Abstract
To combat bacterial resistance, a series of new oxazolidinone-fluoroquinolone hybrids have been synthesized and characterized. All synthetic hybrids were preliminarily evaluated for their in vitro antibacterial activities against 6 standard strains and 3 clinical isolates. The majority of hybrids displayed excellent activities against Gram-positive bacteria, but limited activities against Gram-negative bacteria. Hybrids OBP-4 and OBP-5 were found to be the most promising compounds. Further, in vitro antibacterial activities, mode of action and acute toxicity in mice of hybrids OBP-4 and OBP-5 were investigated. Hybrids OBP-4 and OBP-5 exhibited potent activities against Gram-positive bacteria, including drug-resistant strains. Correspondingly, studies on the mode of action of hybrids OBP-4 and OBP-5 indicated a strong inhibitory activity on protein synthesis by binding the active site of 50S subunit, but a weak inhibitory action on DNA synthesis. In addition, LD50 values of hybrids OBP-4 and OBP-5 in the acute oral toxicity were larger than 2000 mg/kg, suggesting a good safety profile.
1. Introduction
Although the discovery of antibiotics has had a profound positive impact on the improvements of human health and life expectancy, the emergence of antibiotic resistance has severely limited their clinical efficacy and applications [1,2]. Drug-resistant bacteria, especially multi-drug resistant (MDR) bacteria, representing the most common causative agents of nosocomial infections [3,4], are difficult to deal with using currently existing antibiotics. Worse still, the continuous evolution and spread of antibiotic-resistant bacteria have caused enormous financial burden worldwide in terms of increased morbidity, mortality and use of medical resources [5,6,7]. It is estimated that antibiotic-resistant infections could claim 10 million deaths per year and up to 100 trillion US dollars by 2050 if nothing is done [8]. Nowadays, not only is antimicrobial resistance a major public concern, but more worryingly, there is an urgent need for novel antibacterials with improved potency and a low propensity for resistance development.
In recent years, hybrid antibacterials, linking two different pharmacophores by a non-cleavable spacer, were introduced as a promising strategy to fight drug-resistant bacteria and delay resistance development [9,10]. Such a strategy has been applied to the design and synthesis of various hybrid antibacterials [11,12,13,14]. Fluoroquinolone, with its superior dual-target activity, chemical stability and distinct structure-activity relationship (SAR), is the most popular partner in the design of hybrid antibacterials [15]. Oxazolidinone exerted antibacterial activity by inhibiting protein synthesis [16], and has a dissimilar mechanism of action to fluoroquinolone. The covalent combination of oxazolidinone and fluoroquinolone may be a better choice for fighting the prevalent resistance to these two classes of compounds [17,18]. Furthermore, their SAR investigations have shown that oxazolidinone and fluoroquinolone have a good tolerance for structural modification and a common heterocyclic amine was introduced at the 4- and 7- positions of the respective phenyl ring [19], which provide an advantage for their combination. Meanwhile, previous studies have demonstrated that the oxazolidinone-quinolone hybrids with a 4-piperidinemethanol linker exhibited improved antibacterial activities [20] and a low propensity for spontaneous resistance development [21,22].
Inspired by these previous studies, we synthesized a series of new oxazolidinone-fluoroquinolone hybrids with the modifications of the spacer or fluoroquinolone pharmacophore. Here, we report the in vitro antibacterial activities of these hybrids, and the mode of action and acute toxicity of hybrids OBP-4 and OBP-5.
2. Results and Discussion
2.1. Chemistry
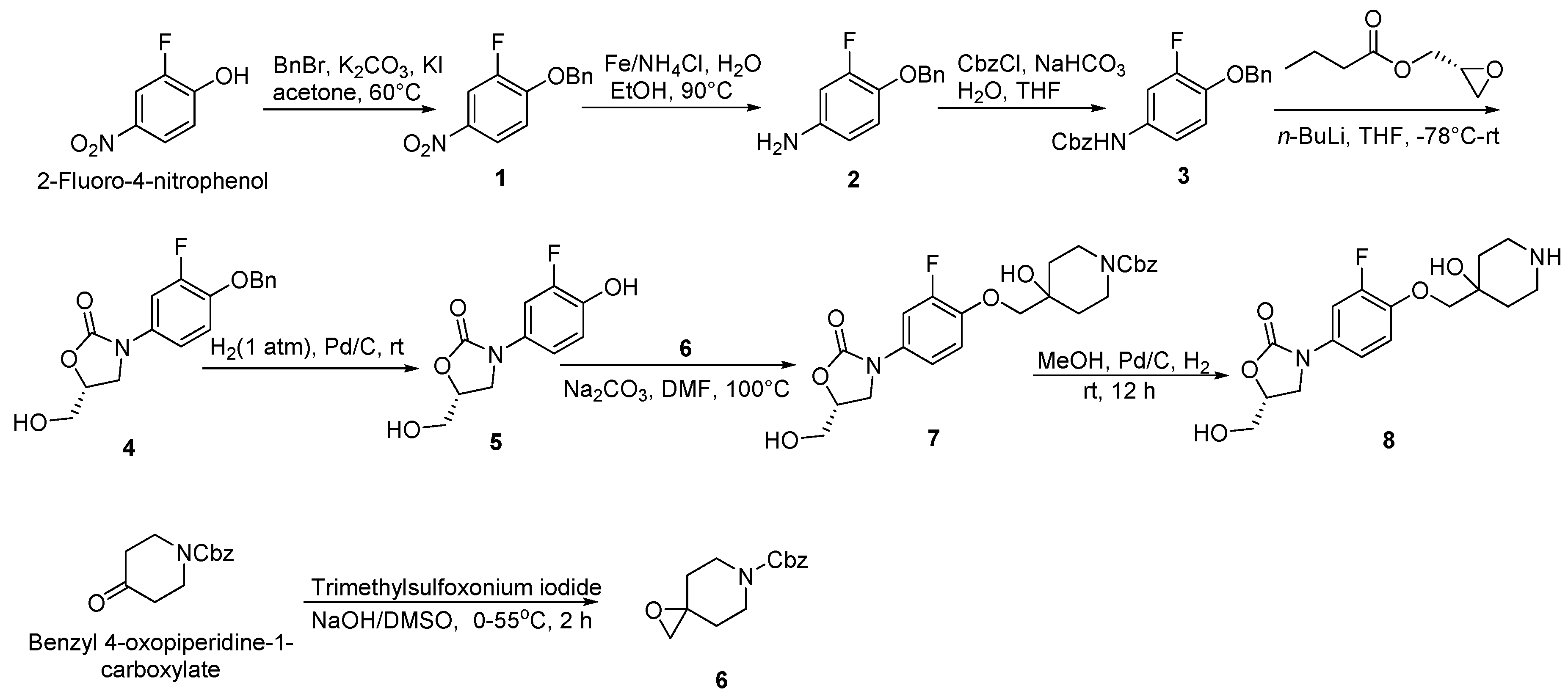
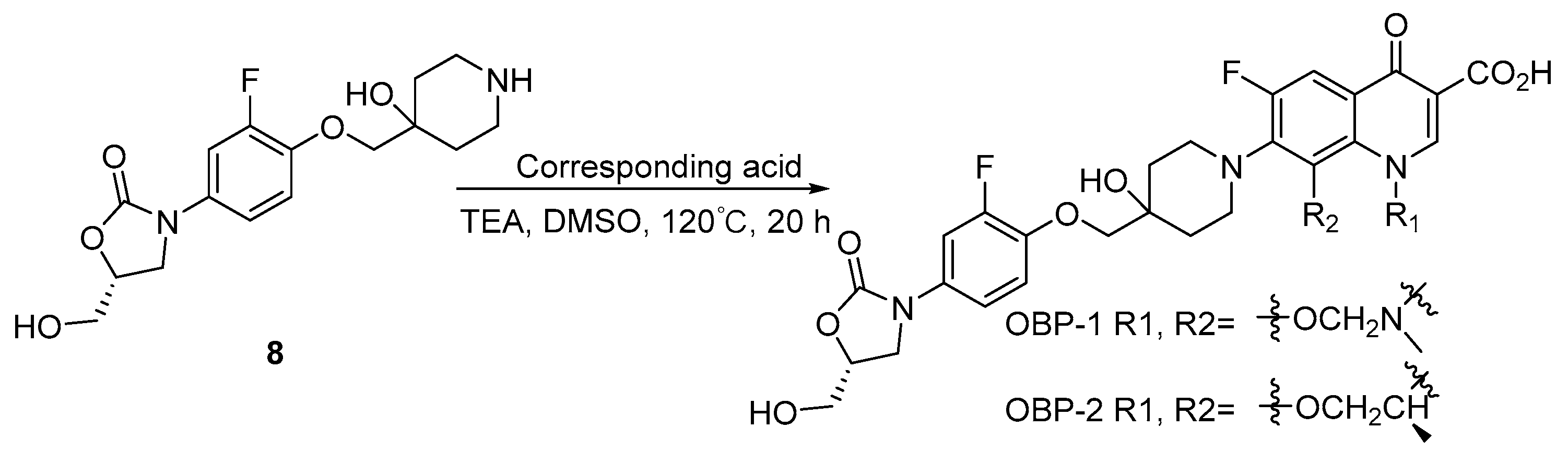
The synthesis of oxazolidinone-fluoroquinolone hybrids OBP-(1–7) was performed as outlined in Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5, Scheme 6 and Scheme 7. The hybrids OBP-(1–5) consisted of two parts, compound 8 and corresponding acids (Scheme 2, Scheme 3, Scheme 4 and Scheme 5). Compound 8 was synthesized according to the following Scheme 1. The corresponding acids of OBP-1 and OBP-2 were obtained from commercial sources, and those of OBP-(3–5) were synthesized by another routes (Scheme 3, Scheme 4 and Scheme 5). Meanwhile, we also prepared hybrids OBP-6 and OBP-7, containing an exo-3-methyl-6-amino-3-azabicyclo [3.1.0] hexane linker between the oxazolidinone and fluoroquinolone elements (Scheme 6 and Scheme 7). The structures of the target hybrids OBP-(1–7) were further characterized (Supplementary Materials).
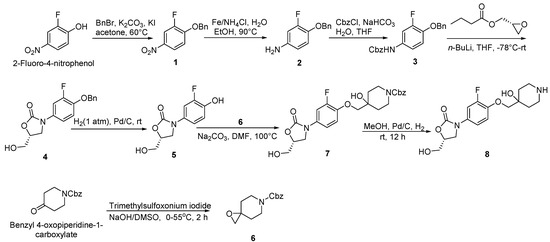
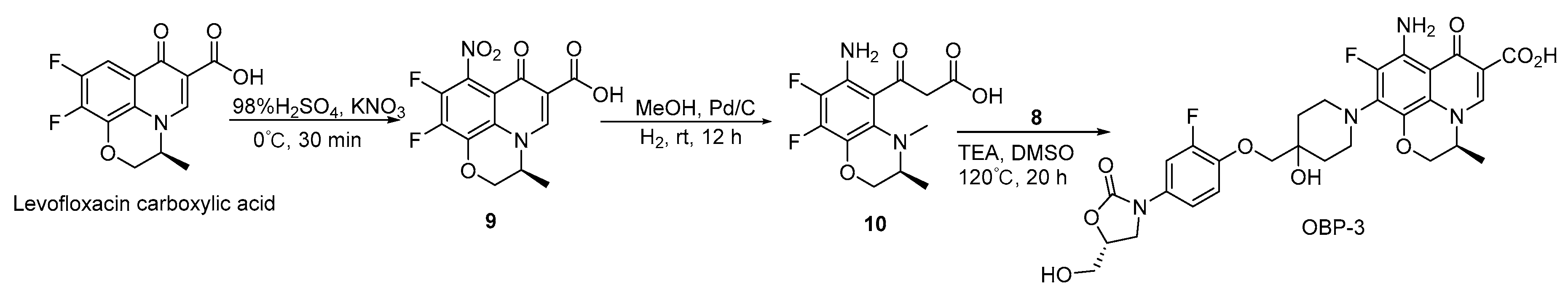
Scheme 1.
Synthesis of compounds 6 and 8.
Scheme 2.
Synthesis of hybrids OBP-1 and OBP-2.
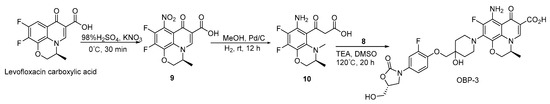
Scheme 3.
Synthesis of hybrid OBP-3.
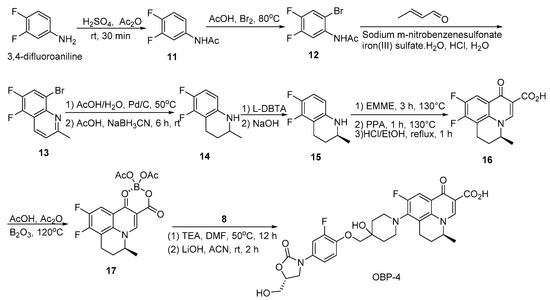
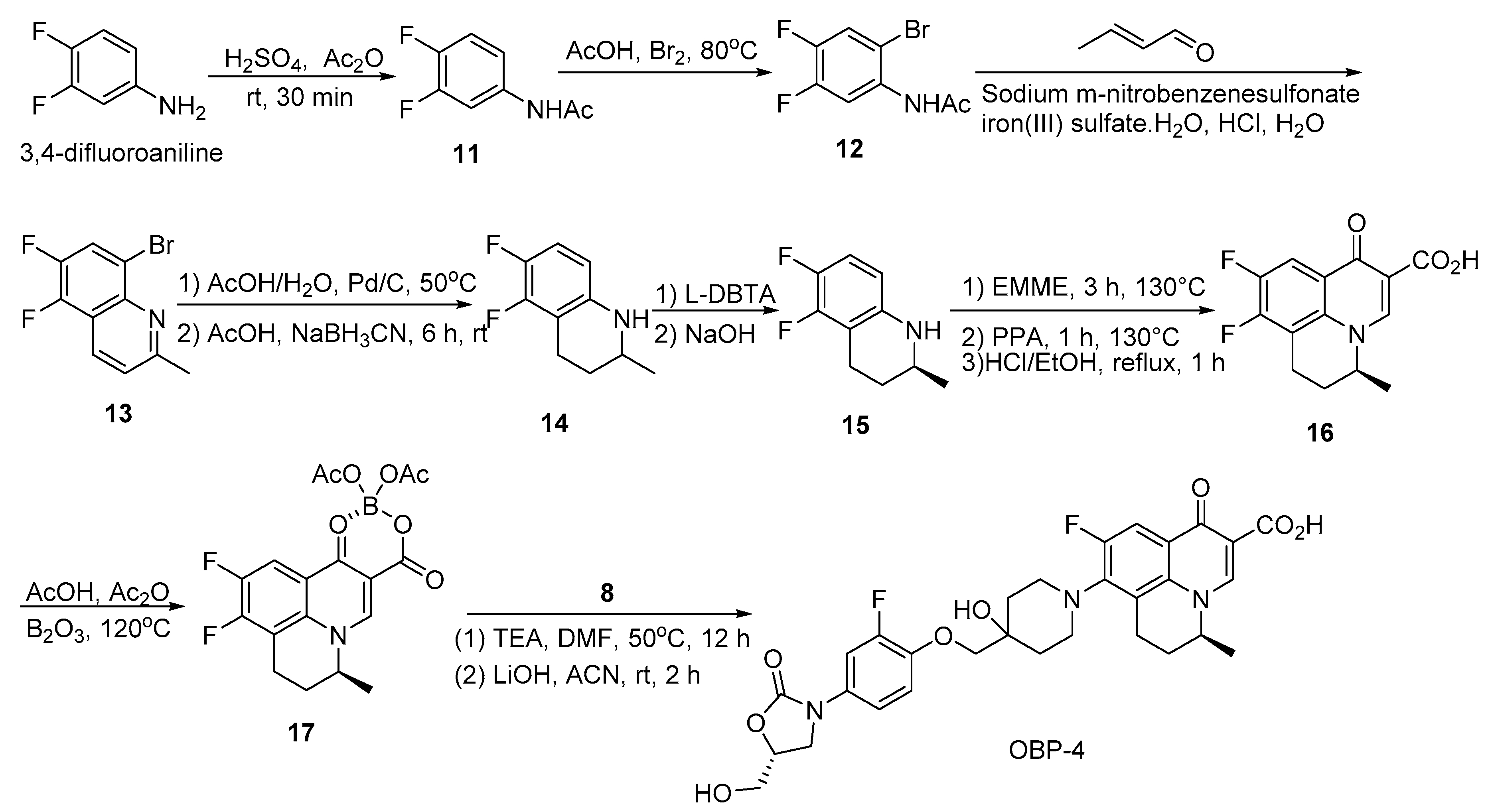
Scheme 4.
Synthesis of hybrid OBP-4.
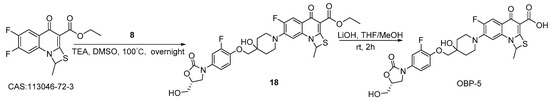
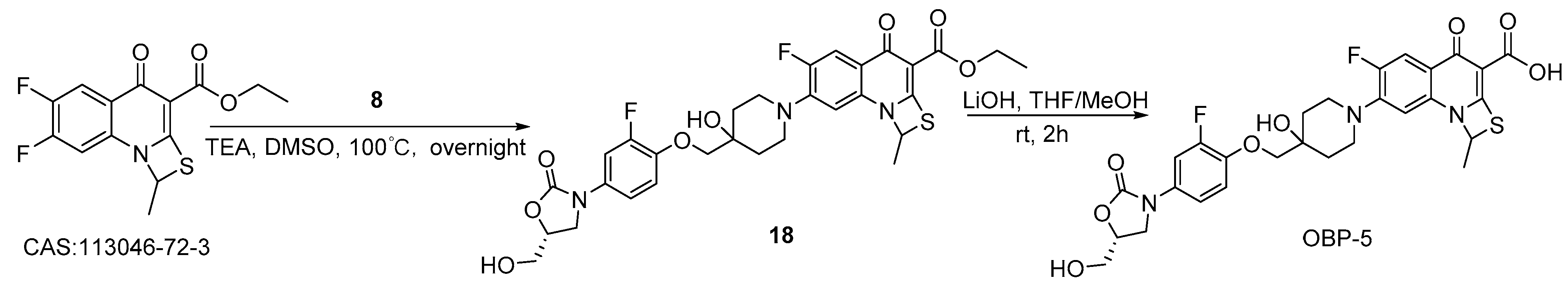
Scheme 5.
Synthesis of hybrid OBP-5.
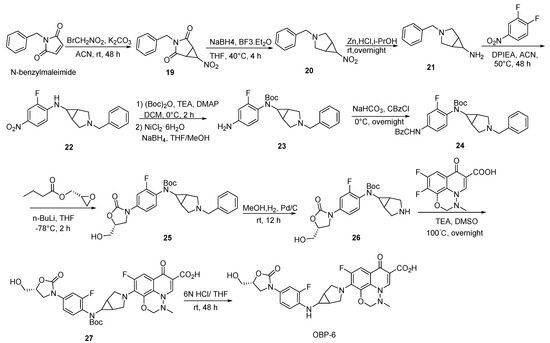
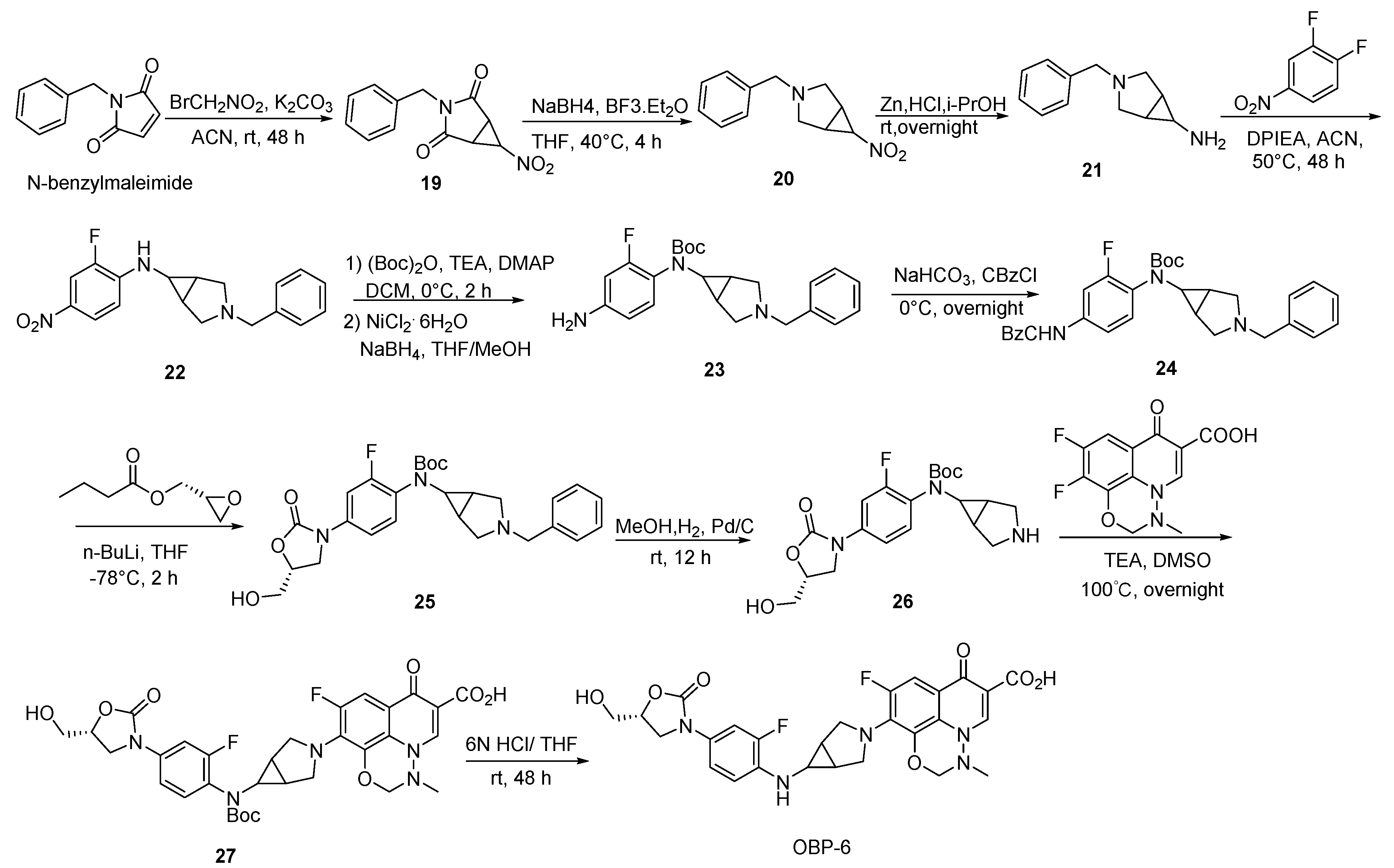
Scheme 6.
Synthesis of hybrid OBP-6.
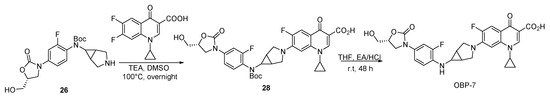
Scheme 7.
Synthesis of hybrid OBP-7.
2.2. Antibacterial Activities
2.2.1. In Vitro Activities of Oxazolidinone-Fluoroquinolone Hybrids
The in vitro antibacterial activities of oxazolidinone-fluoroquinolone hybrids against a panel of well-characterized susceptible and resistant Gram-positive and Gram-negative bacteria were evaluated and the results were presented as MICs expressed in µg/mL (Table 1). CZD (cadazolid), LZD (linezolid) and moxifloxacin (MXF) were used as comparator drugs. As shown in Table 1, MIC values of LZD and MXF against standard strains were within the acceptable range, indicating that the test results were reliable.

Table 1.
In vitro antibacterial activities of oxazolidinone-fluoroquinolone hybrids.
For Gram-positive bacteria, most oxazolidinone-fluoroquinolone hybrids with the exceptions of OBP-6 and OBP-7 exhibited the potent antibacterial activities with MICs ranging from 0.031 µg/mL to 2 µg/mL, even against levofloxacin-resistant Streptococcus agalactiae (S. agalactiae), vancomycin-resistant enterococcus (VRE) and methicillin-resistant Staphylococcus aureus (MRSA). Among these new hybrids, OBP-4 was the most active compound against Gram-positive bacteria (MICs ≤ 0.5 µg/mL), which was twice as active overall than the comparator agent CZD, a oxazolidinone-fluoroquinolone hybrid in phase III clinical trials [23]. The antibacterial efficacy of hybrid OBP-5 against Gram-positive bacteria was comparable to that of CZD, but was approximately 4–16 times stronger than that of LZD. The antibacterial activities of hybrids OBP-1, OBP-2 and OBP-3 against Gram-positive bacteria laid between that of CZD and LZD. Gram-positive bacteria were less susceptible to OBP-6 and OBP-7 than other hybrids, especially resistant strains. For Gram-negative bacteria, only Haemophilus influenzae (H. influenzae) was susceptible to these hybrids, while hybrids OBP-6 and OBP-7 also exhibited good activities against Escherichia coli (E. coli) (Table 1). The above results demonstrated that the antibacterial activities of these oxazolidinone-fluoroquinolone hybrids were mainly restricted to Gram-positive bacteria, suggesting a more pronounced oxazolidinone-type antibacterial spectrum.
Obviously, hybrids OBP-6 and OBP-7 differed greatly from other hybrids in antibacterial activities and spacer, which indicated the spacer was a major factor influencing their antibacterial activities [20]. Meanwhile, the antibacterial activity was more or less changed due to the differences of the substituents at N1-position of fluoroquinolone pharmacophores.
2.2.2. In Vitro Activities of Hybrids OBP-4 and OBP-5 against Clinical Isolates
As can be seen from the above data, OBP-4 and OBP-5 were the two most active oxazolidinone-fluoroquinolone hybrids. Therefore, the susceptibility of OBP-4 and OBP-5 against 263 clinical isolates were further tested. The resulting MICs for OBP-4, OBP-5 and comparator drugs enrofloxacin (ENR), ciprofloxacin (CIP), gatifloxacin (GAFX), nadifloxacin (NAFX), LZD and vancomycin (VAN) were listed in Table 2. For Gram-positive isolates, hybrids OBP-4 and OBP-5 had the lowest MICs among all tested compounds, with MIC90 values of 0.125–0.25 µg/mL for OBP-4 and ≤0.0625–0.25 µg/mL for OBP-5. For Gram-negative isolates (Haemophilus parasuis), hybrids OBP-4 and OBP-5 had relatively weak activities with MIC90 values of 4 µg/mL and 2 µg/mL, respectively (Table 2).
Table 2.
In vitro activities of OBP-4 and OBP-5 against clinical isolates.
For Staphylococcus aureus (S. aureus), Staphylococcus haemolyticus (S. haemolyticus), Staphylococcus epidermidis (S. epidermidis) and Streptococcus suis (S. suis), most clinical isolates (>90%) can be inhibited by hybrids OBP-4 and OBP-5 at 0.25 µg/mL, and MIC90 values of OBP-4 and OBP-5 against these isolates were much lower than those of comparator agents (Table 2 and Table 3). Against Streptococcus pneumoniae (S. pneumoniae) isolates, OBP-4 and OBP-5 retained the excellent activities with MIC90 of 0.25 µg/mL, while comparator drugs were almost inactive with MIC90 ≥ 32 µg/mL except for LZD (Table 2). Furthermore, OBP-4 and OBP-5 showed 2- to 16-fold higher activity against Enterococcus faecalis (E. faecalis), and 16- to 128-fold higher activity against Enterococcus faecium (E. faecium) than comparator agents (Table 2). Although OBP-4 and OBP-5 were less active against H. parasuis than quinolone antimicrobials, they were much better than LZD and VAN (Table 2). Therefore, hybrids OBP-4 and OBP-5 were the most potent compounds against tested clinical isolates. Overall, OBP-5 exhibited 2-fold greater activity than OBP-4, which can be best explained by the difference of the substituents at N1-position of fluoroquinolone pharmacophores between OBP-4 and OBP-5.
Table 3.
MIC frequency distributions for tests of OBP-4 and OBP-5 against clinical isolates.
Meanwhile, hybrids OBP-4 and OBP-5 were also tested against a group of common resistant strains, including MRSA, methicillin-resistant S. epidermidis (MRSE), CIP-resistant S. aureus, CIP-resistant S. haemolyticus, VAN- and quinolone-resistant S. pneumoniae, LZD-resistant E. faecalis, and LZD or VAN- and quinolone-resistant E. faecium. The activities of OBP-4 and OBP-5 against MRSA and MRSE were equivalent to those of quinolone comparator agents ENR, CIP, GAFX and NDFX, but markedly superior to those of LZD and VAN (Table 4). For other resistant organisms in the test, MICs values of OBP-4 and OBP-5 ranged from ≤ 0.0625 to 1 µg/mL, and were much lower than the corresponding MICs values of comparator agents (Table 4). Similarly, OBP-5 displayed higher activities against tested resistant strains than OBP-4. The superior activities of OBP-4 and OBP-5 against resistant strains indicated that they had no cross-resistance with tested oxazolidinones or quinolones.
Table 4.
In vitro activities of OBP-4 and OBP-5 against drug-resistant strains.
Again, the superior antibacterial activities of hybrids OBP-4 and OBP-5 against Gram-positive bacteria including drug-resistant strains were confirmed. The result hinted that OBP-4 and OBP-5 may have a similar mode of action with oxazolidinones. In this study, the reduced membrane- permeability may be a leading cause of poor activities of OBP-4 and OBP-5 against Gram-negative bacteria. Intrinsically, the weak fluoroquinolone-like characteristic of OBP-4 and OBP-5 could be attributed to the large molecular volume and decreased liposolubility caused by the hybridization.
2.3. Mode of Action of Hybrids OBP-4 and OBP-5
2.3.1. In Vitro Enzyme Assays of Hybrids OBP-4 and OBP-5
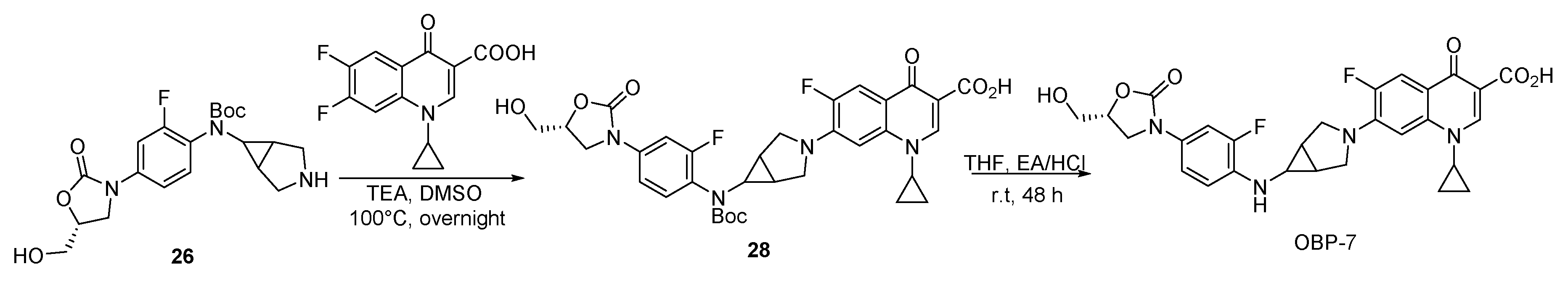
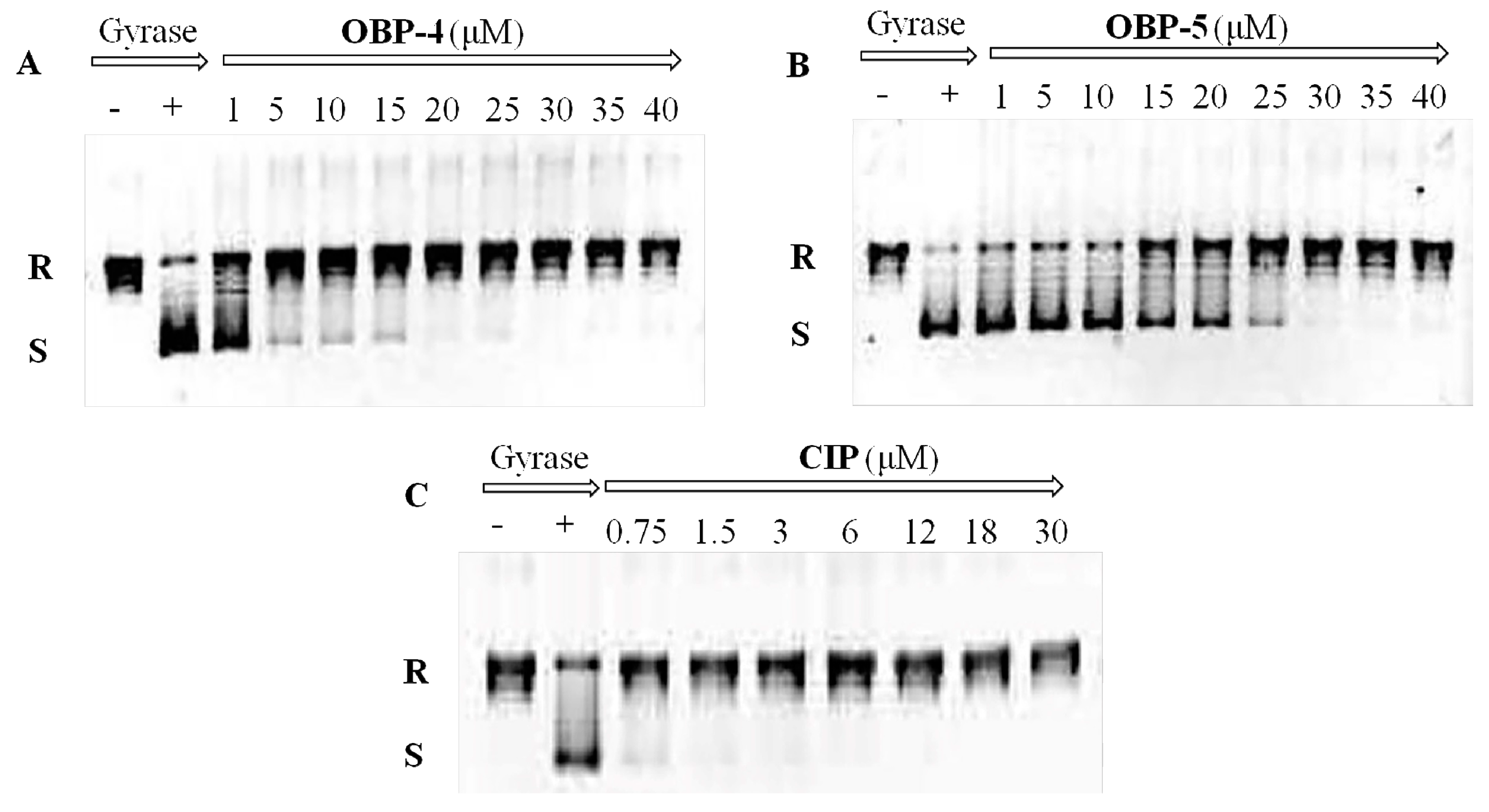
In order to explore the mode of action of OBP-4 and OBP-5, we carried out the enzyme assays for these two hybrids. The results were presented in Figure 1 and Figure 2 and Table 5.
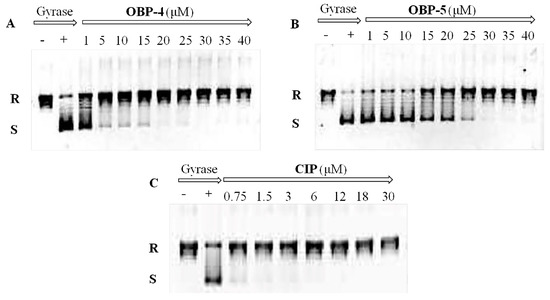
Figure 1.
Inhibition assay of OBP-4 (A), OBP-5 (B) and CIP (C) on DNA gyrase. Line 1(−): no compound and DNA gyrase; Line 2(+): DNA gyrase present and compound absent; Line 3-11(9): DNA gyrase with different concentrations of tested compound. R: relaxed DNA; S: supercoiled DNA.
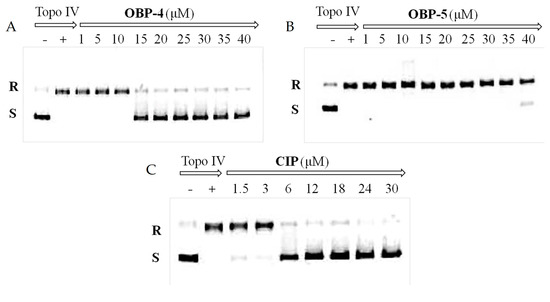
Figure 2.
Inhibition assay of OBP-4 (A), OBP-5 (B) and CIP (C) on Topo IV. Line 1(−): no compound and Topo IV; Line 2(+): Topo IV present and compound absent; Line 3-11(9): Topo IV with different concentrations of tested compound. R: relaxed DNA; S: supercoiled DNA.
Table 5.
Activity of OBP-4 and OBP-5 against DNA gyrase, topo IV, and in an in vitro transcription/translation assay.
DNA gyrase and topoisomerase IV (Topo IV), two well-known targets for quinolone antimicrobials [24], play a vital role in the replication and transcription of DNA [25]. Therefore, the impact of OBP-4 and OBP-5 on DNA gyrase and topo IV was firstly investigated by DNA gyrase supercoiling assay and Topo IV relaxation assay, respectively. As shown in Figure 1, OBP-4 and OBP-5 could inhibit DNA gyrase in a dose-dependent manner with IC50 values being 1–5 μM and 20 μM for OBP-4 and OBP-5, respectively (Table 5). By comparison, the inhibition action of OBP-4 and OBP-5 on DNA gyrase was evidently inferior to the reference drug CIP (IC50, <0.75 μM). Similar results but with higher IC50 values were found for hybrids OBP-4 and OBP-5 in the topo IV relaxation assay. OBP-4 (IC50, 10–15 μM) was less active in inhibiting topo IV compared with CIP (IC50, 3–6 μM) while OBP-5 was the least active (IC50 > 40 μM) (Figure 2, Table 5). Overall, hybrid OBP-4 exhibited a much better inhibitory activity on DNA synthesis than OBP-5, but they were significantly less active than CIP.
Oxazolidinones have a unique mechanism of action involving inhibition of protein synthesis by binding the ribosomal 50S subunit [16]. Thus, the action of hybrids OBP-4, OBP-5 and the reference drug LZD on protein synthesis was investigated with the E. coli-based cell-free protein synthesis system. As expected, OBP-4 and OBP-5 showed potent inhibition of protein synthesis in vitro transcription/translation assay. IC50 values of OBP-4 and OBP-5 were 5 μM for OBP-4 and 2 μM for OBP-5, respectively (Table 5), which were equal or 2.5-fold greater than that of LZD (IC50, 5 μM) (Table 5).
The above data demonstrated that the protein synthesis inhibition was the primary mode of action for OBP-4 and OBP-5 whilst the activity against DNA synthesis was weak, which correlated well with the observed antibacterial activity pattern. The results confirmed our previous speculation. For the imbalanced dual-action mode, the possible explanation was that the introduction of oxazolidinone substructure or spacer in hybrid changed the intrinsic binding affinity of fluoroquinolone in two targets.
2.3.2. Molecular Docking of Hybrids OBP-4 and OBP-5
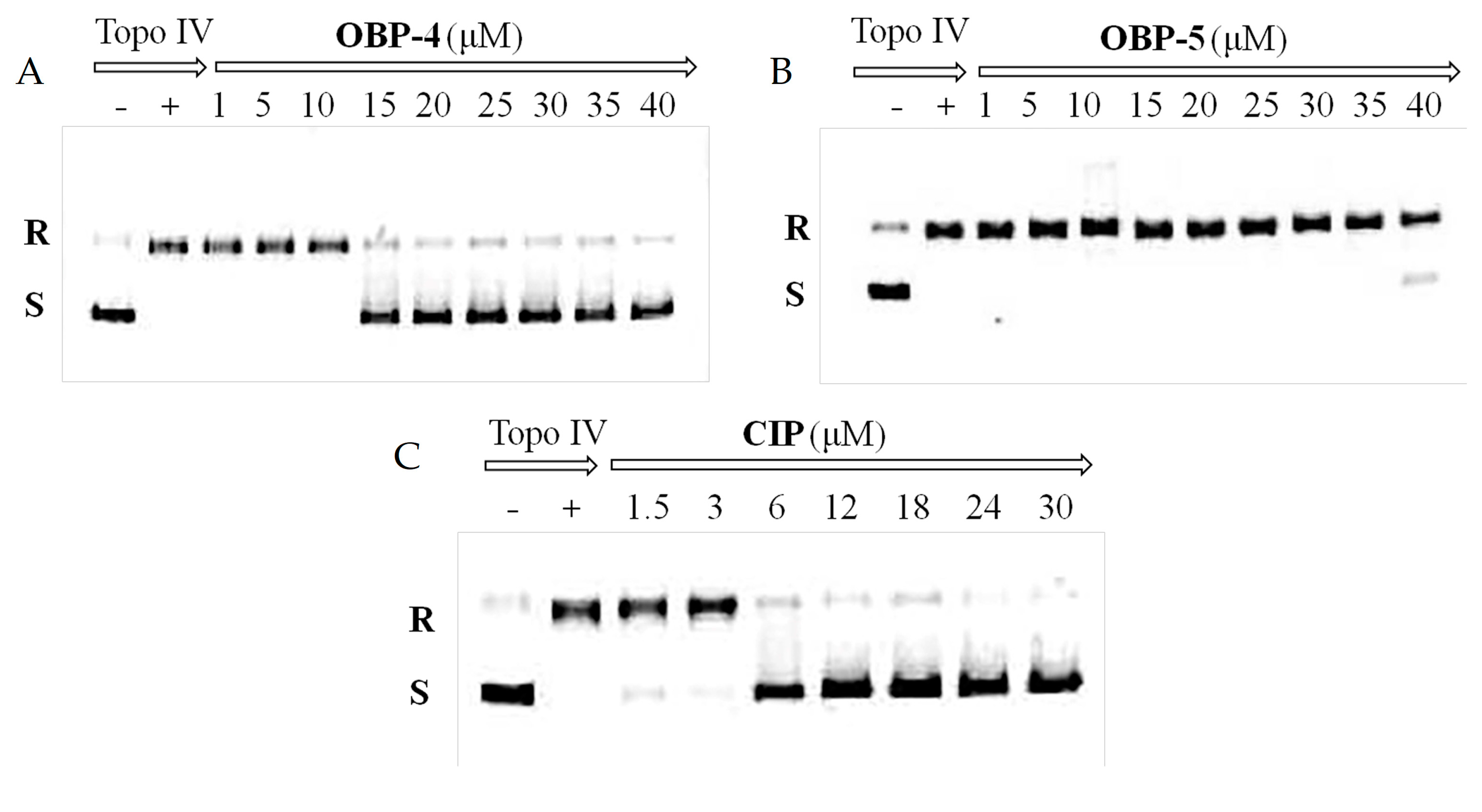
As shown in Table 5, OBP-4 and OBP-5 displayed a strong inhibition action in protein synthesis. To understand the binding mode of OBP-4 and OBP-5, the molecular docking was performed between OBP-4 (or OBP-5) and the active site of 50S subunit. The optimal docking conformations of tested compounds in the active site of 50S subunit were presented in Figure 3. The blue, light blue and red sticks represented the optimal conformations of OBP-4 (Figure 3A), OBP-5 (Figure 3B) and LZD (Figure 3C). As expected, both OBP-4 and OBP-5 can bind to the active site of the 50S subunit. OBP-4 were docked into the active site of 50S subunit, with its oxazolidinone part forming hydrogen bonds with U-2612 and G-2532 (Figure 3A), while OBP-5 were surrounded by G-2580, G-2610, U-2611 and C-2479 (Figure 3B). Furthermore, OBP-4, OBP-5 and LZD have a similarity in the optimal conformations and binding modes to the active site of 50S subunit (Figure 3C). This similarity could explain why OBP-4 and OBP-5 have an oxazolidinone-like antibacterial spectrum and a strong inhibitory activity on protein synthesis. Meanwhile, the better antibacterial activities of OBP-4 and OBP-5 than that of LZD may be attributed to the interactions between fluoroquinolone pharmacophores and the 50S subunit. The further investigations are required to clearly elucidate the mode of action of OBP-4 and OBP-5.
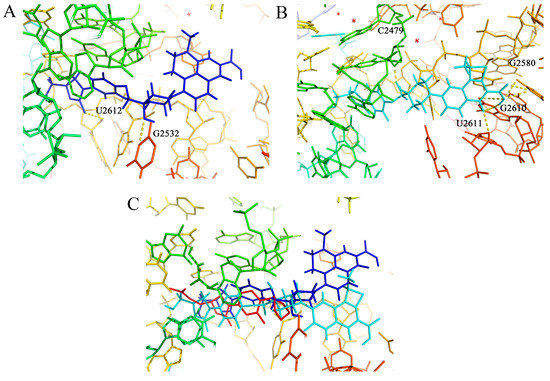
Figure 3.
(A) The optimal docking conformation of OBP-4 (blue sticks) in the active site of 50S subunit (PDB ID: 4WFA). (B) The optimal docking conformation of OBP-5 (light blue sticks) in the active site of 50S subunit (PDB ID: 4WFA). (C) The superposition of OBP-4, OBP-5 and LZD (red sticks) in the active site of 50S subunit (PDB ID: 4WFA). Hydrogen bonds are showed as dashed lines.
2.4. Acute Toxicity Test
The previous study has shown that CZD, structurally similar to hybrids OBP-4 and OBP-5, has a good safety level [26]. Therefore, the oral acute toxicity of hybrids OBP-4 and OBP-5 was directly evaluated with a limit test dose of 2000 mg/kg. No death and any toxicity signs were observed for all animals until the experiment was finished. The oral LD50 values for hybrids OBP-4 and OBP-5 were considered to be higher than 2000 mg/kg, suggesting that hybrids OBP-4 and OBP-5 are likely to be safe for future application, at least with a single oral dose.
3. Materials and Methods
3.1. General
The chemicals and reagents were acquired commercially and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) plates pre-coated with 0.2-mm-thick silica gel GF254 (Qingdao Ocean Chemical Co., Ltd., Shandong, China). The visualized analysis of products was achieved by UV illumination at 254 nm and staining in the solution of bismuth nitrate and 0.05% KMnO4. The determination of the melting point was conducted with an X-4 microscopic melting point apparatus (Beijing Tech Instrument Co., Ltd., Beijing, China). 1H-NMR and 13C-NMR were performed on a Bruker spectrometer (Bruker BioSpin, Zürich, Switzerland) at 400 and 151 MHz, respectively. All NMR spectra were reported in chemical shifts (δ, ppm) with residual solvents as internal standards. Multiplicities were expressed as s (singlet), d (doublet), dd (doublet of doublets), t (triplet) and m(multiplet), and coupling constants were expressed as J in hertz (Hz). Electrospray ionization mass spectra (ESI-MS) were recorded with a Shimadzu mass spectrometer (Shimadzu, Tokyo, Japan) and high resolution mass spectra (HRMS) were obtained with an Agilent Q-TOF 6530 mass spectrometer (Agilent Technologies, Palo Alto, CA, USA).
3.2. Chemistry
3.2.1. Preparation of Hybrids OBP-1, OBP-2 and OBP-3
Compound 1: a mixture of 2-fluoro-4-nitrophenol (10.00 g, 63.69 mmol, 1 eq) [27], benzyl bromide (11.98 g, 70.00 mmol, 1.1 eq), K2CO3 (13.18 g, 95.54 mmol, 1.5 eq) and KI (1.06 g, 6.37 mmol) were added to acetone (0.10 L) and then heated to 60 °C overnight. Upon cooling to room temperature and addition of ice water, yellow solid was precipitated and dried (15.00 g, 95%), m.p., 197–198 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.18 (dd, J = 11.2, 2.8 Hz, 1H), 8.13 (d, J = 9.2 Hz, 1H), 7.53–7.39 (m, 6H), 5.35 (s, 2H). ESI-MS (m/z): 247.9 [M + H]+.
Compound 2: compound 1 (72.80 g, 294.74 mmol, 1 eq), reduced iron powder (99.00 g, 1768.42 mmol, 6 eq) and ammonium chloride (94.59 g, 1768.42 mmol, 6 eq) were mixed with EtOH:H2O (2:1), and refluxed at 90 °C for 2 h. After cooling to room temperature, the mixture was filtered. The filtered and concentrated to give the brown oily substance compound 2 (60.00 g, 94%). 1H-NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.2 Hz, 2H), 7.36 (t, J = 7.2 Hz, 2H), 7.31 (t, J = 7.0 Hz, 1H), 6.80 (t, J = 9.0 Hz, 1H), 6.46 (dd, J = 12.4, 2.8 Hz, 1H), 6.33 (d, J = 8.8 Hz, 1H), 5.03 (s, 2H). ESI-MS (m/z): 217.9 [M + H]+.
Compound 3: a saturated sodium bicarbonate solution (0.50 L) was added to compound 2 (60.00 g, 276.50 mmol, 1 eq) in anhydrous THF (0.50 L) and cooled to 0 °C. The mixed solution was treated dropwise with benzyl carbonochloridate (51.88 g, 304.15 mmol, 1.1 eq) and then stirred for 30 min at 0 °C. The mixture was extracted with EtOAc (3.00 L*2) and brine (2.40 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo to give compound 3 (88.00 g, 91%) as a yellow solid, m.p., 116–118 °C. 1H-NMR (400 MHz, CDCl3) δ 7.48–7.27 (m, 11H), 6.95–6.87 (m, 2H), 6.57 (s, 1H), 5.19 (s, 2H), 5.10 (s, 2H). ESI-MS (m/z): 351.9 [M + H]+.
Compound 4: the suspension of compound 3 (40.00 g, 113.96 mmol, 1 eq) in THF (1.00 L) was cooled to −78 °C, and then n-BuLi (2.50 M in hexane 54.70 mL, 136.75 mmol, 1.2 eq) was added dropwisely and stirred at −78 °C for 0.5 h. Subsequently, (R)-oxiran-2-ylmethyl butyrate (20.35 g, 141.31 mmol, 1.24 eq) in THF (0.50 L) was added dropwisely to the mixed solution within 30 min. The reaction mixture was warmed up slowly and stirred at room temperature overnight. The mixture was extracted with EtOAc (2.00 L*2) and brine (1.50 L). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give compound 4 (31.50 g, 87%) as a yellow solid, m.p., 135–137 °C. 1H-NMR (400 MHz, DMSO-d6) δ 7.58 (dd, J = 13.6, 2.4 Hz, 1H), 7.44 (d, J = 7.2 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.33 (t, J = 6.8 Hz, 1H), 7.26 (t, J = 9.2 Hz, 1H), 7.22–7.16 (m, 1H), 5.22 (t, J = 5.6 Hz, 1H), 5.16 (s, 2H), 4.67 (dd, J = 9.2, 5.6 Hz, 1H), 4.03 (t, J = 9.0 Hz, 1H), 3.78 (dd, J = 8.8, 6.4 Hz, 1H), 3.69–3.62 (m, 1H), 3.58–3.50 (m, 1H). ESI-MS (m/z): 317.9 [M + H]+.
Compound 5: Pd/C (1.50 g, 5%) was carefully added to a solution of compound 4 (31.00 g, 97.79 mmol, 1 eq) in MeOH (0.04 L). The reaction was proceeded with H2 at room temperature for 12 h. The liquid was filtered, concentrated and purified by recrystallization in petroleum ether and ethyl acetate (2:1) to give compound 5 (21.76 g, 98%) as a white solid, m.p., 179–181 °C. 1H-NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 7.47 (d, J = 13.6 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 6.95 (t, J = 9.4 Hz, 1H), 5.20 (s, 1H), 4.65 (s, 1H), 4.01 (t, J = 9.0 Hz, 1H), 3.76 (t, J = 7.4 Hz, 1H), 3.65 (d, J = 11.6 Hz, 1H), 3.54 (d, J = 12.4 Hz, 1H). ESI-MS (m/z): 227.9 [M + H]+.
Compound 6: the sodium hydroxide (60%, 4.37 g, 109.25 mmol, 1.5 eq) was added to a solution of trimethylsulfoxonium iodide (24.06 g, 109.32 mmol, 1.5 eq) in dry DMSO (0.10 L) at 0 °C, and then stirred at 0 °C for 40 min. Benzyl 4-oxopiperidine-1-carboxylate (17.00 g, 72.88 mmol, 1 eq) [28] was added to the mixture and stirred for 2 h at 55 °C. The reaction mixture was extracted with EtOAc (0.18 L) and brine (0.06 L). The organic layer was dried (Na2SO4) and concentrated in vacuo. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel column to give compound 6 (14.50 g, 80%) as a yield oil. 1H-NMR (400 MHz, CDCl3) δ 7.42–7.28 (m, 5H), 5.15 (s, 2H), 3.82 (s, 2H), 3.48 (t, J = 10.8 Hz, 2H), 2.70 (s, 2H), 1.83 (s, 2H), 1.45 (d, J = 10.4 Hz, 2H). ESI-MS (m/z): 247.9 [M + H]+.
Compound 7: the mixed solution of compound 5 (8.00 g, 35.24 mmol, 1 eq) and compound 6 (13.06 g, 52.86 mmol, 1.5 eq) DMF (0.05 L) was treated with Na2CO3 (7.47 g, 70.48 mmol, 2 eq). The resulting solution was heated to 100 °C and stirred for 12 h. The reaction was quenched by H2O and extracted by EtOAc (0.20 L*3). The combined organic layer was washed by brine and dried with anhydrous Na2SO4. After evaporation and recrystallization, compound 7 (17.00 g, 90%) was obtained as a white solid, m.p., 121–123 °C. 1H-NMR (400 MHz, DMSO-d6) δ 7.61–7.54 (m, 1H), 7.40–7.30 (m, 5H), 7.23–7.16 (m, 2H), 5.21 (t, J = 5.6 Hz, 1H), 5.08 (s, 2H), 4.84 (s, 1H), 4.68 (dd, J = 9.2, 5.6 Hz, 1H), 4.04 (t, J = 9.0 Hz, 1H), 3.87–3.81 (m, 4H), 3.81–3.76 (m, 1H), 3.70–3.62 (m, 1H), 3.60–3.49 (m, 1H), 3.17 (d, J = 3.6 Hz, 2H), 1.66–1.50 (m, 4H). ESI-MS (m/z): 475.0 [M + H]+.
Compound 8: Pd/C (1.00 g, 5%) was carefully added to a solution of compound 7 (17.00 g, 35.86 mmol, 1 eq) in MeOH (0.50 L). The reaction was proceeded with H2 at room temperature for 12 h. The mixture was filtered, concentrated and purified by recrystallization to obtained compound 8 as a white solid (11.00 g, 92%), m.p., 179–181 °C. 1H-NMR (400 MHz, DMSO-d6) δ 7.57 (d, J = 13.2 Hz, 1H), 7.25–7.12 (m, 2H), 5.24 (s, 1H), 4.70–4.65 (m, 1H), 4.52 (s, 1H), 4.04 (t, J = 9.0 Hz, 1H), 3.85–3.73 (m, 3H), 3.66 (dd, J = 12.4, 3.2 Hz, 1H), 3.55 (dd, J = 12.4, 4.0 Hz, 1H), 2.81 (t, J = 10.6 Hz, 2H), 2.67 (d, J = 12.0 Hz, 2H), 1.65–1.51 (m, 2H), 1.44 (d, J = 12.8 Hz, 2H). ESI-MS (m/z): 340.9 [M + H]+.
Compound 9: to a solution of (S)-9,10-difluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-de] -1,4-benzoxazine-6-carboxylic acid (1.00 g, 3.56 mmol, 1 eq) in H2SO4 (98%, 0.02 L) [29], KNO3 (0.719 g, 7.12 mmol, 2 eq) was added and then stirred at 0 °C for 30 min. The reaction mixture was diluted with ice water (0.05 L) and filtered. Compound 9 Yield (1.00 g, 86%) was obtained as a yellow solid. 1H-NMR (400 MHz, DMSO-d6) δ 9.15 (s, 1H), 5.08 (d, J = 6.8 Hz, 1H), 4.76 (d, J = 10.8 Hz, 1H), 4.51 (d, J = 9.6 Hz, 1H), 1.47 (d, J = 6.8 Hz, 3H).
Compound 10: Pd/C (0.05 g, 5%) was carefully added to a solution of compound 9 (1.00 g, 3.07 mmol, 1 eq) in MeOH (0.05 L). The reaction proceeded with H2 at room temperature for 12 h. The liquid was filtered and concentrated to give compound 10 (0.50 g, 55%) a yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H), 4.86 (d, J = 6.8 Hz, 1H), 4.47 (d, J = 11.2 Hz, 1H), 4.21 (d, J = 10.4 Hz, 1H), 1.40 (d, J = 6.8 Hz, 3H). ESI-MS (m/z): 300.9 [M + H]+.
General procedure for the synthesis of hybrids OBP-1, OBP-2 and OBP-3.
A suspension of compound 8 (1.1 eq), TEA (5 eq) and corresponding acid (1 eq) in DMSO were mixed and stirred at 120 °C for 20 h. The mixed solution was cooled to room temperature, adjusted pH (<6) with 50% H2SO4, filtered and concentrated in vacuo. Finally, the target hybrids were obtained by recrystallization in petroleum ether and ethyl acetate (2:1).
OBP-1: while solid (0.516 g, 30%), m.p., 256–257 °C. 1H-NMR (400 MHz, DMSO-d6) δ 15.11 (s, 1H), 8.76 (s, 1H), 7.66–7.53 (m, 2H), 7.21 (dd, J = 14.4, 5.2 Hz, 2H), 5.32 (s, 2H), 5.22 (s, 1H), 4.82 (s, 1H), 4.71–4.66 (m, 1H), 4.05 (t, J = 8.8 Hz, 1H), 3.88 (s, 2H), 3.83–3.77 (m, 1H), 3.67 (d, J = 12.8 Hz, 1H), 3.57–3.50 (m, 3H), 3.23 (s, 2H), 3.02 (s, 3H), 1.88 (t, J = 11.0 Hz, 2H), 1.65 (d, J = 12.8 Hz, 2H).13C-NMR (151 MHz, DMSO-d6) δ 176.50 (d, J = 3.02 Hz), 165.98, 155.89 (d, J = 246.13 Hz), 154.88, 151.85 (d, J = 226.50 Hz), 145.73, 143.15 (d, J = 10.57 Hz), 138.54 (d, J = 7.55 Hz), 132.92 (d, J = 15.1 Hz), 132.74, 124.37, 119.51 (d, J = 9.06 Hz), 116.35, 114.27, 107.06 (d, J = 22.65 Hz), 106.69, 104.21 (d, J = 24.16 Hz), 83.22, 78.01, 73.57, 67.56, 62.25, 46.61, 46.51, 43.31, 34.46. ESI-MS (m/z) [M + H]+ calcd. for C28H28F2N4O9: 603.1897, found: 603.1906.
OBP-2: while solid (0.60 g, 56%), m.p., 269–270 °C. 1H-NMR (400 MHz, DMSO-d6) δ 15.26 (s, 1H), 8.96 (s, 1H), 7.58 (d, J = 12.4 Hz, 2H), 7.27–7.18 (m, 2H), 5.22 (t, J = 5.6 Hz, 1H), 4.97–4.86 (m, 1H), 4.79 (s, 1H), 4.72–4.65 (m, 1H), 4.59 (d, J = 10.8 Hz, 1H), 4.39 (s, 1H), 4.05 (t, J = 9.0 Hz, 1H), 3.88 (s, 2H), 3.80 (dd, J = 8.8, 6.4 Hz, 1H), 3.72–3.62 (m, 1H), 3.58–3.46 (m, 3H), 3.24 (d, J = 11.6 Hz, 2H), 1.96–1.79 (m, 2H), 1.64 (d, J = 12.8 Hz, 2H), 1.45 (d, J = 6.8 Hz, 3H). 13C-NMR (151 MHz, DMSO-d6) δ 176.80 (d, J = 3.02 Hz), 166.52, 156.00 (d, J = 247.64 Hz), 154.88, 151.90 (d, J = 243.11 Hz), 146.51, 143.16 (d, J = 12.08 Hz), 140.49 (d, J = 6.04 Hz), 133.28 (d, J = 15.1Hz), 132.73, 125.27, 119.76 (d, J = 9.06 Hz), 116.34, 114.26, 107.05 (d, J = 24.16 Hz), 107.01, 103.70 (d, J = 24.16 Hz), 77.67, 73.57, 68.44, 68.15, 62.07, 55.26, 46.61, 46.55, 34.48, 18.36.ESI-MS (m/z) [M + H]+ calcd. for C29H29F2N3O9: 602.1945, found: 602.1948.
OBP-3: yellow solid (0.70 g, 83%), m.p., 255–256 °C. 1H-NMR (400 MHz, DMSO-d6) δ 15.09 (s, 1H), 8.68 (s, 1H), 7.59 (d, J = 14.4 Hz, 1H), 7.29–7.12 (m, 2H), 6.91 (s, 2H), 5.23 (s, 1H), 4.82–4.61 (m, 3H), 4.38 (d, J = 11.2 Hz, 1H), 4.16–3.99 (m, 2H), 3.87 (s, 2H), 3.83–3.76 (m, 1H), 3.67 (d, J = 11.2 Hz, 1H), 3.55 (d, J = 14.4 Hz, 1H), 3.50–3.42 (m, 2H), 3.24 (s, 2H), 1.93–1.75 (m, 2H), 1.63 (d, J = 12.0 Hz, 2H), 1.38 (d, J = 6.4 Hz, 3H). 13C-NMR (151 MHz, DMSO-d6) δ 180.25 (d, J = 3.02 Hz), 166.62, 154.89, 151.90 (d, J = 243.11 Hz), 146.78, 144.42, 143.17 (d, J = 12.08 Hz), 141.26 (d, J = 235.56 Hz), 134.31 (d, J = 15.1 Hz), 132.68 (d, J = 9.06 Hz), 127.91 (d, J = 6.04 Hz), 126.15, 124.09, 116.34 (d, J = 1.51 Hz), 114.25, 109.99, 107.06 (d, J = 24.16 Hz), 105.67, 77.66, 73.57, 68.22, 67.23, 62.06, 55.74, 46.61, 34.53, 18.31. ESI-MS (m/z) [M + H]+ calcd. for C29H30F2N4O9: 617.2054, found: 617.2063.
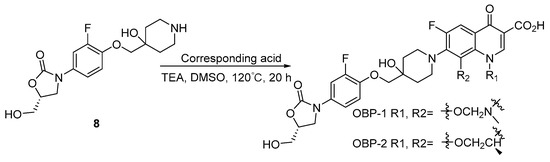
3.2.2. Preparation of Hybrid OBP-4
Compound 11: to a stirred solution of H2SO4 (7.78 mL) in Ac2O (0.778 L, 8.21 mol), 3,4-difluoroaniline (500.00 g, 3.88 mol) [30] was added at about 10 °C, and then stirred at room temperature for 30 min. The mixture was poured into ice water, filtered, the filter cake was washed with water and dried to give an off-white solid (604.00 g, 91%). 1H-NMR (400 MHz, CDCl3) δ 7.63–7.56 (m, 1H), 7.53 (s, 1H), 7.12–7.03 (m, 2H), 2.17 (s, 3H). ESI-MS (m/z): 171.9 [M + H]+.
Compound 12: to a stirred solution of compound 11 (588.00 g, 3.44 mol), NaOAc (338.00 g, 4.12 mol) in AcOH (1.94 L) was added dropwise to a solution of Br2 (0.212 L, 4.12 mol) in AcOH (0.60 L) at 60 °C, then the mixture was stirred at 80 °C until SM was disappeared. After cooling to room temperature, the mixture was poured into ice water, filtered, the filter cake was washed with water and dried to give a white solid (735.00 g, 85%). 1H-NMR (400 MHz, CDCl3) δ 8.40–8.30 (m, 1H), 7.60–7.48 (m, 1H), 7.41–7.35 (m, 1H), 2.24 (s, 3H). ESI-MS (m/z): 249.8 [M + H]+.
Compound 13: a mixture of compound 12 (565.00 g, 2.26 mol), sodium m-nitrobenzensulfonate (508.00 g, 2.26 mol), iron sulfate heptahydrate (63.30 g, 158.00 mmol), boric acid (616.00 g, 9.94 mol), water (2.16 L) and concentrated HCl (2.16 L) was refluxed for 0.5 h. Crotonaldehyde (0.278 L, 3.38 mol) was added dropwise to the mixture at the same temperature and stirring was continued for 1 h under reflux. After cooling to room temperature, the insoluble matter was filtered off and the filtrate was poured into MeOH (0.80 L). The solution was neutralized with 25% (w/v) aqueous NaOH, cooled at ice bath. The resultant precipitates were collected by filtration, the wet solid was dissolved in DCM (2.00 L) and dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated in vacuo and the crude product was recrystallized from hexane to give an off-white solid (330.00 g, 57%). 1H-NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.4 Hz, 1H), 7.94–7.88 (m, 1H), 7.42 (d, J = 8.8 Hz, 1H), 2.82 (s, 3H).
Compound 14: a mixture of compound 13 (330.00 g, 1.28 mol), 10% wet Pd/C (6.60 g), and sodium acetate (105.00 g, 1.28 mol) in 90% (v/v) aqueous AcOH (2.00 L) was hydrogenated at 50 °C under a hydrogen pressure 1–5 atm. After the hydrogen absorption had ceased, the catalyst was filtered off and the filtrate was concentrated under reduced pressure until about half of the solvent was evaporated, the residue was poured into water, the resulting precipitates were collected by filtration and dried to give a light red solid as the first crop (160.00 g), the filtrate was extracted with EA, the combined organic layer was washed with saturated aqueous NaHCO3, dried over Na2SO4 and evaporated in vacuo to give a brown solid as the second crop (65.00 g).The combined crops (225.00 g, 1.26 mol) were dissolved in AcOH (1.10 L), to the stirred solution was added NaBH3CN (198.00 g, 3.15 mol), then the mixture was stirred at room temperature for 6 h, to the mixture was added saturated aqueous Na2CO3 until there was no bubble produced, extracted with EA and the combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated in vacuo, the residue was purified by silica gel column chromatography (1% EA in PE) to give a colorless oil (180.00 g, 77%). 1H-NMR (400 MHz, CDCl3) δ 6.80–6.71 (m, 1H), 6.18–6.09 (m, 1H), 3.64 (brs, 1H), 3.35–3.26 (m, 1H), 2.90–2.80 (m, 1H), 2.70–2.58 (m, 1H), 1.99–1.90 (m, 1H), 1.56–1.44 (m, 1H), 1.20 (d, J = 6.0 Hz, 3H). ESI-MS (m/z): 184.0 [M + H]+.
Compound 15: compound 14 (174.00 g, 951.00 mmol) was added to a stirred solution of L-DBTA (187.00 g, 522.00 mmol) in EA (0.35 L) and the mixture was stirred at room temperature for 2 h. The resulting precipitates were collected by filtration and recrystallized from 60% (v/v) aqueous MeOH. The resulting solid was treated with aqueous NaOH to give a colorless oil (58.00 g, 33%), 100% ee as determined by chiral HPLC. 1H-NMR (400 MHz, CDCl3) δ 6.80–6.71 (m, 1H), 6.18–6.09 (m, 1H), 3.67 (s, 1H), 3.35- 3.26 (m, 1H), 2.90–2.80 (m, 1H), 2.70–2.58 (m, 1H), 1.99–1.90 (m, 1H), 1.56- 1.44 (m, 1H), 1.20 (d, J = 6.4 Hz, 3H).
Compound 16: A mixture of compound 15 (58.00 g, 317.00 mmol) and EMME (103.00 g, 477.00 mmol) was heated at 130 °C with stirring for 3 h and the reaction mixture was added dropwise to the pre-heated PPA (161.00 g) at 130 °C. When the addition was complete, the mixture was stirred at the same temperature for 1 h. Then EtOH (0.875 L), concentrated HCl (0.058 L) and water (0.58 L) were added, the mixture was refluxed for 1 h. After cooling, water (1.13 L) was added to the mixture and the resulting precipitates were collected by filtration, the solid was washed with water then ether and dried to give an off-white solid (62.00 g, 70%). 1H-NMR (400 MHz, DMSO-d6)) δ 14.96 (s, 1H), 9.06 (s, 1H), 8.13–8.06 (m, 1H), 5.02–4.89 (m, 1H), 3.16–3.08 (m, 1H), 3.07–2.95 (m, 1H), 2.23–2.07 (m, 2H), 1.39 (d, J = 6.8 Hz, 3H). ESI-MS (m/z): 279.9 [M + H]+.
Compound 17: A mixture of boric anhydride (2.74 g, 39.40 mmol), Ac2O (25.58 g, 251.00 mmol), AcOH (12.04 g, 201.00 mmol) was refluxed for 3 h. Compound 16 (20.00 g, 71.6 mmol) was added to the resulting solution and the mixture was refluxed for 2 h, then toluene (0.12 L) was added. After cooling to room temperature, the resulting precipitates were collected by filtration to give an off-white solid (25.00 g, 86%). 1H-NMR (400 MHz, CDCl3) δ 9.21 (s, 1H), 8.27–8.21 (m, 1H), 5.05–4.93 (m, 1H), 3.39–3.30 (m, 1H), 3.17–3.06 (m, 1H), 2.38–2.31 (m, 2H), 2.07 (s, 3H), 2.01 (s, 3H), 1.64 (d, J = 6.8 Hz, 3H). ESI-MS (m/z): 405.9 [M + H]+.
OBP-4: A mixture of compound 17 (14.00 g, 34.40 mmol), compound 8 (15.20 g, 44.70 mmol) and TEA (10.40 g, 0.103 mol) in DMF (0.25 L) was stirred at 50 °C for 12 h. After cooling to room temperature, the mixture was poured into water (2.50 L), the resulting precipitates were collected by filtration and dried to give a yellow solid (21.60 g, 86%), the yellow solid was added to a mixture of 1.5 N aqueous LiOH (0.08 L) and ACN (0.04 L). The mixture was stirred at room temperature for about 2 h and then concentrated HCl was added until PH is around 2, the resulting precipitates were collected by filtration, the obtained solid was suspended in 50% (v/v) aqueous ethanol and refluxed for 6 h, after cooling to room temperature, the mixture was filtered to give a white solid, which was recrystallized with ACN and EtOH to give a white solid (10.25 g, 50%), m.p., 225–226 °C. 1H-NMR (400 MHz, DMSO-d6) δ 15.32 (s, 1H), 8.96 (s, 1H), 7.87 (d, J = 12.5 Hz, 1H), 7.59 (d, J = 13.8 Hz, 1H), 7.27–7.21 (m, 2H), 5.23 (t, J = 5.5 Hz, 1H), 4.88 (br s, 1H), 4.81 (s, 1H), 4.69 (br s, 1H), 4.05 (t, J = 8.9 Hz, 1H), 3.91 (s, 2H), 3.82–3.78 (m, 1H), 3.74–3.66 (m, 2H), 3.58–3.53 (m, 1H), 3.29 (s, 1H), 3.16–2.94 (m, 4H), 2.16–1.94 (m, 3H), 1.80 (d, J = 10.3 Hz, 1H), 1.68 (t, J = 13.2 Hz, 2H), 1.42 (d, J = 6.5 Hz, 3H). 13C-NMR (151 MHz, DMSO-d6) δ176.02 (d, J = 3.02 Hz), 166.26, 163.59, 154.87, 152.29 (d, J = 246.13 Hz), 151.91 (d, J = 243.11 Hz), 145.97 (d, J = 10.57 Hz), 143.11 (d, J = 10.57 Hz), 136.33, 132.75 (d, J = 9.06 Hz), 117.26 (d, J = 7.55 Hz), 116.32, 114.23, 112.43 (d, J = 22.65 Hz), 107.06 (d, J = 24.16 Hz), 102.72, 101.96, 77.35, 73.57, 72.72, 67.98, 62.08, 46.62, 45.91, 45.76, 33.52, 33.46, 20.77. ESI-MS (m/z) [M + H]+ calcd. for C30H31F2N3O8: 600.2152, found: 600.2159.
3.2.3. Preparation of Hybrid OBP-5
Compound 18: to a mixture of ethyl 6,7-difluoro-1-methyl-4-oxo-4H-(1,3)-thiazeto (3,2-a) quinoline-3-carboxylate (3.78 g, 12.10 mmol) [31], compound 8 (4.00 g, 11.80 mmol), TEA (3.57 g, 35.30 mmol) and DMSO (0.06 L) was stirred at 100 °C overnight. The reaction was quenched with water (0.12 L), the mixture was filtered to give compound 18 (6.15 g, 83%) as a white solid. 1H-NMR (400 MHz, DMSO-d6) δ 7.66–7.70 (d, J = 16 Hz,1H), 7.57–7.61 (d, J = 16 Hz,1H), 6.84–6.85 (d, J = 4 Hz, 1H), 6.17–6.21 (q, J = 12, 4Hz, 1H), 5.23–5.25 (t, J = 8 Hz, 1H), 4.86 (s, 1H), 4.68–4.70 (dd, J = 8, 4 Hz, 1H), 4.18 (q, J = 16 Hz, 2H), 4.06 (t, J = 12 Hz, 1H), 3.90 (s, 2H), 3.83 (s, 1H), 3.67 (m,1H), 3.55–3.59 (m, 1H), 3.43–3.46 (m, 2H), 3.17–3.25 (dd, J = 24, 12 Hz, 2H), 2.06 (d, J = 4 Hz, 3H), 1.88–1.93(t, J = 12 Hz,2H), 1.69–1.72 (d, J = 12 Hz, 2H), 1.24–1.26 (t, J = 8 Hz, 3H). MS (ESI): mass calcd.. for C30H31F2N3O8S 631.18, m/z found: 632.1 [M + H]+.
OBP-5: compound 18 (1.58 g, 2.50 mmol, 1eq) was dissolved in a mixture of THF/MeOH (0.02 L mL/0.02 L), and then lithium hydroxide (2.00M, 0.02 L) was slowly added and stirred at room temperature for 2 h. The reaction mixture was filtered to give the crude of OBP-5 as a solid after adjusting its pH (<6) with 50% H2SO4. The crude was separated on a silica gel column (DCM: MeOH=1:1) to obtain light white solid OBP-5 (0.38 g, 25%), m.p., 250–251 °C. 1H-NMR (400 MHz, DMSO-d6) δ 14.70 (s, 1H), 7.79 (d, J = 13.6 Hz, 1H), 7.58 (d, J = 14.4 Hz, 1H), 7.27–7.17 (m, 2H), 6.96 (d, J = 6.4 Hz, 1H), 6.40 (s, 1H), 5.23 (t, J = 5.6 Hz, 1H), 4.90 (s, 1H), 4.68 (dd, J = 8.8, 5.4 Hz, 1H), 4.04 (t, J = 8.8 Hz, 1H), 3.90 (s, 2H), 3.82–3.75 (m, 1H), 3.71–3.62 (m, 1H), 3.57–3.50 (m, 3H), 3.29–3.23 (m, 2H), 2.11 (d, J = 5.6 Hz, 3H), 1.90 (t, J = 10.8 Hz, 2H), 1.71 (d, J = 12.8 Hz, 2H).13C-NMR (151 MHz, DMSO-d6) δ176.98 (d, J = 3.02 Hz), 166.55, 154.88, 151.99 (d, J = 242.11 Hz), 147.81, 143.31 (d, J = 12.08 Hz), 143.22 (d, J = 10.57 Hz), 133.45(d, J = 199.32 Hz), 132.73, 122.32, 116.56 (d, J = 6.04 Hz), 114.25 (d, J = 3.02 Hz), 109.40 (d, J = 22.65 Hz), 107.06 (d, J = 24.16 Hz), 106.94, 73.57, 68.19, 62.09, 57.54, 46.87, 46.62, 34.86, 34.33, 25.35, 20.03, 18.99. ESI-MS (m/z) [M + H]+ calcd. for C28H27F2N3O8S: 604.1560, found: 604.1565.
3.2.4. Preparation of Hybrids OBP-6 and OBP-7
Compound 19: to a mixture of N-benzylmaleimide (75.00 g, 401.07 mmol, 1 eq) [32] and BrCH2NO2 (45.00 g, 320.09 mmol, 0.8 eq), K2CO3 (44.00 g, 320.09 mmol, 0.8 eq) and ACN (0.40 L) were slowly added and then stirred at 25 °C for 48 h. The reaction mixture was extracted with DCM (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4 and concentrated to give compound 19 (17.00 g, 17%) as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 7.36–7.27 (m, 5H), 4.54 (s, 2H), 4.50–4.46 (m, 1H), 3.35 (s, 2H). ESI-MS (m/z): 246.9 [M + H]+.
Compound 20: to a mixed solution of compound 19 (16.30 g, 66.26 mmol, 1 eq) and NaBH4 (6.29 g, 165.65 mmol, 2.5 eq) in THF (0.40 L), BF3·Et2O (8.36 mL, 66.26 mmol, 1 eq) was added dropwise at 0 °C and maintained for 1 h. The mixture was heated to 40 °C and kept for 4 h. After cooling, the reaction mixture was treated with EtOH (0.40 L) under reflux for 4 h. The mixture was extracted with DCM (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, compound 20 (9.00 g, 60%) was obtained by flash chromatography as a colorless oil. 1H-NMR (400 MHz, CDCl3) δ 7.35–7.20 (m, 5H), 4.63 (s, 1H), 3.59 (s, 2H), 3.14 (d, J = 9.2 Hz, 2H), 2.51 (d, J = 8.0 Hz, 4H). ESI-MS (m/z): 218.9 [M + H]+.
Compound 21: Zn (39.20 g, 599.48 mmol, 20 eq) was slowly added to a solution of compound 20 (6.54 g, 300.00 mmol, 1 eq) and 1N HCl (0.30 L, 0.30 mol, 10 eq) in i-PrOH (0.60 L) within 1 h, and kept overnight. Excess hydrochioric acid was neutralized using NaHCO3 until pH > 7. The reaction was extracted with DCM (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, compound 21 (5.56 g crude) was obtained by flash chromatography as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 7.31–7.18 (m, 5H), 3.55 (s, 2H), 2.95 (d, J = 8.8 Hz, 2H), 2.66 (s, 1H), 2.38 (d, J = 8.4 Hz, 2H), 1.34 (s, 2H).
Compound 22: DIPEA (7.63 g, 0.06 mol, 2 eq) was added dropwise to a solution of compound 21 (5.56 g, 0.03 mol, 1 eq) and 2,4-difluoronitrobenzene in ACN (0.10 L) and stirred at 50 °C for 48 h. After cooling to room temperature, the reaction mixture was extracted with DCM (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, compound 22 (9.10 g, 93%) was obtained by flash chromatography as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.8 Hz, 1H), 7.86 (dd, J = 11.6, 2.4 Hz, 1H), 7.38–7.28 (m, 5H), 6.93 (t, J = 8.8 Hz, 1H), 4.85 (s, 1H), 3.79–3.60 (m, 2H), 3.25 (s, 2H), 3.03 (s, 1H), 2.61 (s, 2H), 1.26 (s, 2H).
Compound 23: to a mixture of compound 22 (9.10 g, 0.03 mol, 1 eq), (Boc)2O (9.11 g, 0.04 mol, 1.5 eq) and TEA (5.60 g, 56.00 mmol, 2 eq) in DCM (0.20 L), DMAP (340.00 mg, 3.00 mmol, 0.1 eq) was added at 0 °C and stirred for 2 h. The reaction mixture was extracted with DCM (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4 and then concentrated to give a yellow oil. NaBH4 (3.20 g, 80.00 mmol, 3 eq) and NiCl2·6H2O (20.00 g, 80.00 mmol, 3eq) were separately added to the oil in THF/MeOH (0.05 L/0.05 L) at 0 °C and stirred for 2 h. The reaction mixture was extracted with EA (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, compound 23 (10.00 g, 91%) was obtained by flash chromatography as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 7.31–7.22 (m, 5H), 6.87 (t, J = 8.2 Hz, 1H), 6.38 (d, J = 9.6 Hz, 2H), 3.72 (s, 2H), 3.55 (s, 2H), 3.25 (s, 1H), 3.03 (d, J = 8.8 Hz, 2H), 2.37 (d, J = 7.6 Hz, 2H), 1.53 (s, 2H), 1.41 (s, 9H).
Compound 24: the saturated NaHCO3 solution was added to compound 23 (10.00 g, 30.00 mmol, 1 eq) in THF (0.50 L). CBzCl (5.16 g, 36.00 mmol, 1.2 eq) was slowly added to the mixed solution at 0 °C and stirred overnight. The reaction mixture was extracted with EA (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, compound 24 (10.00 g, 75%) was obtained by flash chromatography as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 7.41–7.38 (m, 3H), 7.26 (s, 10H), 5.20 (s, 2H), 3.54 (s, 2H), 3.28 (s, 1H), 3.02 (d, J = 8.8 Hz, 2H), 2.35 (d, J = 8.8 Hz, 2H), 2.02 (s, 1H), 1.56 (s, 9H), 1.34 (s, 2H).
Compound 25: the suspension of compound 24 (10.00 g, 18.83 mmol, 1 eq) in 0.30 L of THF was cooled to −78 °C. n-BuLi (2.50 M, 23.00 mL, 56.50 mmol, 3 eq) was added dropwisely to the mixed solution within 30 min. After stirring at −78 °C for 30 min, (R)-oxiran-2-ylmethyl butyrate in THF (0.30 L) was added dropwisely within 30 min and stirred at −78 °C for 30 min. After being restored to room temperature, the reaction solution was continuously stirred for 12 h. The resulting mixture was extracted with EA (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous MgSO4. After evaporation, compound 25 (3.50 g, 37%) was obtained by flash chromatography as a yellow oil. 1H-NMR (400 MHz, CDCl3) δ 7.51 (d, J = 11.2 Hz, 1H), 7.36–7.27 (m, 5H), 7.16 (s, 2H), 4.75 (s, 1H), 4.10–3.98 (m, 3H), 3.76 (d, J = 12.4 Hz, 1H), 3.64 (s, 2H), 3.34 (s, 1H), 3.09 (s, 2H), 2.50 (s, 2H), 1.42 (s, 9H), 1.34 (s, 2H).
Compound 26: Pd/C (0.35 g, 10%) was carefully added to a solution of compound 25 (3.50 g, 7.04 mmol, 1 eq) in MeOH (0.05 L). The reaction was proceeded with H2 at room temperature for 12 h. The liquid was filtered and concentrated to afford compound 26 (2.50 g crude, 87%) as a yellow oil. ESI-MS (m/z): 408.0 [M + H]+.
Compound 27: to a mixture of compound 26 (1.20 g, 2.95 mmol, 1 eq) and 9,10-difluoro-3- methyl-7-oxo-2,3-dihydro-7H-[1,3,4] oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid (0.831 g, 2.95 mmol, 1 eq) [33] in DMSO (0.02 L), TEA (1.50 g, 14.74 mmol, 5 eq) was added and stirred at 100 °C overnight. Excess TEA was neutralized using 50% H2SO4 until pH < 6. The filtrate was concentrated and the residue was purified by silica gel column to afford compound 27 (0.35 g, 18%) as a yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ 15.13 (s, 1H), 8.71 (s, 1H), 7.58 (t, J = 13.6 Hz, 2H), 7.36–7.30 (m, 2H), 5.26 (s, 2H), 4.71 (s, 1H), 4.10 (t, J = 8.8 Hz, 1H), 3.91–3.81 (m, 2H), 3.71–3.60 (m, 3H), 3.57 (d, J = 3.6 Hz, 1H), 3.54 (d, J = 3.6 Hz, 1H), 3.50 (s, 2H), 2.99 (s, 3H), 1.74 (s, 2H), 1.38 (s, 9H).
Compound 28: to a mixture of compound 26 (1.30 g, 3.19 mmol, 1 eq) and 1-cyclopropyl-6,7- difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (0.846 g, 3.19 mmol, 1 eq) [34] in DMSO (0.02 L), TEA (1.63 g, 15.97 mmol, 5 eq) was added and stirred at 100 °C overnight. After cooling to room temperature, excess TEA was neutralized using 50% H2SO4 until pH < 6. After concentration of the solution, the residue was purified by silica gel column to afford compound 28 (0.35 g, 17%) as a white solid. 1H-NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 7.80 (d, J = 14.4 Hz, 1H), 7.61 (d, J = 12.4 Hz, 1H), 7.40 (t, J = 8.6 Hz, 1H), 7.34 (d, J = 10.4 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 5.26 (d, J = 5.2 Hz, 1H), 4.77–4.70 (m, 1H), 4.10 (t, J = 9.0 Hz, 1H), 3.8–3.80 (m, 3H), 3.75–3.64 (m, 4H), 3.58–3.50 (m, 3H), 1.93 (s, 2H), 1.39 (s, 9H), 1.23 (s, 2H), 1.12 (s, 2H).
OBP-6: to a solution of compound 27 (0.35 g, 0.52 mmol, 1 eq) in THF (0.03 L), 6N HCl (0.03 L) was added dropwise at 0 °C and then stirred for 48 h at room temperature. The saturated NaHCO3 solution was added in ice bath to adjust pH (>7). The mixture was extracted with EA (0.30 L*3) and brine (0.30 L). The combined organic layers were dried with anhydrous Na2SO4. After evaporation, the residue was purified by silica gel column to afford hybrid OBP-6 as a yellow solid (0.28 g crude, 94%), m.p., 188–189 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.51 (d, J = 13.6 Hz, 1H), 7.39 (d, J = 13.6 Hz, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.91 (t, J = 9.2 Hz, 1H), 5.92 (s, 1H), 5.21 (s, 3H), 4.64 (s, 1H), 3.98 (d, J = 8.8 Hz, 2H), 3.79–3.70 (m, 3H), 3.65 (d, J = 11.6 Hz, 2H), 3.54 (d, J = 11.2 Hz, 2H), 2.99 (s, 3H), 1.78 (s, 2H). 13C-NMR (151 MHz, DMSO-d6) δ 176.28 (d, J = 3.02 Hz), 166.03, 154.97, 154.37 (d, J = 206.87 Hz), 150.55 (d, J = 241.60 Hz), 145.42, 136.44 (d, J = 9.06 Hz), 133.49 (d, J = 12.08 Hz), 128.65 (d, J = 4.53 Hz), 124.37, 115.51, 113.13 (d, J = 4.53 Hz), 106.52 (d, J = 24.16 Hz), 104.35 (d, J = 24.16Hz), 82.48, 73.45, 62.11, 52.11, 46.85, 43.16, 43.01, 33.84, 31.12, 24.35, 21.58. ESI-MS (m/z) [M + H]+ calcd. for C27H25F2N5O7: 570.1795, found: 570.1804.
OBP-7: to a solution of compound 28 (0.35 g, 0.54 mmol, 1 eq) in THF (0.01 L), EA/HCl (0.03 L) was added dropwise and then stirred for 48 h at room temperature. The reaction mixture was filtered and the yellow solid OBP-7 was obtained (0.25 g crude, 84%) by recrystallization in PE and EA (1:1), m.p., 268–270 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.58 (s, 1H), 7.80 (d, J = 14.4 Hz, 1H), 7.41 (d, J = 14.4 Hz, 1H), 7.13 (d, J = 5.6 Hz, 2H), 6.99 (t, J = 9.2 Hz, 1H), 4.69–4.61 (m, 1H), 4.42–4.25 (m, 1H), 4.03–4.01 (m, 3H), 3.77 (d, J = 7.6 Hz, 4H), 3.69–3.61 (m, 2H), 3.61–3.48 (m, 2H), 1.96 (s, 2H), 1.33 (d, J = 6.8 Hz, 2H), 1.16 (s, 2H). 13C-NMR (151 MHz, DMSO-d6) δ 176.21 (d, J = 3.02 Hz), 166.54, 154.97, 150.53 (d, J = 238.58 Hz), 150.20 (d, J = 247.64 Hz), 148.18, 141.65 (d, J = 12.08 Hz), 139.96, 133.28 (d, J = 12.08 Hz), 128.79 (d, J = 9.06 Hz), 115.21, 115.18, 113.37 (d, J = 4.53 Hz), 111.12 (d, J = 24.16 Hz), 109.99, 106.62, 106.50 (d, J = 24.16 Hz), 102.02, 73.46, 62.12, 52.09, 46.83, 36.49, 36.09, 25.62, 8.21. ESI-MS (m/z) [M + H]+ calcd. for C28H26F2N4O6: 553.1893, found: 553.1901.
3.3. Antibacterial Activities
3.3.1. Bacterial Strains
6 standard strains and 3 clinical isolates were used to the preliminary evaluation of in vitro antibacterial activities of oxazolidinone-fluoroquinolone hybrids. For the further evaluation of hybrids OBP-4 and OBP-5, 263 clinical isolates including S. aureus (46), S. haemolyticus(32), S. epidermidis (10), E. faecalis (72), E. faecium (47), S. pneumoniae (13), H. parasuis (34), S. suis (9), and 8 resistant strains including MRSA, MRSE, CIP-resistant S. aureus and S. haemolyticus, VAN and quinolone-resistant S. pneumoniae, LZD-resistant E. faecalis, LZD/VAN and quinolone-resistant E. faecium were collected and tested.
3.3.2. Susceptibility Testing
Susceptibility testing of oxazolidinone-fluoroquinolone hybrids against various bacteria was performed by the broth microdilution method as outlined by the Clinical and Laboratory Standards Institute (CLSI) [35]. Three types of media, including cation-adjusted Mueller Hinton broth, Haemophilus Test Medium and cation-adjusted Mueller Hinton broth that contained 3%–5% lysed horse blood, were used to culture the corresponding bacteria. All compounds including comparator drugs were dissolved in an appropriate solvent at a concentration of 2.048 mg/mL and then serially diluted two-fold to provide the required concentrations. A final inoculum of 5 × 105 CFU/mL was used and plates were read after incubation for 16–24 h at 35 °C. The MIC was defined as the lowest concentration of the antibacterial at which visible growth was completely inhibited. Standard quality control strains (ATCC) were included throughout. Each determination was repeated in triplicate.
3.4. Modes of Action
3.4.1. DNA Gyrase Supercoiling Assay
The E. coli DNA Gyrase Supercoiling Assay Kit (Inspiralis Co., Ltd.) was used to investigate the inhibition activities of hybrids OBP-4 and OBP-5 against DNA gyrase. DNA gyrase is prepared from E. coli. One unit of DNA gyrase can convert 0.5 μg of relaxed pBR322 to the supercoiled forms in 30 min. The final reaction system (30 μL) consisted of 0.5 μg relaxed pBR322 and 1 unit DNA gyrase with or without compounds in assay buffer containing 35 mM Tris·HCl (pH 7.5), 24 mM KCl, 4 mM MgCl2, 2 mM DTT, 1.8 mM spermidine, 1 mM ATP, 6.5% (w/v) glycerol and 0.1 mg/mL albumin, and then was incubated at 37 °C for 30 min. The reaction was terminated by adding 30 μL of STEB and 30 μL of chloroform/isoamyl alcohol (v:v, 24:1), and then the products were resolved by gel electrophoresis on 1% (w/v) agarose gel at 80 V for 2 h in the absence of ethidium bromide. The gels were stained with ethidium bromide (1 μg/mL in distilled water) for 15 min, and destained (5–10 min) in distilled water. DNA bands were visualized with UV light in Bio-Rad® Chemidoc xrs and analyzed by using Quantity One software (Version 4.6, Bio-Rad). IC50 is defined as the concentration of compound that inhibits the enzyme activity by 50%. CIP was used as a positive control.
3.4.2. Topo IV Relaxation Assay
The E. coli Topo IV Relaxation Assay Kit (Inspiralis Co., Ltd.) was used to evaluate the effects of hybrids OBP-4 and OBP-5 against topo IV. Topo IV was prepared by the overexpressing E. coli. One unit of topo IV will relax 0.4 μg of supercoiled pBR322. In the assay, the final reaction mixtures (30 μL) consisted of 0.4 μg supercoiled pBR322 and one unit DNA Topo IV with or without compounds in assay buffer with 40 mM Hepes-KOH (pH 7.6), 100 mM potassium glutamate, 10 mM magnesium acetate, 10 mM DTT, 1 mM ATP and 50 μg/mL albumin, and then was incubated at 37 °C for 30 min. The reaction was terminated by adding 30 μL of STEB and 30 μL of chloroform/isoamyl alcohol (v:v, 24:1) after incubation for 30 min. Then, the reaction products were processed as described in a DNA gyrase supercoiling assay.
3.4.3. In Vitro Transcription/Translation Assay
The influence of hybrids OBP-4 and OBP-5 against protein synthesis was confirmed by the S30 T7 High-Yield Protein Expression System (Promega Co., Ltd.). The S30 T7 High-Yield Protein Expression System is an E. coli extract-based cell-free protein expression system, which can effectively transcribe and translate DNA fragments with a T7 promoter. The reaction system containing 1 μg plasmid DNA, 20 μL S30 Premix Plus and 18 μL T7 S30 Extract with or without compounds was incubated for 1 h at 37 °C with a rotational speed of 250 rpm, and terminated by cooling in ice for 5 min in a 96-well white multi-well plate. Then, 1.25 μL of the translation reaction mixture was added to 48.75 μL of Renilla Luciferase Assay Lysis Buffer, and mixed with 50 μL of Renilla Luciferase Assay Reagent (cat no. E2810; Promega). Renilla luciferase activity was measured on EnSpire® Multimode Plate Reader (PerkinElmer, Waltham, MA, USA). IC50 is defined as the concentration of compound that inhibits the protein synthesis by 50%. LZD was used as a positive control.
3.4.4. Molecular Docking Stusy
The molecular docking of hybrids OBP-4 and OBP-5 was carried out with the Discovery Studio 4.5 software. The crystal structure of the large ribosomal subunit (50S subunit) of S. aureus (PDB: 4WFA) [36] was selected as the template to perform the docking study. Prior to docking, the structures of ligands were drawn and energy was minimized. During the process of receptor preparation, water molecules were removed and hydrogen atoms were added [37]. Hit compounds were docked into the active site of the receptor with an acceptable target flexibility and the docking score was calculated. The docking complexes with the highest score were kept for further analyses. The presentation of ligand-receptor interactions was depicted by PyMol 1.5.03 [38].
3.5. Acute Toxicity Test
Female Kunming mice (SPF grade, 18–22 g) were obtained from laboratory animal center of Lanzhou Veterinary Research Institute (Lanzhou, China). Mice were maintained under the standard conditions of 12 h light/dark cycle, room temperate (22 ± 3 °C), 50–60% relative humidity. They were fed normally with an unlimited supply of feed and water for seven days and deprived of food but not water for 4 h prior to the experiment. The toxicity test was supported by the Animal Ethics Committee of Lanzhou Institute of Husbandry and Pharmaceutical Sciences.
The acute toxicity of hybrids OBP-4 and OBP-5 was conducted by the limit test described in the OECD guideline 425 [39]. The mice were divided randomly into two groups, five animals in each group. One group of mice received a single oral dose of hybrids OBP-4 or OBP-5 at 2000 mg/kg·bw, and the other group of mice was given the equal volume of 0.5% CMC-Na solution (0.1 mL/10 g of body weight). Food was restored until 4 h after administration. The observation of each animal occurred according to the OECD guideline 425 [27], meaning continuously during the first 30 min, periodically during the next 24 h, and then daily thereafter until 14 d after dosing. The weight changes and mortality were calculated and recorded. The mice that survived were humanely killed at the end of the experiment.
4. Conclusions
A series of new oxazolidinone-fluoroquinolone hybrids incorporating the structural elements of oxazolidinone and fluoroquinolone with a linker were synthesized and evaluated for their in vitro antibacterial activities against various Gram-positive and Gram-negative bacteria. The antibacterial activities of most hybrids against Gram-positive bacteria were significantly higher than that of LZD, and hybrids OBP-4 and OBP-5 were the most active two hybrids. The mode of action for hybrids OBP-4 and OBP-5 was more directed to protein synthesis inhibition instead of DNA synthesis inhibition, showing a good consistency with the antibacterial profile. Meanwhile, a good safety profile was also demonstrated for hybrids OBP-4 and OBP-5 with LD50 values greater than 2000 mg/kg. These combined data suggested that hybrids OBP-4 and OBP-5 may be promising therapeutic options for severe nosocomial infections caused by Gram-positive bacteria.
Supplementary Materials
1H-NMR,13C-NMR and HRMS spectra for oxazolidinone-fluoroquinolone hybrids OBP-(1–7) are available online.
Author Contributions
Conceptualization, J.Z., L.L. and J.L.; Data curation, L.L., L.S. and H.C.; Formal analysis, L.L., L.S., J.L., H.C., B.L., X.Z. and P.L.; Funding acquisition, J.Z.; Investigation, L.L., L.S., J.L., H.C., B.L., X.Z. and P.L.; Project administration, J.L.; Resources, B.L. and X.Z.; Supervision, J.Z.; Validation, L.L., L.S., J.L. and H.C.; Visualization, L.L. and L.S.; Writing—original draft, L.L., L.S. and J.Z.; Writing—review & editing, J.Z.
Funding
This study was supported by the Key Projects in the National Science & Technological Pillar Program during the Twelfth Five-year Plan Period (2015BAD11B01) and the embarked fund for the China Agriculture Research System (CARS-37).
Acknowledgments
We are grateful to Hongsheng Li and Zhiyong Ma for supplying strains.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zinner, S.H. Antibiotic use: Present and future. New Microbiol. 2007, 30, 321–325. [Google Scholar] [PubMed]
- Li, B.; Webster, T.J. Bacteria antibiotic resistance: New challenges and opportunities for implant-associated orthopedic infections. J. Orthop. Res. 2018, 36, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-resistant bacteria and alternative methods to control them: An overview. Microb. Drug Resist. 2019. [Google Scholar] [CrossRef]
- Watkins, R.R.; Bonomo, R.A. Overview: Global and local impact of antibiotic resistance. Infect. Dis. Clin. N. Am. 2016, 30, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ciorba, V.; Odone, A.; Veronesi, L.; Pasquarella, C.; Signorelli, C. Antibiotic resistance as a major public health concern: Epidemiology and economic impact. Ann. Iq. 2015, 27, 562–579. [Google Scholar]
- Thabit, A.K.; Crandon, J.L.; Nicolau, D.P. Antimicrobial resistance: Impact on clinical and economic outcomes and the need for new antimicrobials. Expert Opin. Pharmacother. 2015, 16, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, G.S.; Zinner, S.H. Burden of antimicrobial resistance in an era of decreasing susceptibility. Expert Rev. Anti Infect. Ther. 2017, 15, 663–676. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the number of human and bacteria cells in the body. PLoS. Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Parkes, A.L.; Yule, I.A. Hybrid antibiotics—Clinical progress and novel designs. Expert Opin. Drug Discov. 2016, 11, 665–680. [Google Scholar] [CrossRef]
- Barbachyn, M.R. Recent advances in the discovery of hybrid antibacterial agents. Annu. Rep. Med. Chem. 2008, 43, 281–290. [Google Scholar]
- Robertson, G.T.; Bonventre, E.J.; Doyle, T.B.; Du, Q.; Duncan, L.; Morris, T.W.; Roche, E.D.; Yan, D.; Lynch, A.S. In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: Studies of the mode of action in Staphylococcus aureus. Antimicrob. Agents. Chemother. 2008, 52, 2313–2323. [Google Scholar] [CrossRef]
- Pokrovskaya, V.; Belakhov, V.; Hainrichson, M.; Yaron, S.; Baasov, T. Design, synthesis, and evaluation of novel fluoroquinolone-aminoglycoside hybrid antibiotics. J. Med. Chem. 2009, 52, 2243–2254. [Google Scholar] [CrossRef]
- Blais, J.; Lewis, S.R.; Krause, K.M.; Benton, B.M. Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents. Chemother. 2012, 56, 1584–1587. [Google Scholar] [CrossRef]
- Louie, T.; Nord, C.E.; Talbot, G.H.; Wilcox, M.; Gerding, D.N.; Buitrago, M.; Kracker, H.; Charef, P.; Cornely, O.A. Multicenter, double-blind, randomized, phase 2 study evaluating the novel antibiotic cadazolid in patients with Clostridium difficile infection. Antimicrob. Agents. Chemother. 2015, 59, 6266–6273. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaya, V.; Baasov, T. Dual-acting hybrid antibiotics: A promising strategy to combat bacterial resistance. Expert Opin. Drug Discov. 2010, 5, 883–902. [Google Scholar] [CrossRef]
- Shaw, K.J.; Barbachyn, M.R. The oxazolidinones: Past, present, and future. Ann. N. Y. Acad. Sci. 2011, 1241, 48–70. [Google Scholar] [CrossRef]
- Bender, J.K.; Fleige, C.; Lange, D.; Klare, I.; Werner, G. Rapid emergence of highly variable and transferable oxazolidinone and phenicol resistance gene optrA in German Enterococcus spp. clinical isolates. Int. J. Antimicrob. Agents 2018, 52, 819–827. [Google Scholar] [CrossRef] [PubMed]
- De Lastours, V.; Fantin, B. Resistance to fluoroquinolones in 2013: What are the consequences in internal medicine? Rev. Med. Interne 2014, 35, 601–608. [Google Scholar] [CrossRef]
- Hubschwerlen, C.; Specklin, J.L.; Sigwalt, C.; Schroeder, S.; Locher, H.H. Design, synthesis and biological evaluation of oxazolidinone-quinolone hybrids. Bioorg. Med. Chem. 2003, 11, 2313–2319. [Google Scholar] [CrossRef]
- Hubschwerlen, C.; Specklin, J.L.; Baeschlin, D.K.; Borer, Y.; Haefeli, S.; Sigwalt, C.; Schroeder, S.; Locher, H.H. Structure-activity relationship in the oxazolidinone-quinolone hybrid series: Influence of the central spacer on the antibacterial activity and the mode of action. Bioorg. Med. Chem. Lett. 2003, 13, 4229–4233. [Google Scholar] [CrossRef] [PubMed]
- Sabine Schubert, A.D. Low propensity for development of resistance to MCB3681, the active moiety of oxaquin (MCB3837), in Gram-positive bacteria with vancomycin-, linezolid-, methicillin- and/or ciprofloxacin resistances. In Proceedings of the 46th ICAAC, San Francisco, CA, USA, 27–30 September 2006. [Google Scholar]
- Locher, H.H.; Caspers, P.; Bruyere, T.; Schroeder, S.; Pfaff, P.; Knezevic, A.; Keck, W.; Ritz, D. Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents. Chemother. 2014, 58, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N.; Hecht, D.W.; Louie, T.; Nord, C.E.; Talbot, G.H.; Cornely, O.A.; Buitrago, M.; Best, E.; Sambol, S.; Osmolski, J.R.; et al. Susceptibility of Clostridium difficile isolates from a Phase 2 clinical trial of cadazolid and vancomycin in C. difficile infection. J. Antimicrob. Chemother. 2016, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Poeta, P.; Hebraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, H.; DiGate, R.J.; Marians, K.J. Decatenating activity of Escherichia coli DNA gyrase and topoisomerases I and III during oriC and pBR322 DNA replication in vitro. J. Biol. Chem. 1994, 269, 2093–2099. [Google Scholar] [PubMed]
- Baldoni, D.; Gutierrez, M.; Timmer, W.; Dingemanse, J. Cadazolid, a novel antibiotic with potent activity against Clostridium difficile: Safety, tolerability and pharmacokinetics in healthy subjects following single and multiple oral doses. J. Antimicrob. Chemother. 2014, 69, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Clewley, R.G.; Cross, G.G.; Fischer, A.; Henderson, G.N. Formation of 4-halo-4- nitrocyclohexa-2,5-dienones on nitration of p-halophenols and p-halophenyl acetates. Tetrahedron 1989, 45, 1299–1310. [Google Scholar] [CrossRef]
- Wessig, P.; Möllnitz, K.; Eiserbeck, C. Oligospiroketals as novel molecular rods. Chemistry 2007, 13, 4859–4872. [Google Scholar] [CrossRef]
- Yang, Y.; Ji, R.; Hu, Z.; Chen, K. A new practical route to stereospecific synthesis of (S)-(-)-ofloxacin. Acta Pharm. Sin. 1998, 33, 828–831. [Google Scholar]
- Kose, E.; Karabacak, M.; Atac, A. The spectroscopic and quantum chemical studies of 3,4-difluoroaniline. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 143, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Segawa, J.; Kitano, M.; Kazuno, K.; Matsuoka, M.; Shirahase, I.; Ozaki, M.; Matsuda, M.; Tomii, Y.; Kise, M. Studies on pyridonecarboxylic acids. 1. Synthesis and antibacterial evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acids. J. Med. Chem. 1992, 35, 4727–4738. [Google Scholar] [CrossRef]
- Matuszak, N.; Muccioli, G.G.; Labar, G.; Lambert, D.M. Synthesis and in vitro evaluation of N-substituted maleimide derivatives as selective monoglyceride lipase inhibitors. J. Med. Chem. 2009, 52, 7410–7420. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.H.; Kee, J.L. Baylis-Hillman route to several quinolone antibiotic intermediates. Chem. Inform. 2006, 37, 963–968. [Google Scholar]
- Egawa, H.; Kataoka, M.; Shibamori, K.I.; Miyamato, T.; Nakano, J.; Matsumoto, J.I. A new synthetic route to 7-halo-1-cyclopropyl-6-fluoro-1,4-dihydro-4oxoquinoline-3- carboxylic acid, an intermediate for the synthesis of quinolone antibacterial agents. J. Heterocyclic. Chem. 1987, 18, 181–185. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; 26th Informational Supplement; CLSI document M100-S26; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2016. [Google Scholar]
- Eyal, Z.; Matzov, D.; Krupkin, M.; Wekselman, I.; Paukner, S.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Yonath, A. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2015, 112, E5805–E5814. [Google Scholar] [CrossRef]
- Taher, E.S.; Ibrahim, T.S.; Fares, M.; Al-Mahmoudy, A.M.M.; Radwan, A.F.; Orabi, K.Y.; El-Sabbagh, O.I. Novel benzenesulfonamide and 1,2-benzisothiazol-3(2H)-one-1,1- dioxide derivatives as potential selective COX-2 inhibitors. Eur. J. Med. Chem. 2019, 171, 372–382. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- OECD 425 Guideline for Testing of Chemical. Acute Oral Toxicity-Up-and-Down Procedure; OECD: Paris, France, 2001. [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).