
Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component





Abstract
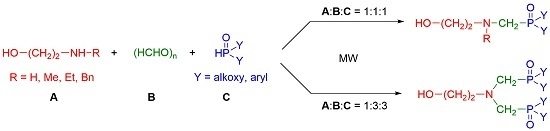
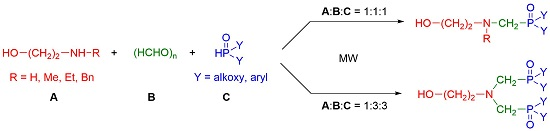
:
1. Introduction
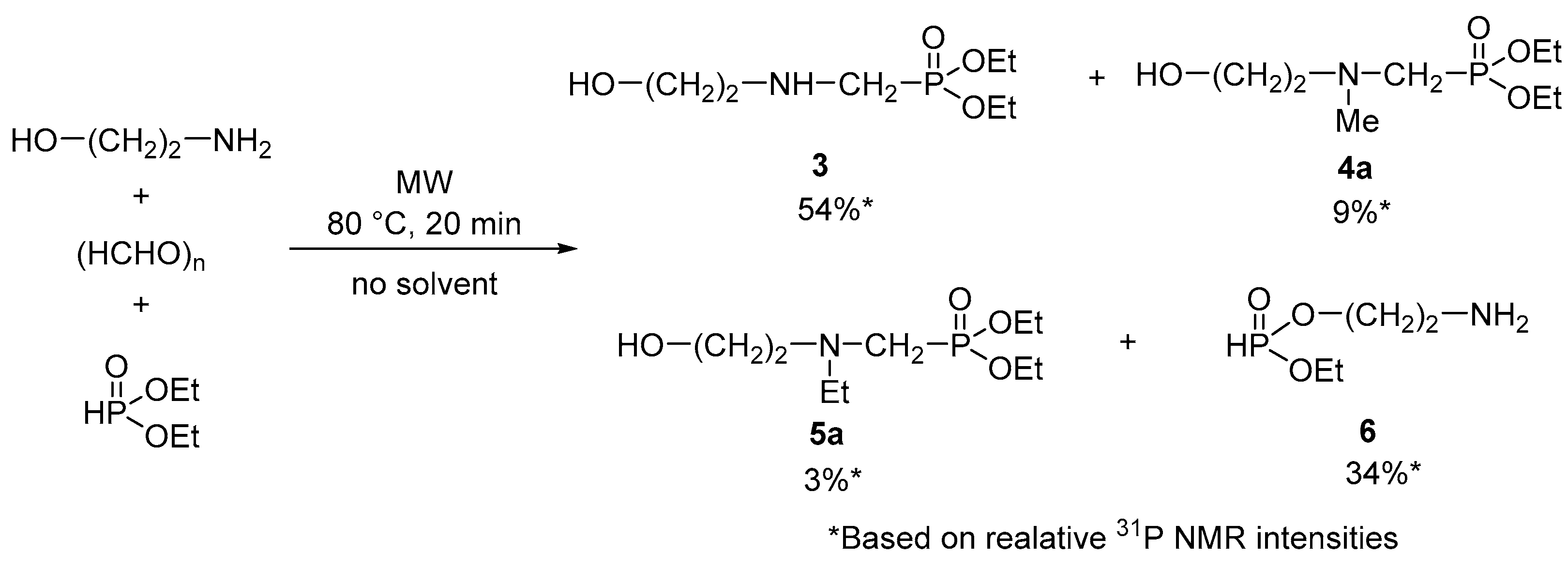
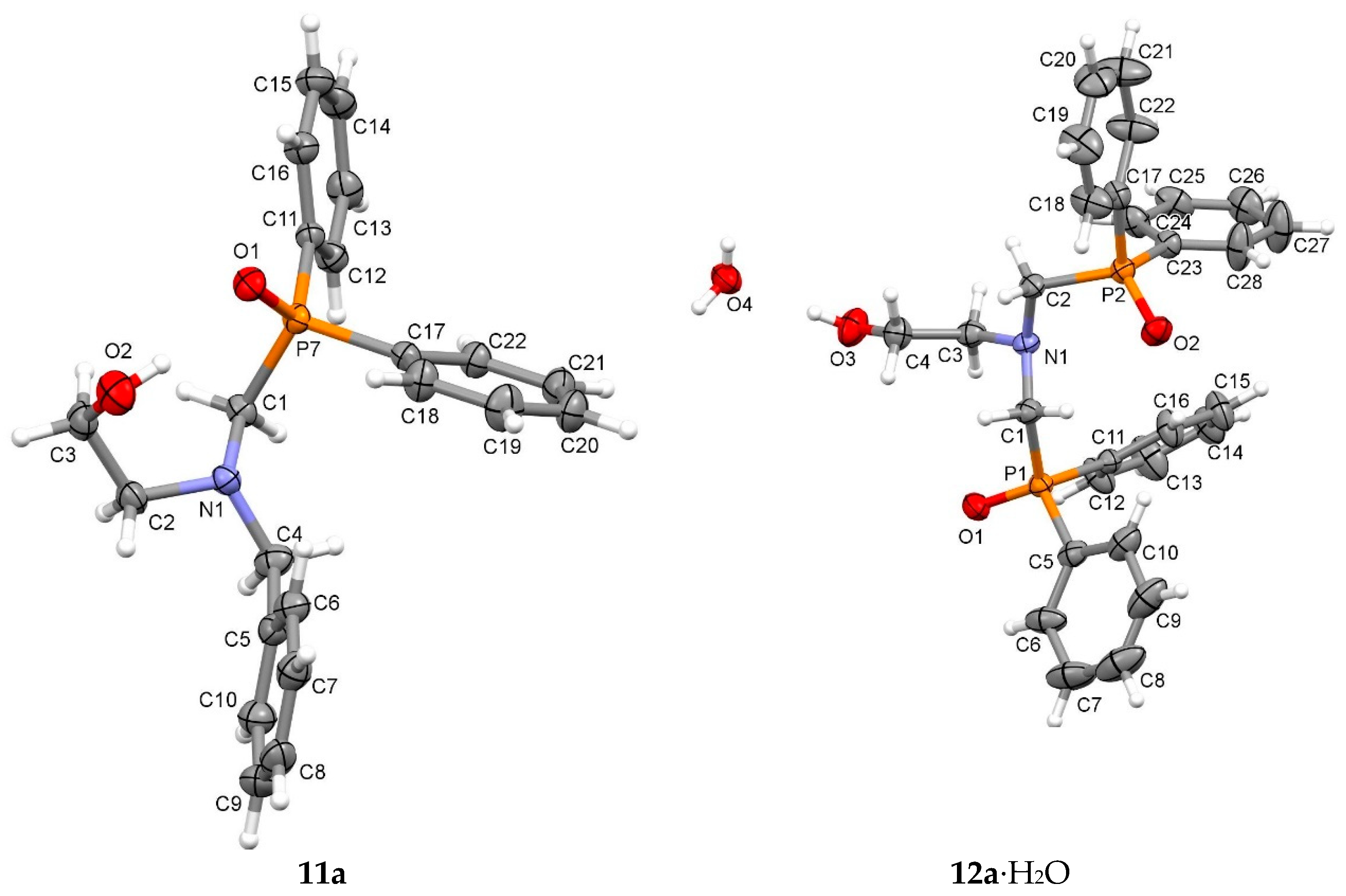
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of the 2-Hydroxyethyl-α-aminophosphonates and -α-Aminophosphinates
3.3. General Procedure for the Synthesis of the 2-Hydroxyethyl-α-aminophosphine oxides
3.4. General Procedure for the Synthesis of the N,N-Bis(diarylphosphinoylmethyl)ethanolamines
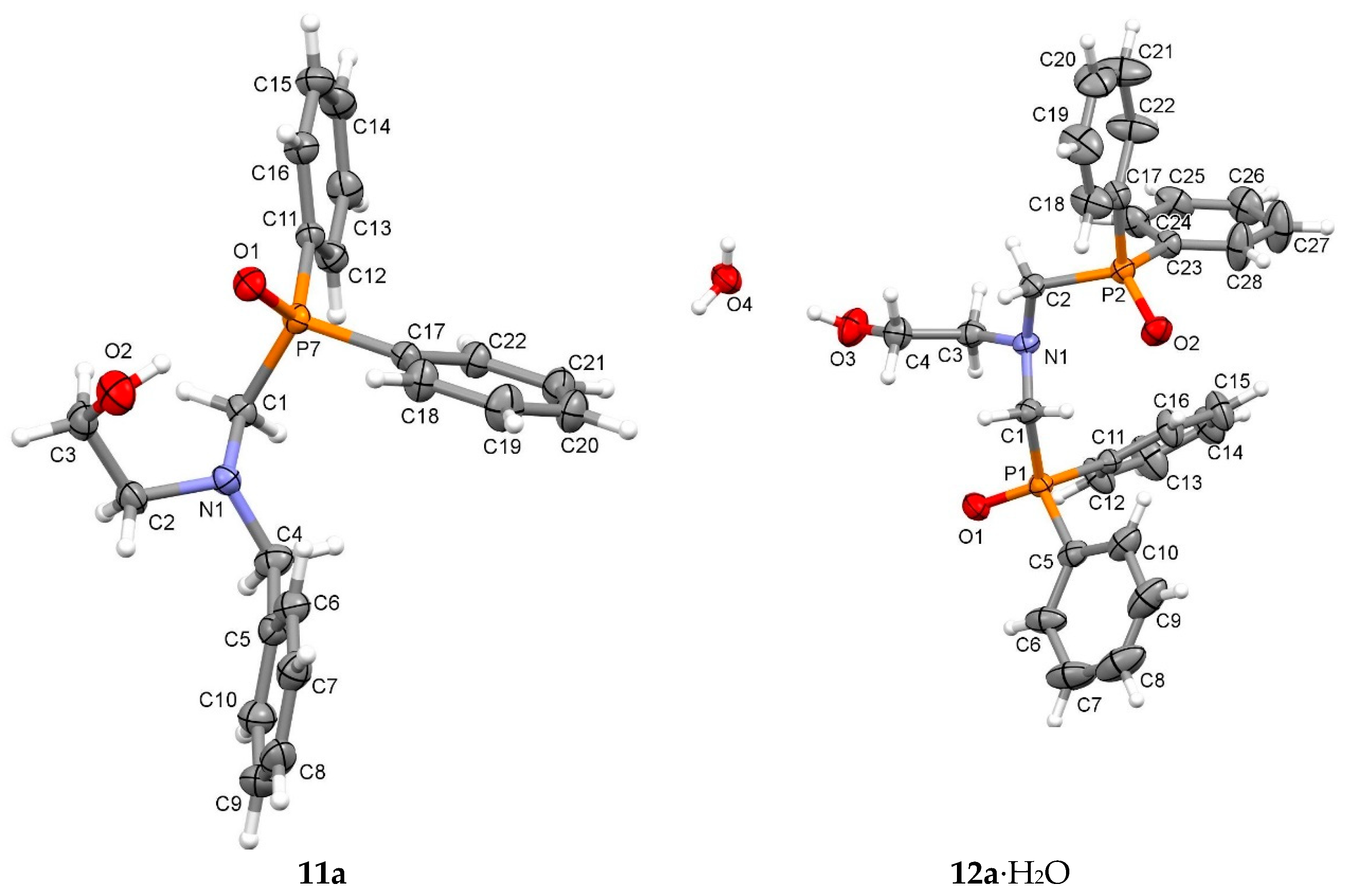
3.5. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hudson, H.R.; Kukhar, V.P. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Wiley: Chichester, UK, 2000; ISBN 978-0-471-89149-9. [Google Scholar]
- Tajti, Á.; Keglevich, G. The Importance of Organophosphorus Compounds as Biologically Active Agents; Organophosphorus Chemistry, Keglevich, G., Eds.; Walter de Gruyter GmbH: Berlin, Germany, 2018; pp. 53–65. ISBN 978-3-11-053453-5. [Google Scholar]
- Bálint, E.; Tajti, Á.; Tripolszky, A.; Keglevich, G. Synthesis of platinum, palladium and rhodium complexes of α-aminophosphine ligands. Dalton Trans. 2018, 47, 4755–4778. [Google Scholar] [CrossRef] [PubMed]
- Ben-Aroya, B.B.-N.; Portnoy, M. Solid-phase synthesis of an α-aminophosphine library. J. Comb. Chem. 2001, 3, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Ben-Aroya, B.B.-N.; Portnoy, M. Preparation of α-aminophosphines on solid support: Model studies and parallel synthesis. Tetrahedron 2002, 58, 5147–5158. [Google Scholar] [CrossRef]
- Patai, S. The Hydroxyl Group; Wiley: Hoboken, NJ, USA, 1971; ISBN 9780471669395. [Google Scholar]
- Patai, S. Carboxylic Acids and Esters; Wiley: Hoboken, NJ, USA, 1969; ISBN 9780471669197. [Google Scholar]
- Bálint, E.; Fazekas, E.; Drahos, L.; Keglevich, G. The Synthesis of N,N-Bis(dialkoxyphosphinoylmethyl)- and N,N-Bis(diphenylphosphinoylmethyl)glycine Esters by the Microwave-Assisted Double Kabachnik–Fields Reaction. Heteroatom Chem. 2013, 24, 510–515. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Kóti, J.; Keglevich, G. Synthesis of N,N-Bis(dialkoxyphosphinoylmethyl)- and N,N-Bis(diphenylphosphinoylmethyl)-β- and γ-amino acid Derivatives by the Microwave-Assisted Double Kabachnik–Fields Reaction. Heteroatom Chem. 2015, 26, 106–115. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Medved, T.Y. New synthesis of aminophosphonic acids. Dokl. Akad. Nauk SSSR 1952, 83, 689–692. [Google Scholar]
- Fields, E.K. The synthesis of esters of substituted amino phosphonic acids. J. Am. Chem. Soc. 1952, 74, 1528–1531. [Google Scholar] [CrossRef]
- Pudovik, A.N. Addition of dialkyl phosphites to imines. New method of synthesis of esters of amino phosphonic acids. Dokl. Akad. Nauk SSSR 1952, 83, 865–869. [Google Scholar]
- Keglevich, G.; Szekrenyi, A. Eco-friendly accomplishment of the extended Kabachnik-Fields reaction; a solvent-and catalyst-free microwave-assisted synthesis of α-aminophosphonates and α-aminophosphine oxides. Lett. Org. Chem. 2008, 5, 616–622. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Tripolszky, A. Synthesis of α-aminophosphonates by the Kabachnik–Fields Reaction and by the Pudovik Reaction. In Organophosphorus Chemistry; Keglevich, G., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2018; pp. 108–147. ISBN 978-3-11-053453-5. [Google Scholar]
- Bálint, E.; Tóth, R.E.; Keglevich, G. Synthesis of alkyl α-aminomethyl-phenylphosphinates and N,N-bis(alkoxyphenylphosphinylmethyl)amines by the microwave-assisted Kabachnik–Fields reaction. Heteroatom Chem. 2016, 27, 323–335. [Google Scholar] [CrossRef]
- Bálint, E.; Tripolszky, A.; Jablonkai, E.; Karaghiosoff, K.; Czugler, M.; Mucsi, Z.; Kollár, L.; Pongrácz, P.; Keglevich, G. Synthesis and use of α-aminophosphine oxides and N,N-bis(phosphinoylmethyl)amines—A study on the related ring platinum complexes. J. Organomet. Chem. 2016, 801, 111–121. [Google Scholar] [CrossRef]
- Tajti, Á.; Bálint, E.; Keglevich, G. Synthesis of ethyl octyl α-aminophosphonate derivatives. Curr. Org. Synth. 2016, 13, 638–675. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Kalocsai, D.; Mátravölgyi, B.; Konstantin, K.; Czugler, M.; Keglevich, G. Synthesis and utilization of optically active α-aminophosphonate derivatives by Kabachnik-Fields reaction. Tetrahedron 2017, 73, 5659–5667. [Google Scholar] [CrossRef]
- Barsukov, A.V.; Zhadanov, B.V.; Matkovskaya, T.A.; Kaslina, N.A.; Polyakova, I.A.; Yaroshenko, G.F.; Kessenikh, A.V.; Dyatlova, N.M. Synthesis of New Complexons of the Aliphatic Series and Investigation of the Mechanism of Acidic Dissociation. Zh. Obshch. Khim. 1985, 55, 1594–1600. [Google Scholar]
- Ju, Z.; Zou, R.; Ye, Y.; Zhao, Y. A Facile and Clean Procedure for Preparation of α-Aminophosphonates via a Rotary Evaporator Equipped with Circulating Water Vacuum Pumps. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 898–902. [Google Scholar] [CrossRef]
- Kaboudin, B.; Nazari, R. Microwave-assisted synthesis of 1-aminoalkyl phosphonates under solvent-free conditions. Tetrahedron Lett. 2001, 42, 8211–8213. [Google Scholar] [CrossRef]
- Bálint, E.; Keglevich, G. The Spread of the Application of the Microwave Technique in Organic Synthesis. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Basel, Switzerland, 2016; p. 110. ISBN 978-3-319-30632-2. [Google Scholar]
- Zamorano-Octaviano, J.; Hernández-Martínez, A.; Ortega-Guevara, A.; Linzaga-Elizalde, I.; Höpfl, H. Linear and cyclic aminomethanephosphonic acid esters derived from benzaldehyde derivatives, 3-aminopropanol, and diethyl phosphite. Heteroatom Chem. 2006, 17, 75–80. [Google Scholar] [CrossRef]
- Floch, V.; Le Bolc’h, G.; Gable-Guillaume, C.; Le Bris, N.; Yaouanc, J.-J.; des Abbayes, H.; Férec, C.; Clément, J.-C. Phosphonolipids as non-viral vectors for gene therapy. Eur. J. Med. Chem. 1998, 33, 923–934. [Google Scholar] [CrossRef]
- Krchová, T.; Herynek, V.; Gálisová, A.; Blahut, J.; Hermann, P.; Kotek, J. Eu (III) Complex with DO3A-amino-phosphonate Ligand as a Concentration-Independent pH-Responsive Contrast Agent for Magnetic Resonance Spectroscopy (MRS). Inorg. Chem. 2017, 56, 2078–2091. [Google Scholar] [CrossRef]
- Martel, S.; Clément, J.-L.; Muller, A.; Culcasi, M.; Pietri, S. Synthesis and 31P NMR characterization of new low toxic highly sensitive pH probes designed for in vivo acidic pH studies. Bioorg. Med. Chem. 2002, 10, 1451–1458. [Google Scholar] [CrossRef]
- Heydari, A.; Karimian, A.; Ipaktschi, J. Lithium perchlorate/diethylether catalyzed aminophosphonation of aldehydes. Tetrahedron Lett. 1998, 39, 6729–6732. [Google Scholar] [CrossRef]
- Chougrani, K.; Boutevin, B.; David, G.; Boutevin, G. New N,N-amino-diphosphonate-containing methacrylic derivatives, their syntheses and radical copolymerizations with MMA. Eur. Polym. J. 2008, 44, 1771–1781. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Garifzyanov, A.R.; Talan, A.S.; Davletshin, R.R.; Kurnosova, N.V. Synthesis of new liophilic functionalized aminomethylphosphine oxides and their acid-base and membrane-transport properties toward acidic substrates. Russ. J. Gen. Chem. 2009, 79, 1835–1849. [Google Scholar] [CrossRef]
- Gancarz, R. Alkylating Properties of Dialkyl Phosphites. Phosphorus, Sulfur Silicon Relat. Elem. 1994, 92, 193–199. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E.; Tajti, Á.; MÁtravölgyi, B.; Balogh, G.T.; Bálint, M.; Ilia, G. Microwave-assisted alcoholysis of dialkyl phosphites by ethylene glycol and ethanolamine. Pure Appl. Chem. 2014, 86, 1723–1728. [Google Scholar] [CrossRef]
- Tajti, Á.; Keglevich, G.; Bálint, E. Microwave-assisted alcoholysis of dialkyl H-phosphonates by diols and amino alcohols. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 769–775. [Google Scholar] [CrossRef]
- CrysAlisPro, version 1.171.39.46e; Rigaku Oxford Diffraction: Yarnton, UK, 2018.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystalstructure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4a–c, 5a–c, 8–11a–c and 12a–c are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
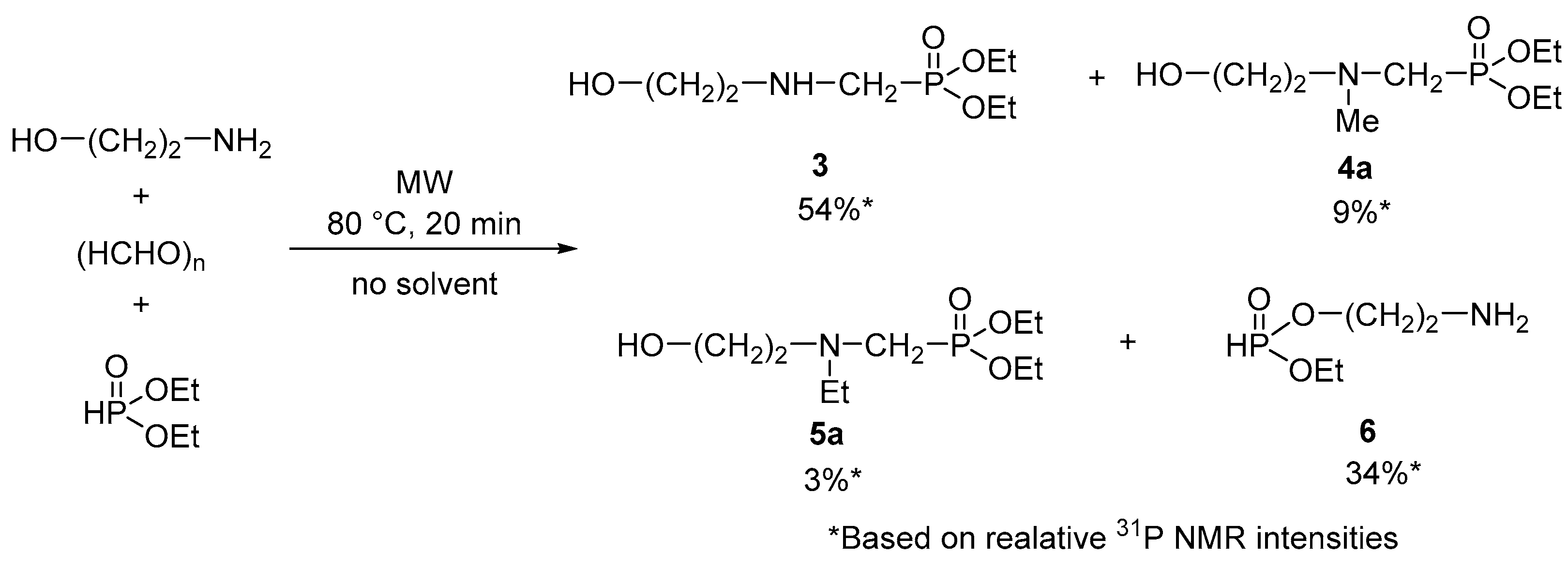
| Entry | Mode of Heating | T [°C] | t [min] | Conversion [%] a | Product Composition [%] a | |
|---|---|---|---|---|---|---|
| 4a | 7 | |||||
| 1 | MW | 60 | 20 | 85 | 96 | 4 |
| 2 | MW | 80 | 20 | 100 | 95 | 5 |
| 3 | Δ | 80 | 20 | 96 | 81 | 19 |
| 4 | MW | 100 | 10 | 100 | 90 | 10 |

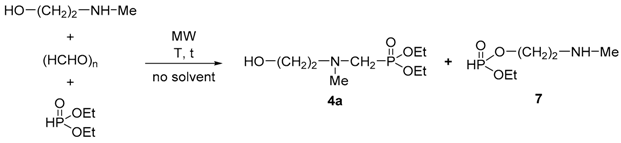
| Entry | R | Y1 | Y2 | Yield [%] a |
|---|---|---|---|---|
| 1 | Me | OEt | OEt | 78 (4a) |
| 2 | Me | OBu | OBu | 84 (4b) |
| 3 | Me | OEt | Ph | 67 (4c) |
| 4 | Et | OEt | OEt | 72 (5a) |
| 5 | Et | OBu | OBu | 79 (5b) |
| 6 | Et | OEt | Ph | 64 (5c) |
| Entry | T [°C] | t [min] | Conversion [%] a |
|---|---|---|---|
| 1 | 80 | 20 | 58 |
| 2 | 80 | 30 | 86 |
| 3 | 80 | 40 | 89 |
| 4 | 100 | 20 | 92 |
| 5 | 100 | 30 | 100 |
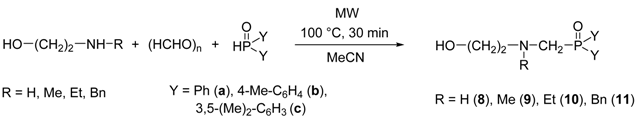
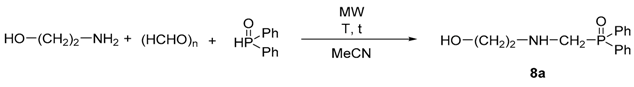
| Entry | R | Y | Yield [%] a |
|---|---|---|---|
| 1 | H | Ph | 96 (8a) |
| 2 | H | 4-Me-C6H4 | 89 (8b) |
| 3 | H | 3,5-(Me)2-C6H3 | 95 (8c) |
| 4 | Me | Ph | 95 (9a) |
| 5 | Me | 4-Me-C6H4 | 96 (9b) |
| 6 | Me | 3,5-(Me)2-C6H3 | 93 (9c) |
| 7 | Et | Ph | 95 (10a) |
| 8 | Et | 4-Me-C6H4 | 93 (10b) |
| 9 | Et | 3,5-(Me)2-C6H3 | 88 (10c) |
| 10 | Bn | Ph | 90 (11a) |
| 11 | Bn | 4-Me-C6H4 | 89 (11b) |
| 12 | Bn | 3,5-(Me)2-C6H3 | 92 (11c) |
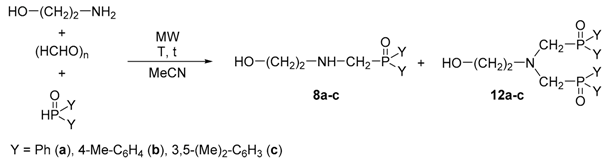
| Entry | Y | (HCHO)n [equiv] | Y2P(O)H [equiv] | T [°C] | t [h] | Product Composition [%] a | Yield b [%] | |
|---|---|---|---|---|---|---|---|---|
| 8a–c | 12a–c | |||||||
| 1 | Ph | 2 | 2 | 100 | 1 | 75 | 25 | - |
| 2 | Ph | 2 | 2 | 120 | 1 | 66 | 34 | - |
| 3 | Ph | 2 | 2 | 120 | 1.5 | 64 | 36 | - |
| 4 | Ph | 2 | 2.5 | 120 | 1 | 62 | 38 | - |
| 5 | Ph | 2.5 | 2.5 | 120 | 1 | 25 | 75 | - |
| 6 | Ph | 3 | 3 | 120 | 1 | 0 | 100 | 95 (12a) |
| 7 | 4-Me-C6H4 | 3 | 3 | 120 | 1 | 0 | 100 | 93 (12b) |
| 8 | 3,5-(Me)2-C6H3 | 3 | 3 | 120 | 1 | 0 | 100 | 91 (12c) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajti, Á.; Szatmári, E.; Perdih, F.; Keglevich, G.; Bálint, E. Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component. Molecules 2019, 24, 1640. https://doi.org/10.3390/molecules24081640
Tajti Á, Szatmári E, Perdih F, Keglevich G, Bálint E. Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component. Molecules. 2019; 24(8):1640. https://doi.org/10.3390/molecules24081640
Chicago/Turabian StyleTajti, Ádám, Enikő Szatmári, Franc Perdih, György Keglevich, and Erika Bálint. 2019. "Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component" Molecules 24, no. 8: 1640. https://doi.org/10.3390/molecules24081640
APA StyleTajti, Á., Szatmári, E., Perdih, F., Keglevich, G., & Bálint, E. (2019). Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component. Molecules, 24(8), 1640. https://doi.org/10.3390/molecules24081640