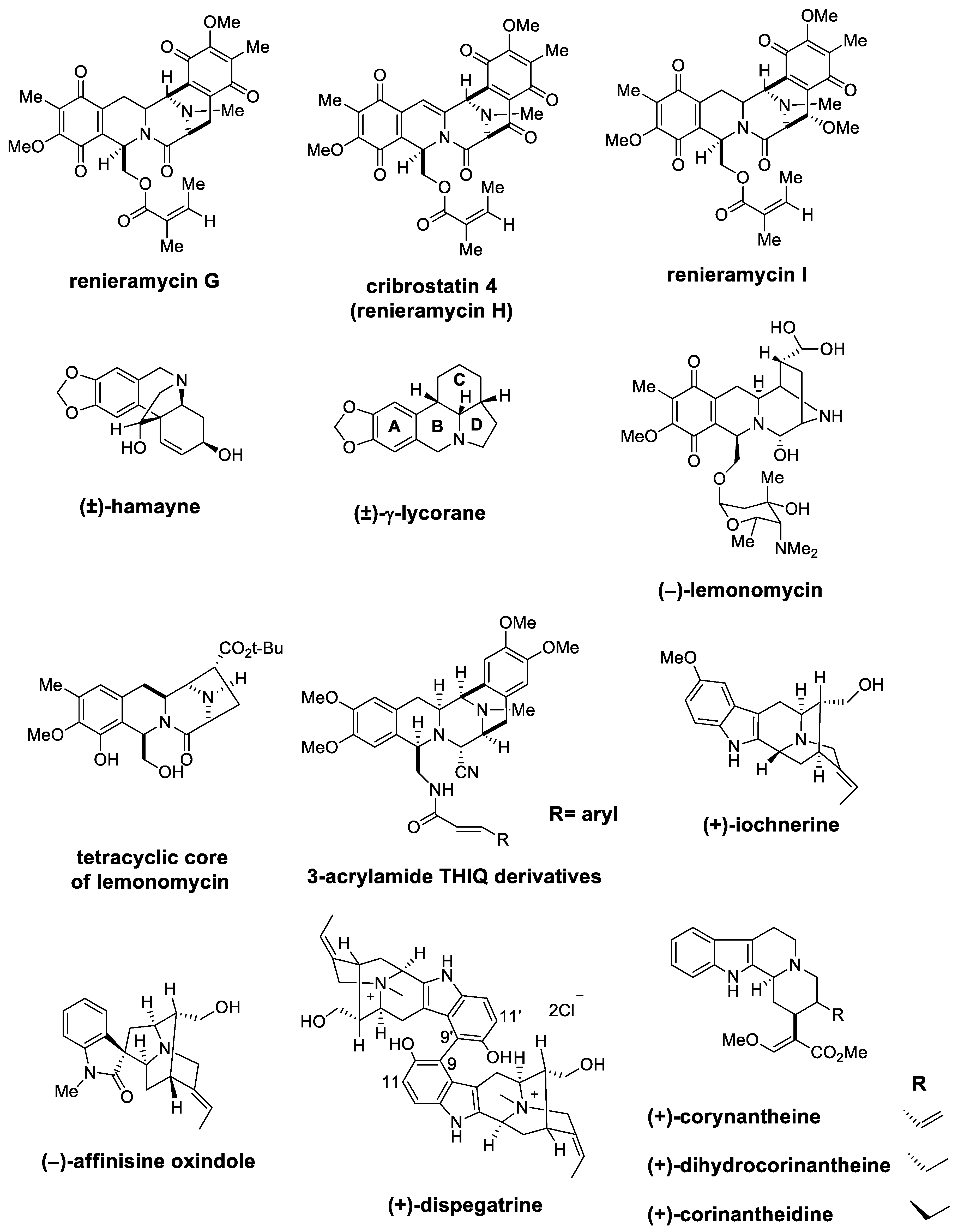
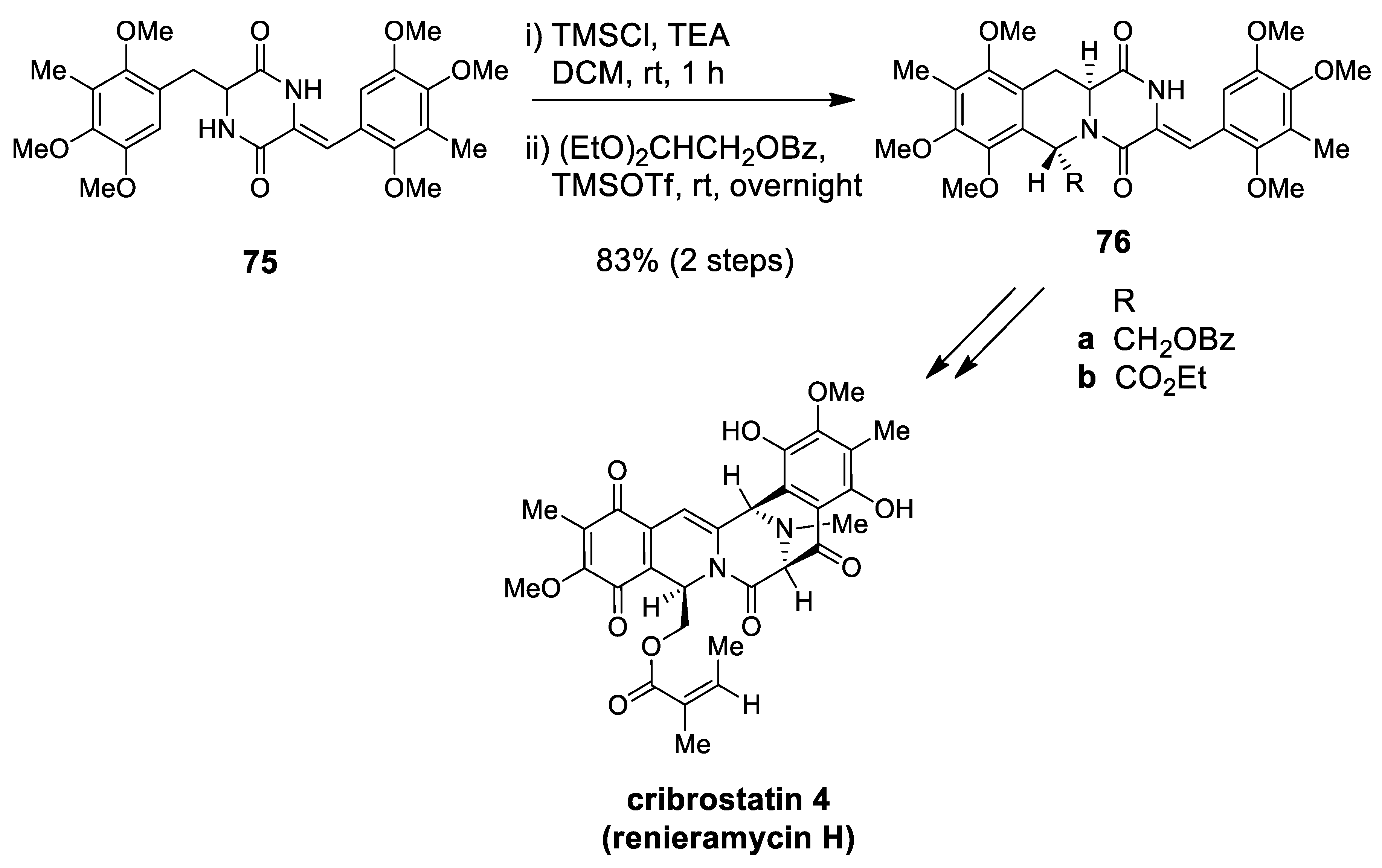
| (±)-cribrostatin 4 (±)-renieramycin G | Scheme 20 Figure 3 | (EtO)2CHCH2OBz TMSOTf | cytotoxic studies | [82,83,84,85] |
| (±)-hamayne | Figure 3 | CH2O/HCO2H | crinine type alkaloids | [86] |
| (±)-lycorane | Figure 3 | CH2O/HCl | Amaryllidaceae family | [87] |
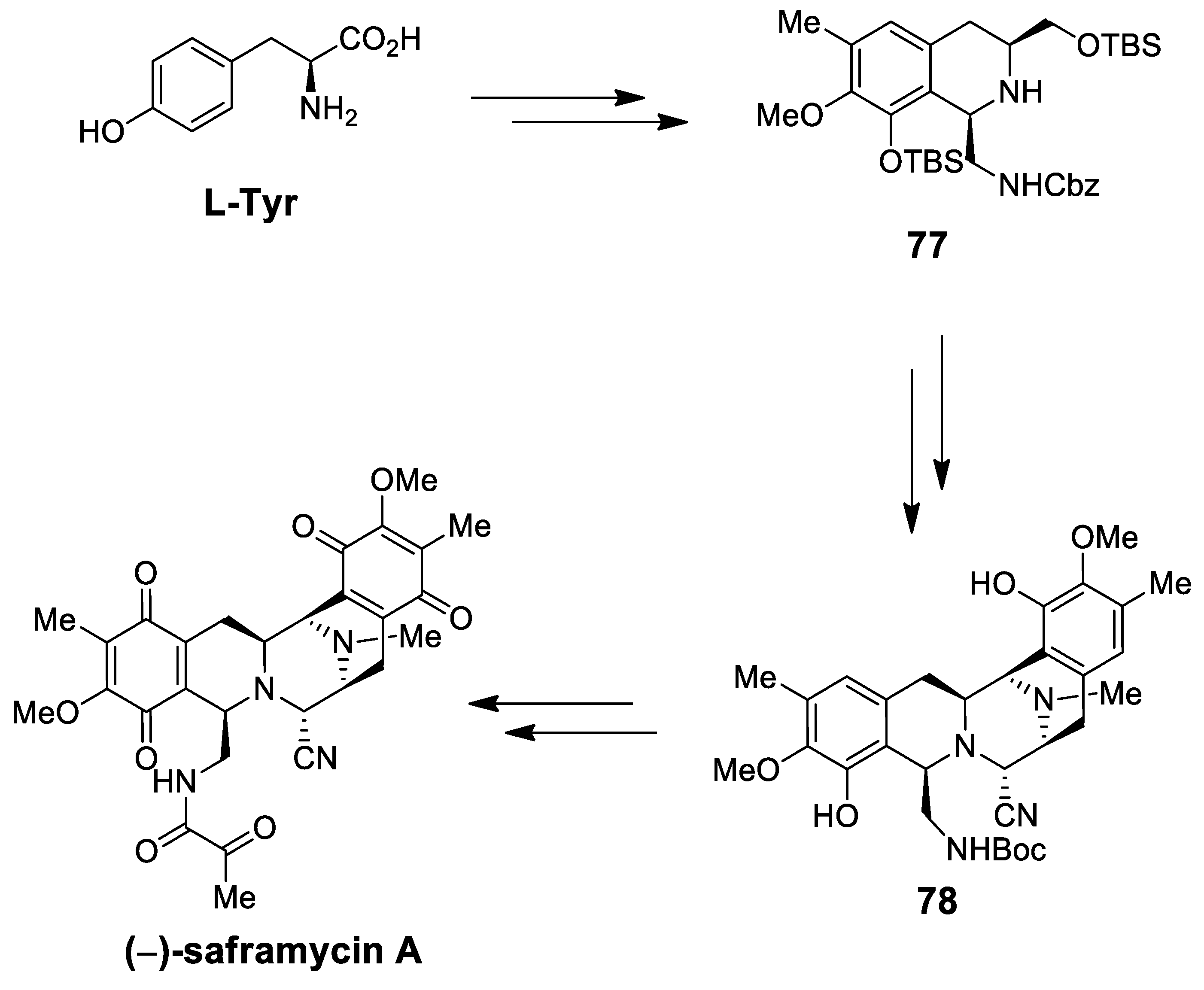
| (−)-saframycin A | Scheme 21 | OHC-CH2NHCbz CF3CH2OH, AcOH, 4 Å MS | intermolecular P-S (C-1) intramolecular P-S (C-11) | [88] |
| mitragynine, paynantheine, speciogynine | Scheme 22 | Aldehyde 80 Thiourea-derived catalysts | Mitragyna yohimbinoid alkaloids | [89] |
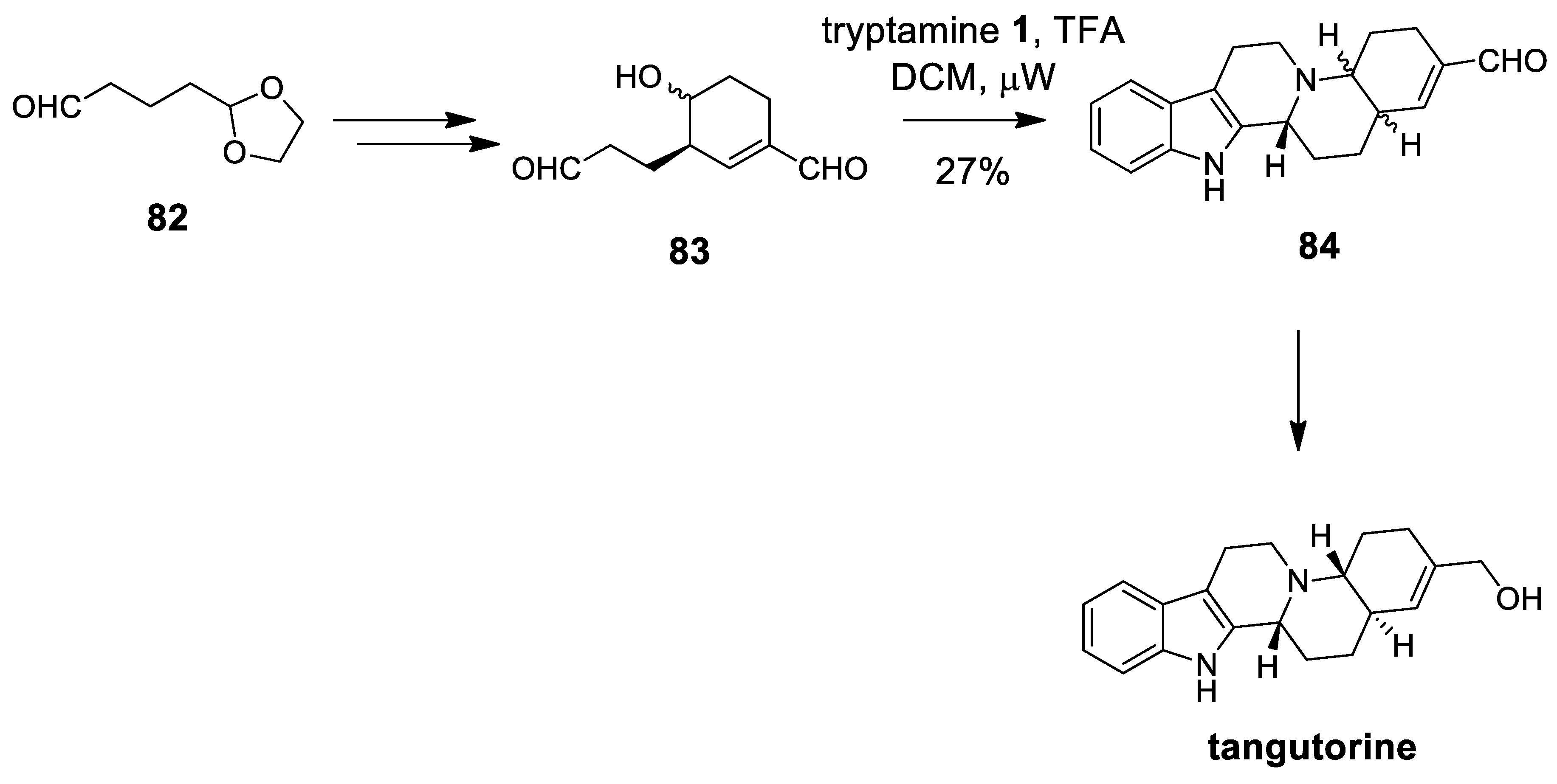
| (±)-tangutorine | Scheme 23 | Aldehyde 83 micRowave | the same cytotoxic activity for racemate and pure enantiomers | [90] |
| (−)-lemonomycin | Figure 3 | Cinnamaldehyde CSA, TMSCN | potent activity against drug-resistant cocci | [91] |
| tetracyclic core of lemonomycin | Figure 3 | OHC-CO2Et CF3CH2OH, AcOH, 4 Å MS | substrate-induced stereocontrol strategy | [92] |
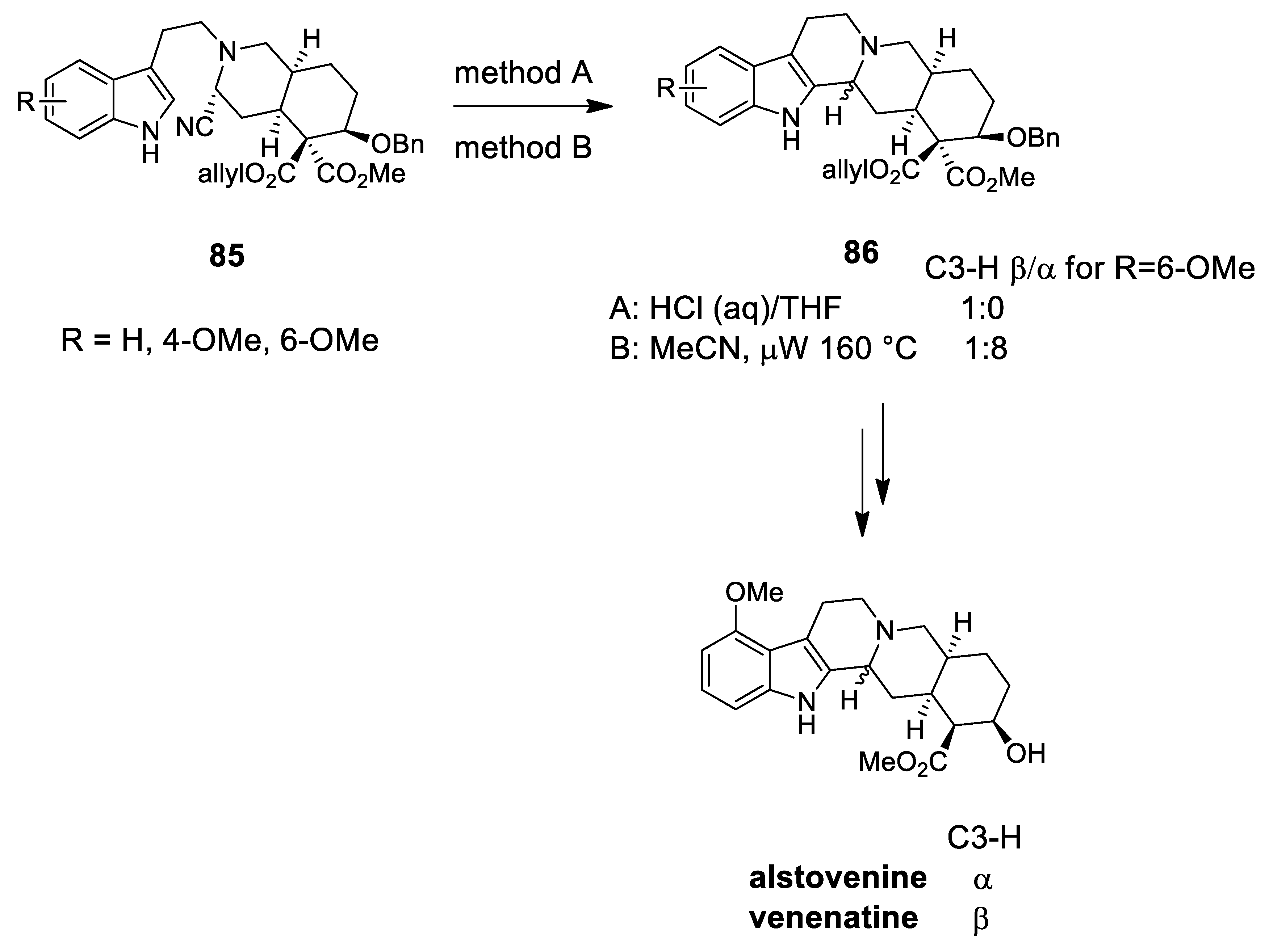
| venenatine alstovenine | Scheme 24 | Compound 86 HCl (aq) oR DMAP oR NaI | yohimbinoid alkaloids C-3 stereochemistry | [93] |
| 3-arylacrilamide (C-1)-side chain derivatives | Figure 3 | OHC-CH2NHCbz NaOAc/AcOH | saframycin/ecteinascidin type compounds | [94,95] |
| (+)-lochnerine (+)-dispegatrine | Figure 3 | OHC(CH2)2CO2Me AcOH | sarpagine type | [96] |
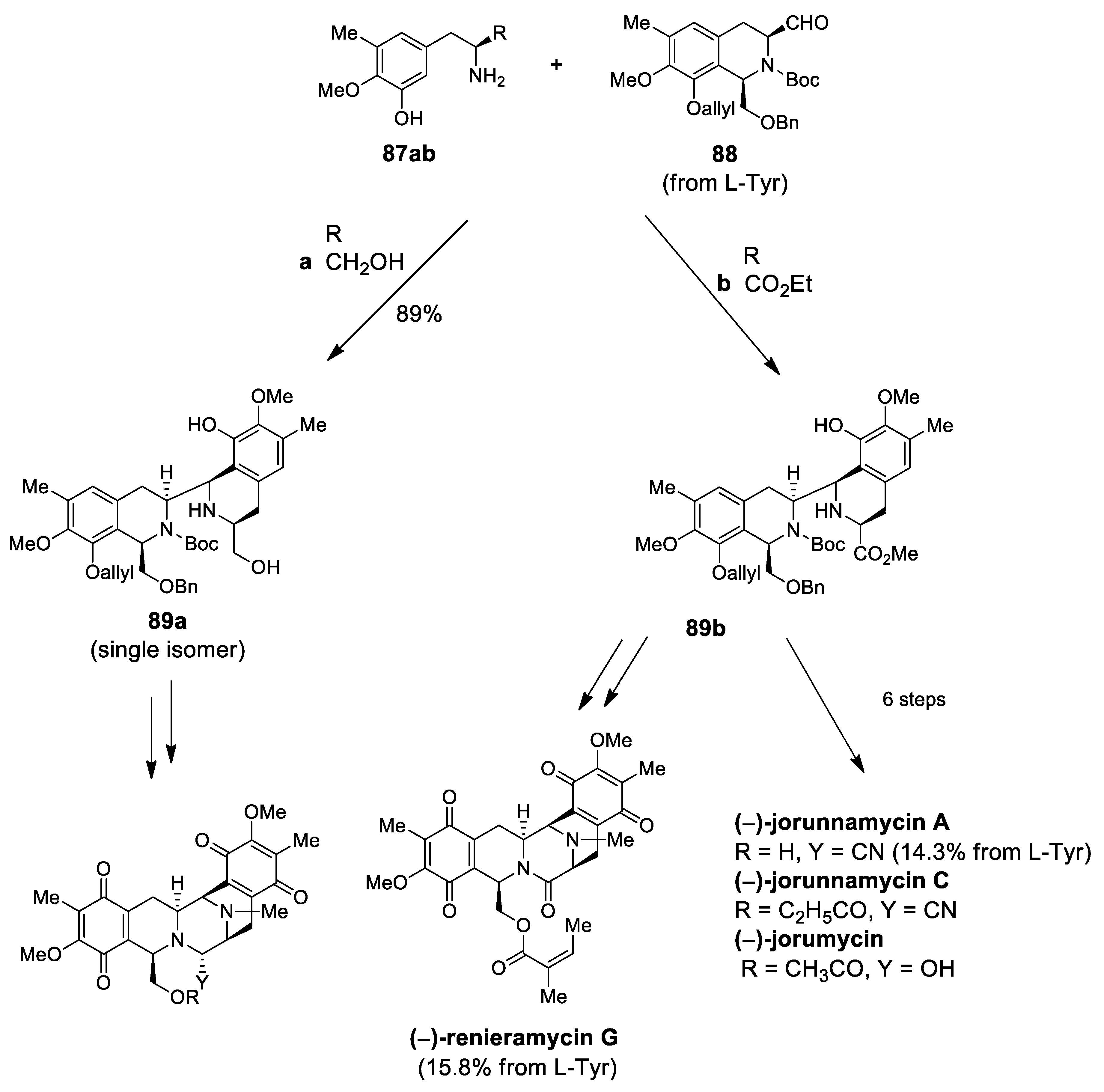
| (−)-jorunnamycins A, C (−)-jorumycin | Scheme 26 | Compound 89 CF3CH2OH, AcOH | renieramycin type | [97] |
| (−)-renieramycin G | Scheme 26 | Compound 89 CF3CH2OH, AcOH | renieramycin type | [98] |
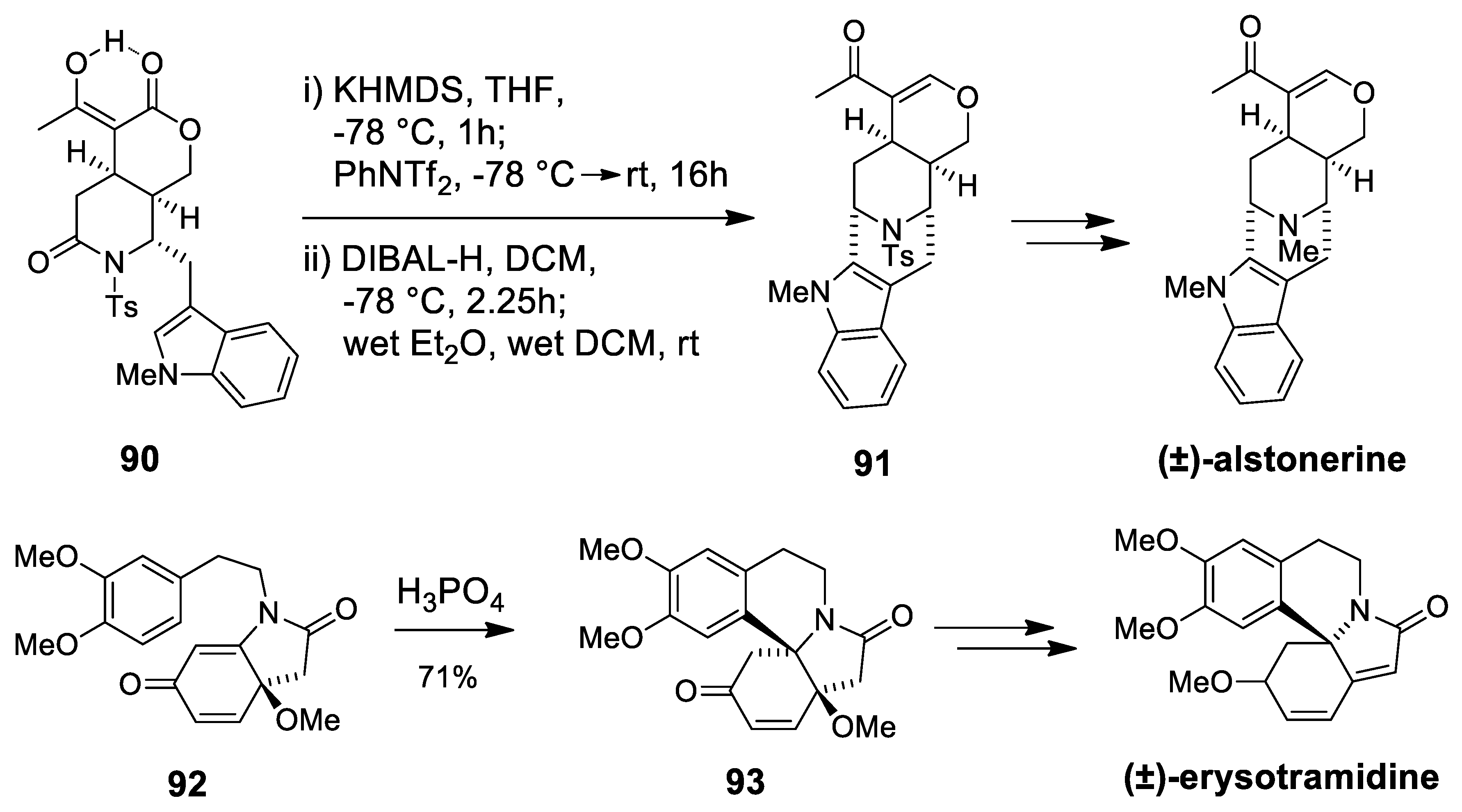
| (±)-alstonerine | Scheme 25 | Compound 91 Wet CH2Cl2 | macroline/sarpagine type | [99] |
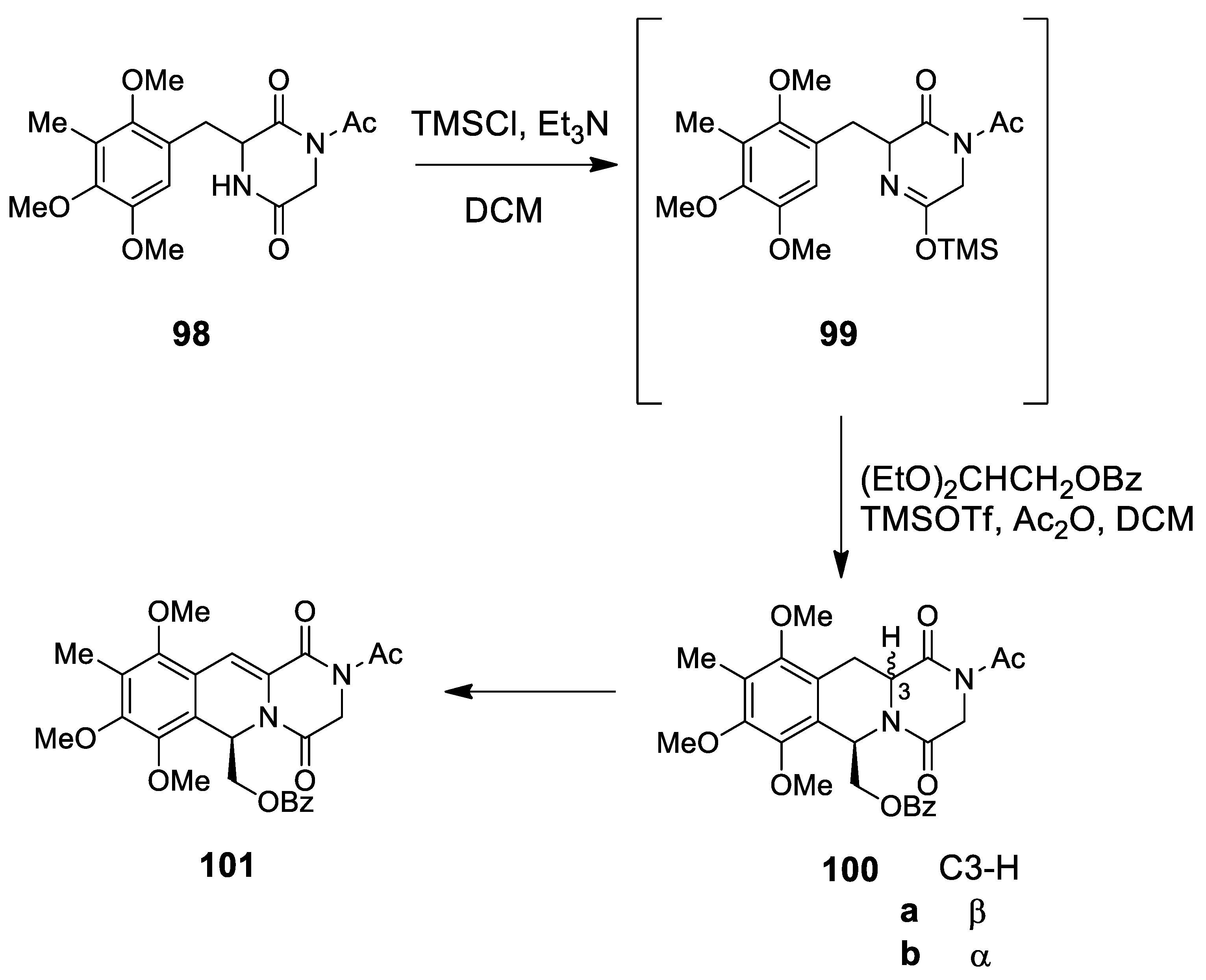
| erysotramidine | Scheme 25 | Compound 93 H3PO4 | erythrina alkaloids | [100,101] |
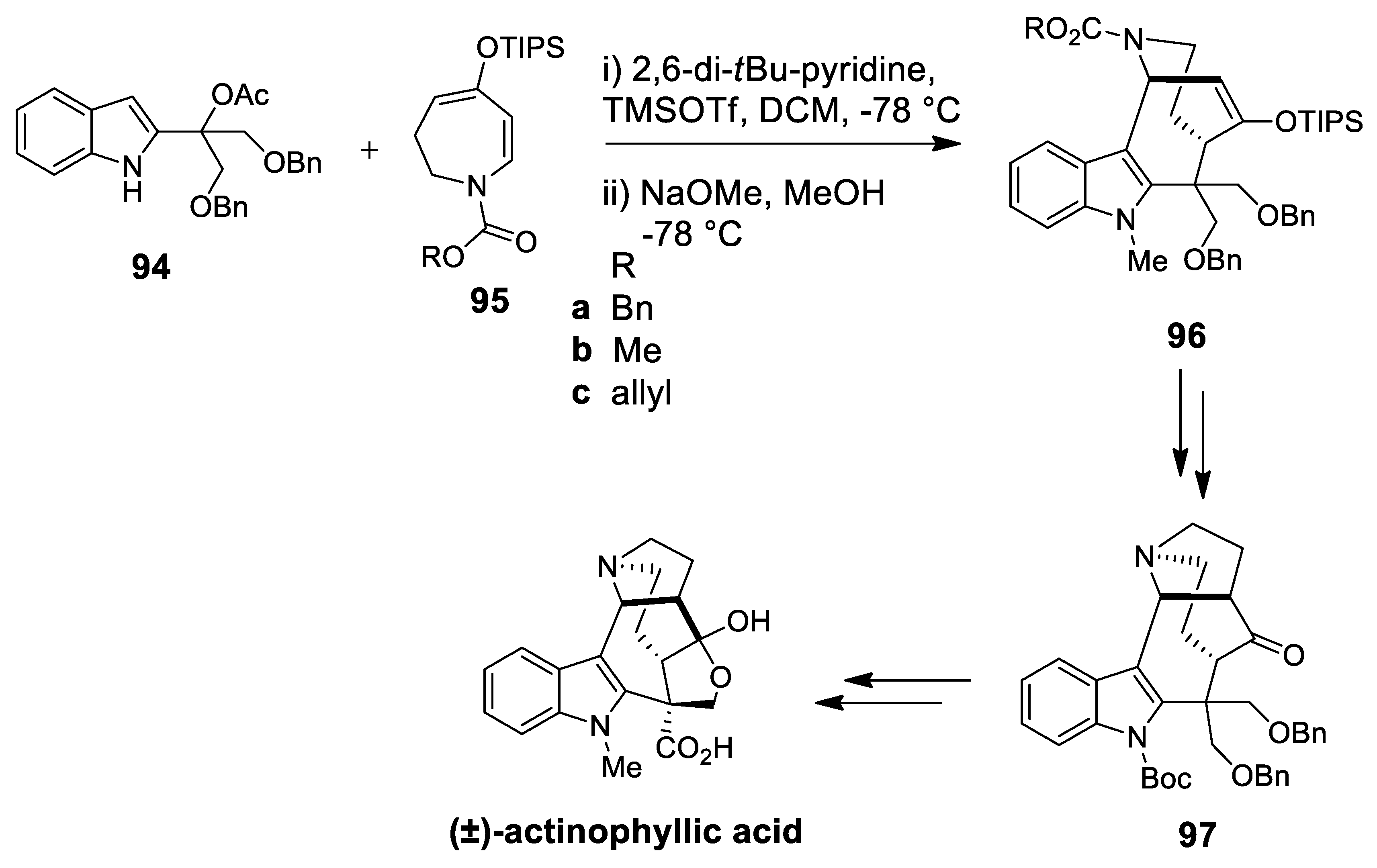
| (±)-actinophyllic acid | Scheme 27 | Different form of P-S | indolohydroazocine | [102] |
| cribrostatin 4 renieramycin I | Scheme 28 | (EtO)2CHCH2OBz TMSOTf/Ac2O | synthesis of left-half renieramycin model compound | [103,104] |
| (+)-yohimbine | Figure 3 | Reported in [65] | - | [105] |
| (−)-corynantheidine | Figure 3 | Reported in [65] | corynanthe alkaloids | [106] |
| (−)-corynantheine (−)-dihydrocorynantheine | Figure 3 | Reported in [65] | corynanthe alkaloids | [107] |
| (−)-affinisine oxindole | Figure 3 | Reported in [66] | - | [108] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calcaterra, A.; Mangiardi, L.; Delle Monache, G.; Quaglio, D.; Balducci, S.; Berardozzi, S.; Iazzetti, A.; Franzini, R.; Botta, B.; Ghirga, F. The Pictet-Spengler Reaction Updates Its Habits. Molecules 2020, 25, 414. https://doi.org/10.3390/molecules25020414
Calcaterra A, Mangiardi L, Delle Monache G, Quaglio D, Balducci S, Berardozzi S, Iazzetti A, Franzini R, Botta B, Ghirga F. The Pictet-Spengler Reaction Updates Its Habits. Molecules. 2020; 25(2):414. https://doi.org/10.3390/molecules25020414
Chicago/Turabian StyleCalcaterra, Andrea, Laura Mangiardi, Giuliano Delle Monache, Deborah Quaglio, Silvia Balducci, Simone Berardozzi, Antonia Iazzetti, Roberta Franzini, Bruno Botta, and Francesca Ghirga. 2020. "The Pictet-Spengler Reaction Updates Its Habits" Molecules 25, no. 2: 414. https://doi.org/10.3390/molecules25020414
APA StyleCalcaterra, A., Mangiardi, L., Delle Monache, G., Quaglio, D., Balducci, S., Berardozzi, S., Iazzetti, A., Franzini, R., Botta, B., & Ghirga, F. (2020). The Pictet-Spengler Reaction Updates Its Habits. Molecules, 25(2), 414. https://doi.org/10.3390/molecules25020414