
First Synthesis of (−)-Altenuene-D3 Suitable as Internal Standard for Isotope Dilution Mass Spectrometry



Abstract
:
1. Introduction
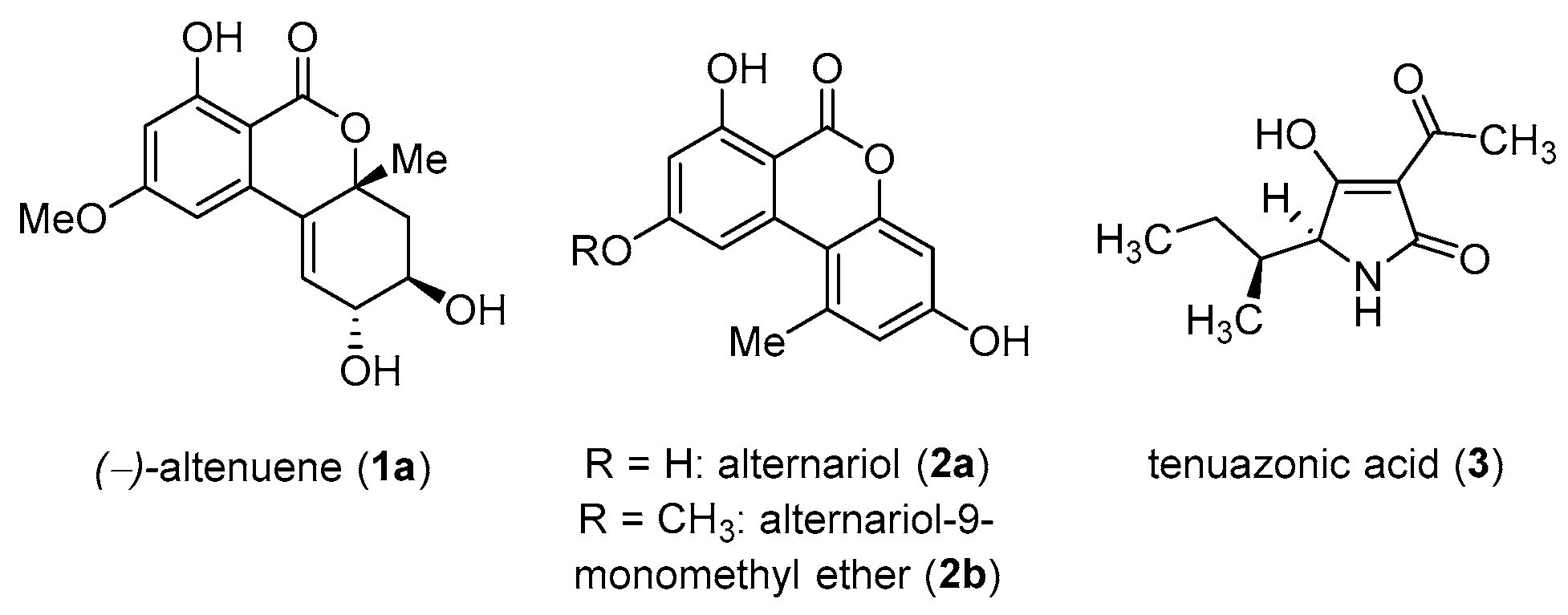
1.1. Alternaria Mycotoxins
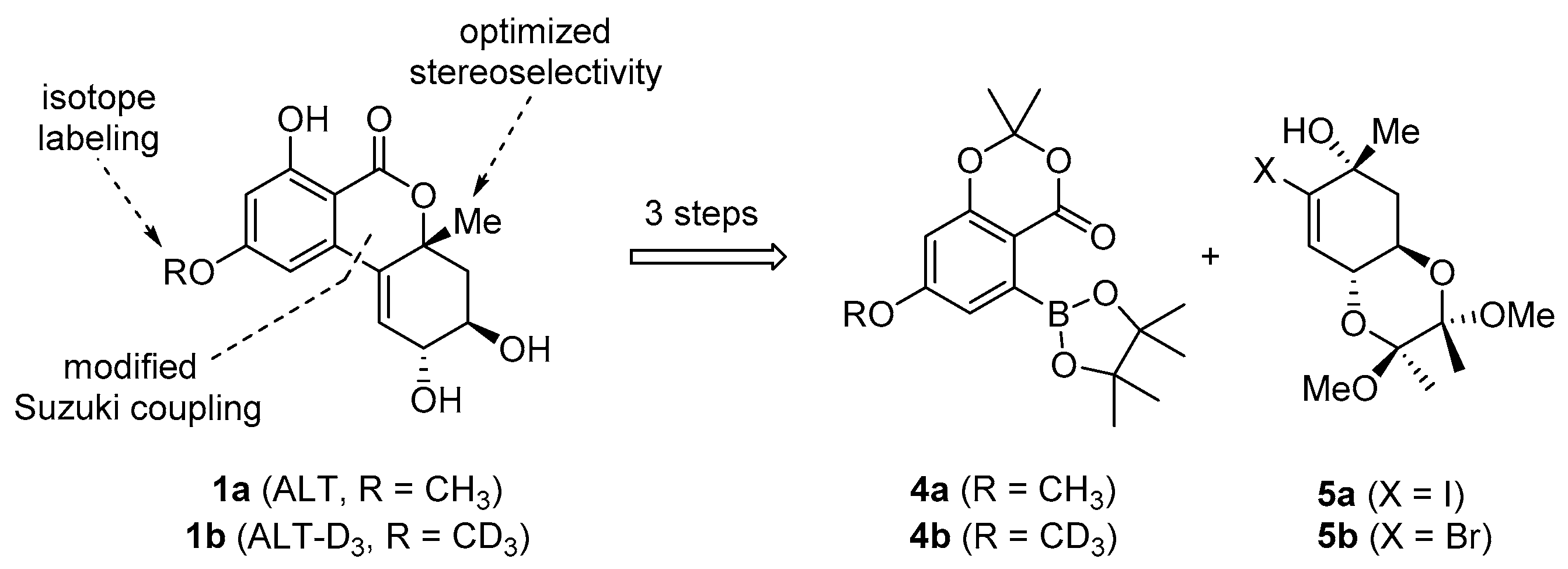
1.2. Retrosynthetic Analysis
2. Results and Discussion
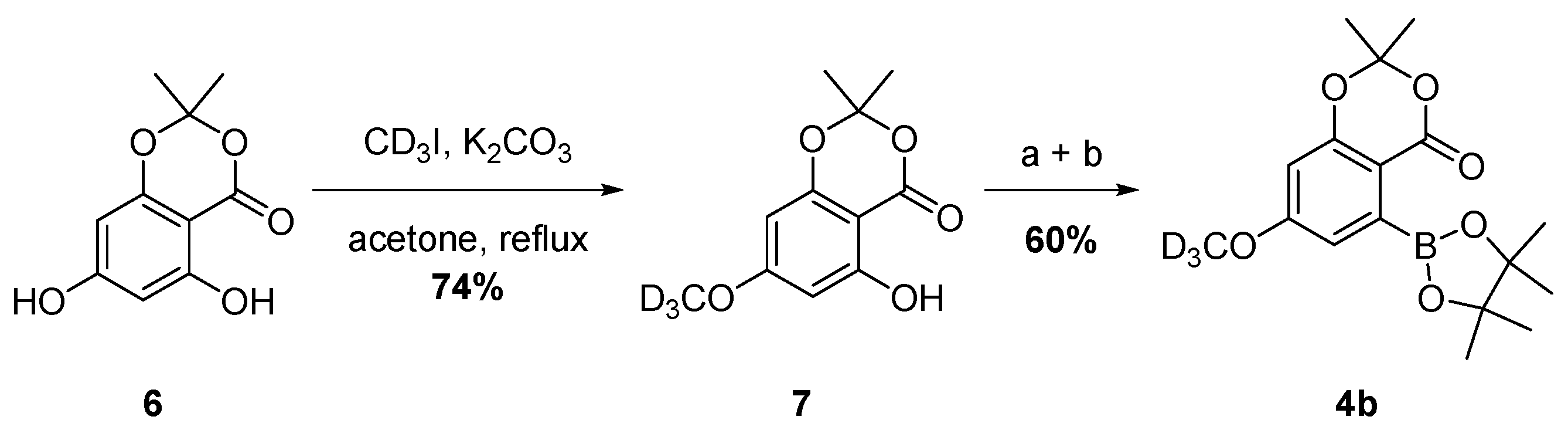
2.1. Synthesis of Deuterated Boronate 4b
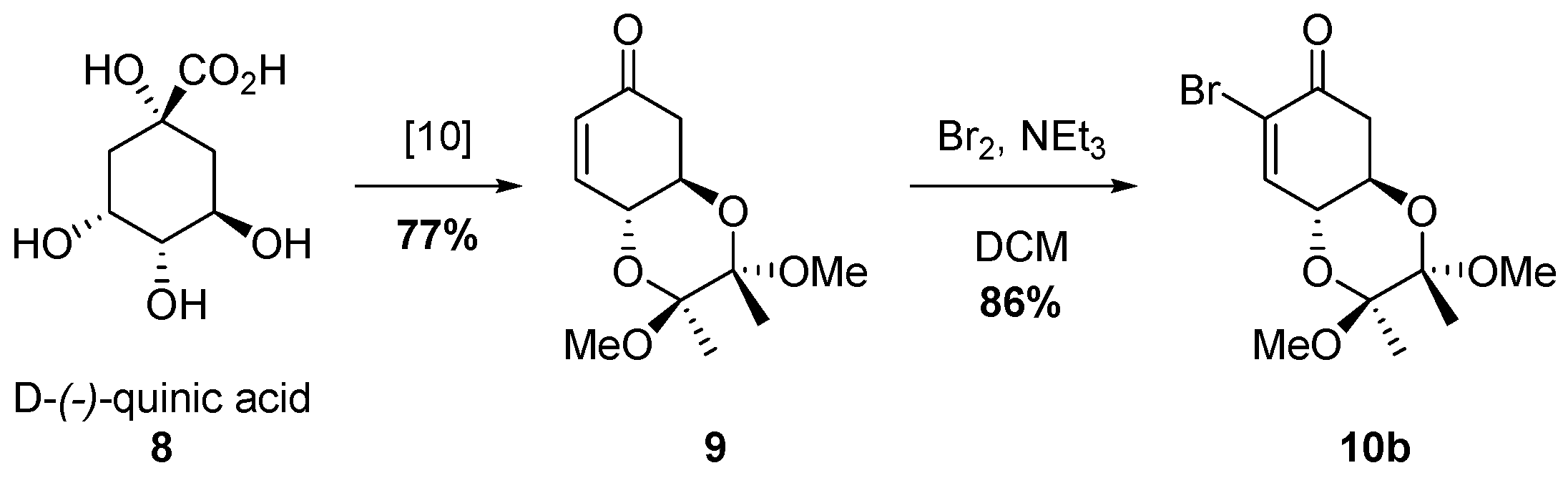
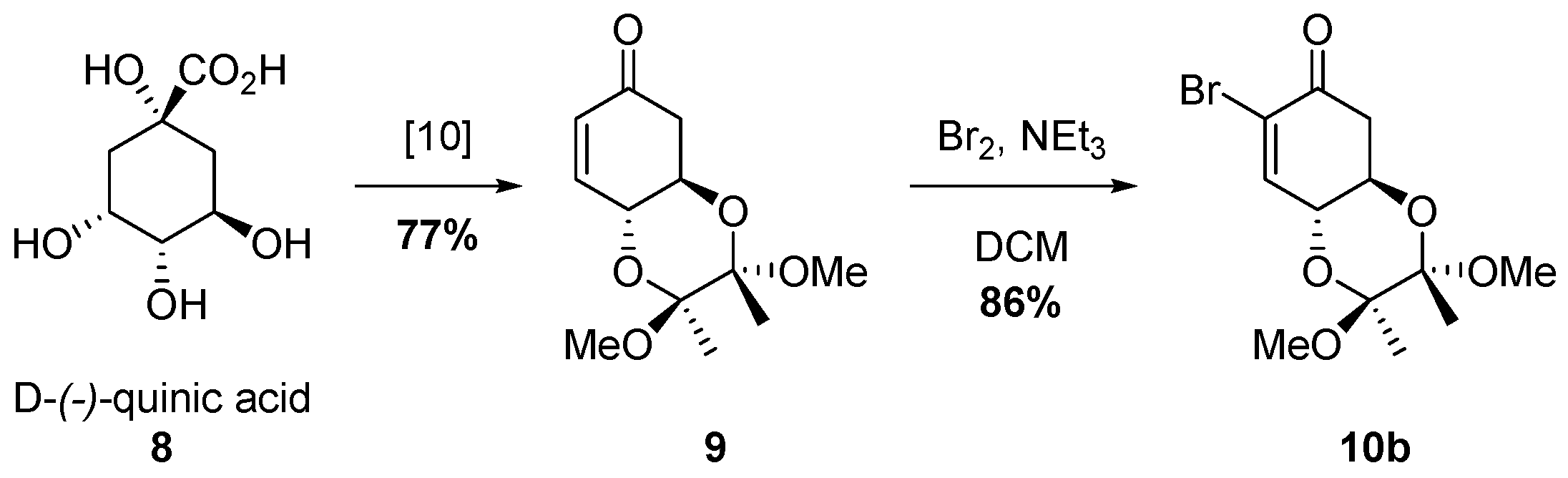
2.2. Synthesis of Bromo Alcohol 5b
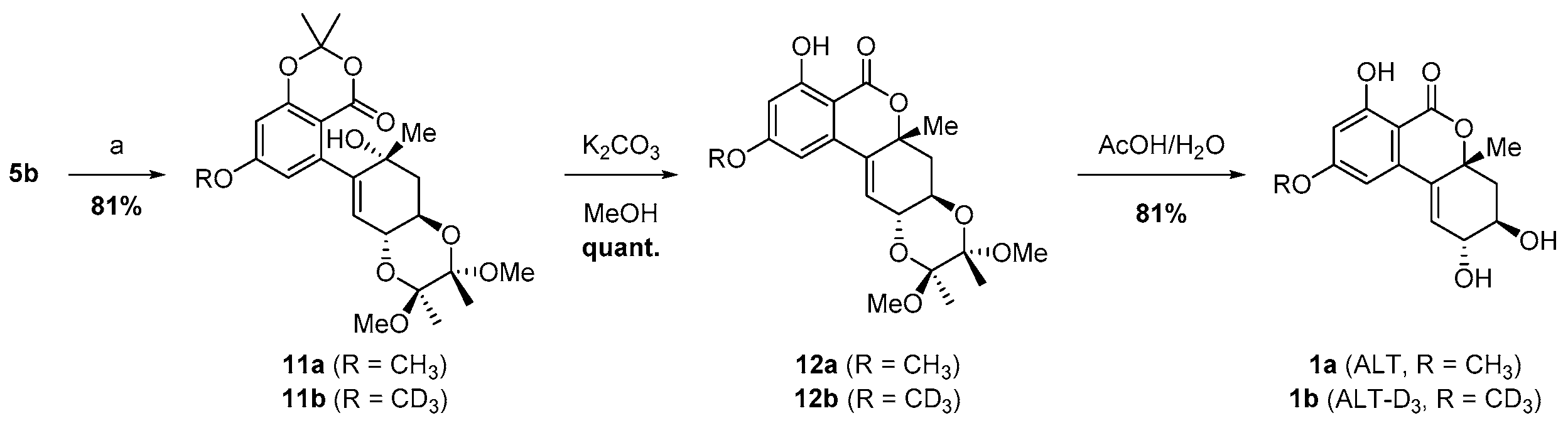
2.3. Suzuki Coupling of Bromo Alcohol 5b and Boronate 4b
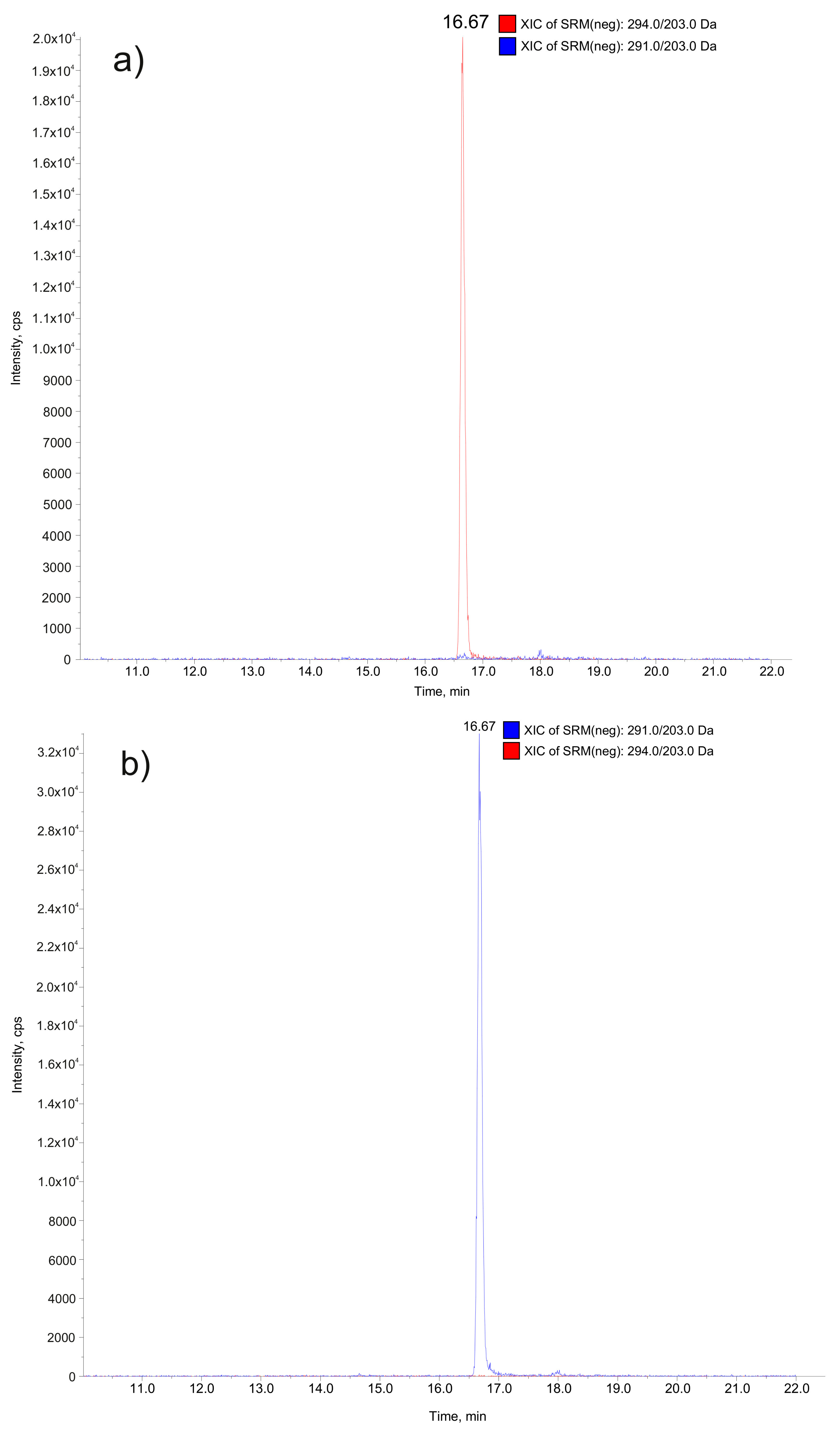
2.4. Implementation of the ALT-D3 Standard (1b) in a LC-MS/MS Method
3. Materials and Methods
3.1. Synthesis of (2S,3S,4aR,8aR)-7-bromo-2,3,4a,5-tetrahydro-2,3-dimethoxy-2,3-dimethylbenzo[b][1,4]dioxin-6(8aH)-one (10b)
3.2. Synthesis of (2S,3S,4aR,6R,8aR)-7-Bromo-2,3,4a,5,6,8a-hexahydro-2,3-dimethoxy-2,3,6-trimethylbenzo[b][1,4]dioxin-6-ol (5b)
3.3. Synthesis of 5-Hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one-D3 (7)
3.4. Synthesis of 7-Methoxy-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-5-yl trifluoromethanesulfonate-D3
3.5. Synthesis of 7-methoxy-2,2-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4H-benzo[d][1,3]dioxin-4-one-D3 (4b)
3.6. Synthesis of 5-((2S,3R,4aR,6R,8aR)-2,3,4a,5,6,8a-hexahydro-6-hydroxy-2,3-dimethoxy-2,3,6-trimethylbenzo[b][1,4]dioxin-7-yl)-7-methoxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one-D3 (11b)
3.7. Synthesis of (6aR,7aR,9S,10S,11aR)-4-Hydroxy-2,9,10-trimethoxy-7a,9,10-trimethyl-6a,7,7a,9,10,11a-hexahydro-5H-benzo[c][1,4]dioxino[2,3-g]-chromen-5-one-D3 (12b)
3.8. Synthesis of (2R,3R,4aR)-2,3,4,4a-Tetrahydro-2,3,7-trihydroxy-9-methoxy-4a-methylbenzo[c]chromen-6-one-D3 ((−)-altenuene-D3, 1b)
3.9. Synthesis of (2R,3R,4aR)-2,3,4,4a-tetrahydro-2,3,7-trihydroxy-9-methoxy-4a-methylbenzo[c]chromen-6-one ((−)-altenuene, 1a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Ostry, V. Alternaria mycotoxins: An overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008, 1, 175–188. [Google Scholar] [CrossRef]
- Lou, J.; Fu, L.; Peng, Y.; Zhou, L. Metabolites from Alternaria Fungi and Their Bioactivities. Moleclues 2013, 18, 5891–5935. [Google Scholar] [CrossRef] [PubMed]
- EFSA on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 2407. [Google Scholar] [CrossRef]
- Pero, R.W.; Owens, R.G.; Dale, S.W.; Harvan, D. Isolation and identification of a new toxin, altenuene, from the fungus Alternaria tenuis. Biochim. et Biophys. Acta 1971, 230, 170–179. [Google Scholar] [CrossRef]
- Asam, S.; Rychlik, M. Recent developments in stable isotope dilution assays in mycotoxin analysis with special regard to Alternaria toxins. Anal. Bioanal. Chem. 2015, 407, 7563–7577. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, Q.; Gao, Y.Q.; Tang, J.J.; Zhang, A.L.; Gao, J.M. Secondary metabolites from the endophytic botryosphaeria dothiadea of melia azedarach and their antifungal, antibacterial, antioxidant, and cytotoxic activities. J. Agric. Food Chem. 2014, 62, 3584–3590. [Google Scholar] [CrossRef] [PubMed]
- Altemöller, M.; Podlech, J.; Fenske, D. Total Synthesis of Altenuene and Isoaltenuene. Eur. J. Org. Chem. 2006, 2006, 1678–1684. [Google Scholar] [CrossRef]
- Kamisuki, S.; Takahashi, S.; Mizushina, Y.; Hanashima, S.; Kuramochi, K.; Kobayashi, S.; Sugawara, F. Total synthesis of dehydroaltenusin. Tetrahedron Lett. 2004, 60, 5695–5700. [Google Scholar] [CrossRef]
- Murray, L.M.; O’Brien, P.; Taylor, R.J.; Wünnemann, S. Lithium enolates from a (−)-quinic acid-derived cyclohexanone with a β-alkoxy leaving group: Regioselective preparation and evaluation of enolate stability towards β-elimination. Tetrahedron Lett. 2004, 45, 2597–2601. [Google Scholar] [CrossRef]
- Feringa, B.L.; Badorrey, R.; Peña, D.; Harutyunyan, S.R.; Minnaard, A.J. Copper-catalyzed asymmetric conjugate addition of Grignard reagents to cyclic enones. Proc. Natl. Acad. Sci. 2004, 101, 5834–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siewert, J.; Sandmann, R.; von Zezschwitz, P. Rhodium-Catalyzed enantioselective 1, 2-addition of aluminum organyl compounds to cyclic enones. Angew. Chem. Int. Ed. 2007, 46, 7122–7124. [Google Scholar] [CrossRef] [PubMed]
- Alexakis, A.; Albrow, V.; Biswas, K.; D’Augustin, M.; Prieto, O.; Woodward, S. Highly enantioselective copper(I)-phosphoramidite-catalysed additions of organoaluminium reagents to enones. Chem. Commun. 2005, 2843–2845. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; García, C.; Walsh, P.J. Catalytic asymmetric addition of diphenylzinc to cyclic α,β-unsaturated ketones. Proc. Natl. Acad. Sci. 2004, 101, 5425–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, P.; Banwell, M.G.; Willis, A.C. Chemoenzymatic total syntheses of ribisins A, B, and D, Polyoxygenated benzofuran derivatives displaying NGF-potentiating properties. J. Org. Chem. 2014, 79, 2829–2842. [Google Scholar] [CrossRef]
- Hickert, S.; Bergmann, M.; Ersen, S.; Cramer, B.; Humpf, H.-U. Survey of Alternaria toxin contamination in food from the German market, using a rapid HPLC-MS/MS approach. Mycotoxin Res. 2015, 32, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Gambacorta, L.; Magistà, D.; Perrone, G.; Murgolo, S.; Logrieco, A.F.; Solfrizzo, M. Co-occurrence of toxigenic moulds, aflatoxins, ochratoxin A, Fusarium and Alternaria mycotoxins in fresh sweet peppers (Capsicum annuum) and their processed products. World Mycotoxin J. 2018, 11, 159–174. [Google Scholar] [CrossRef]
- Sun, D.; Qiu, N.; Zhou, S.; Lyu, B.; Zhang, S.; Li, J.; Zhao, Y.; Wu, Y. Development of sensitive and reliable UPLC-MS/MS methods for food analysis of emerging mycotoxins in china total diet study. Toxins 2019, 11, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: ALT and ALT-D3 are available at HPC Standards (www.hpc-standards.com). |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
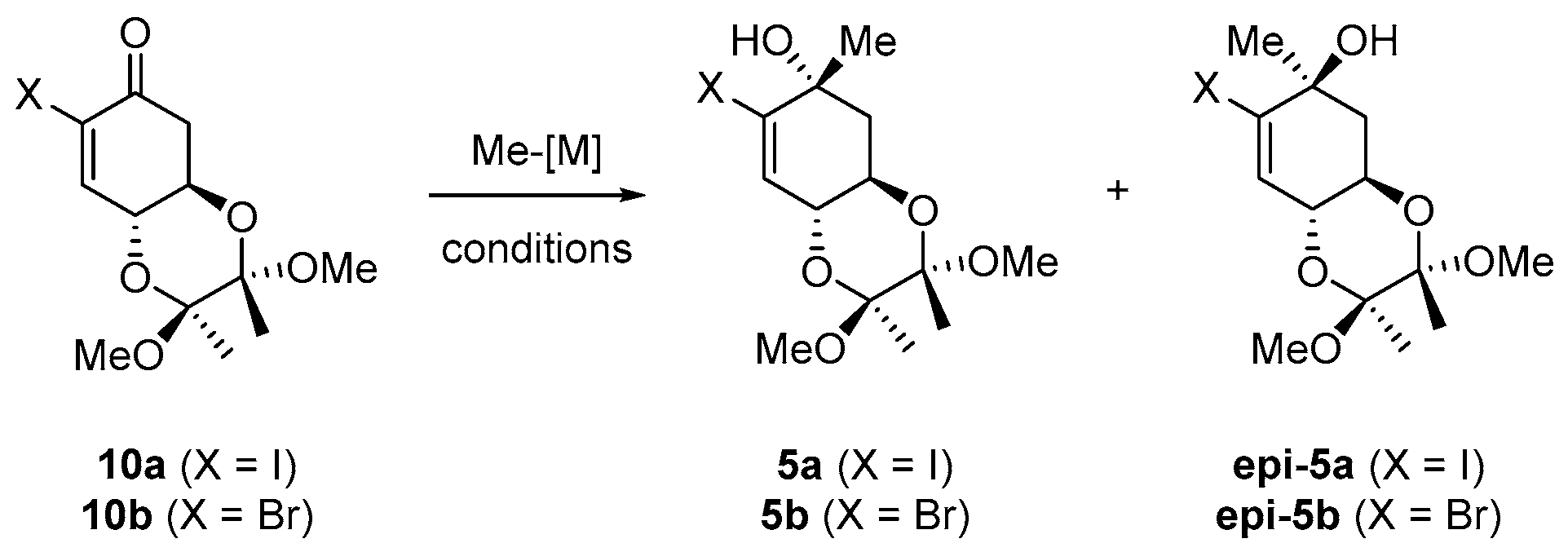
| Entry | X | M | Conditions [a] | d.r. 5/epi-5 [b] |
|---|---|---|---|---|
| 1 | I | MgI | −40 °C / THF | ~1:6 |
| 2 | Br | MgI | −40 °C / THF | ~1:4 |
| 3 | Br | MgI | −40 °C / CeCl3 / THF | ~1:2 |
| 4 | Br | MgBr | r.t. / Fe-Josiphos / CuBr-SMe2 / MTBE | only 1,4-addition |
| 5 | Br | MgBr | −78 °C / Fe-Josiphos CuBr-SMe2 / THF | 1,4-addition + traces of 5b |
| 6 | Br | Li | −40 °C / THF | ~1.4:1 |
| 7 | Br | Li | −40 °C / CeCl3 / THF | ~1:1 |
| 8 | Br | Li | −78 °C / THF | 1.7:1 |
| 9 | I | Li | −78 °C / THF | decomposition |
| 10 | Br | AlMe3 | 0 °C / THF | ~1:3 |
| 11 | Br | AlMe3 | 0 °C / [Rh(cod)Cl2]2 / BINAP (rac.) / THF / n-heptane | only 1,4-addition |
| 12 | Br | DABAL | 0 °C / [Rh(cod)Cl2]2 / BINAP (rac.) / THF / n-heptane | no reaction |
| 13 | Br | ZnMe2 | r.t. / THF | no reaction |
| 14 | Br | ZnMe2 | r.t. / Ti(iPrO)4 / toluene | traces |
| Substance | Q1 Mass (Da) | Q3 Mass (Da) Quantifier | Q3 Mass (Da) Qualifier |
|---|---|---|---|
| ALT (1a) | 291.0 | 203.0 | 248.0 |
| ALT-D3 (1b) | 294.0 | 203.0 | 248.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebald, M.A.; Gebauer, J.; Sommerfeld, T.; Koch, M. First Synthesis of (−)-Altenuene-D3 Suitable as Internal Standard for Isotope Dilution Mass Spectrometry. Molecules 2019, 24, 4563. https://doi.org/10.3390/molecules24244563
Sebald MA, Gebauer J, Sommerfeld T, Koch M. First Synthesis of (−)-Altenuene-D3 Suitable as Internal Standard for Isotope Dilution Mass Spectrometry. Molecules. 2019; 24(24):4563. https://doi.org/10.3390/molecules24244563
Chicago/Turabian StyleSebald, Michael A., Julian Gebauer, Thomas Sommerfeld, and Matthias Koch. 2019. "First Synthesis of (−)-Altenuene-D3 Suitable as Internal Standard for Isotope Dilution Mass Spectrometry" Molecules 24, no. 24: 4563. https://doi.org/10.3390/molecules24244563
APA StyleSebald, M. A., Gebauer, J., Sommerfeld, T., & Koch, M. (2019). First Synthesis of (−)-Altenuene-D3 Suitable as Internal Standard for Isotope Dilution Mass Spectrometry. Molecules, 24(24), 4563. https://doi.org/10.3390/molecules24244563