
Comparing Gly11/dAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
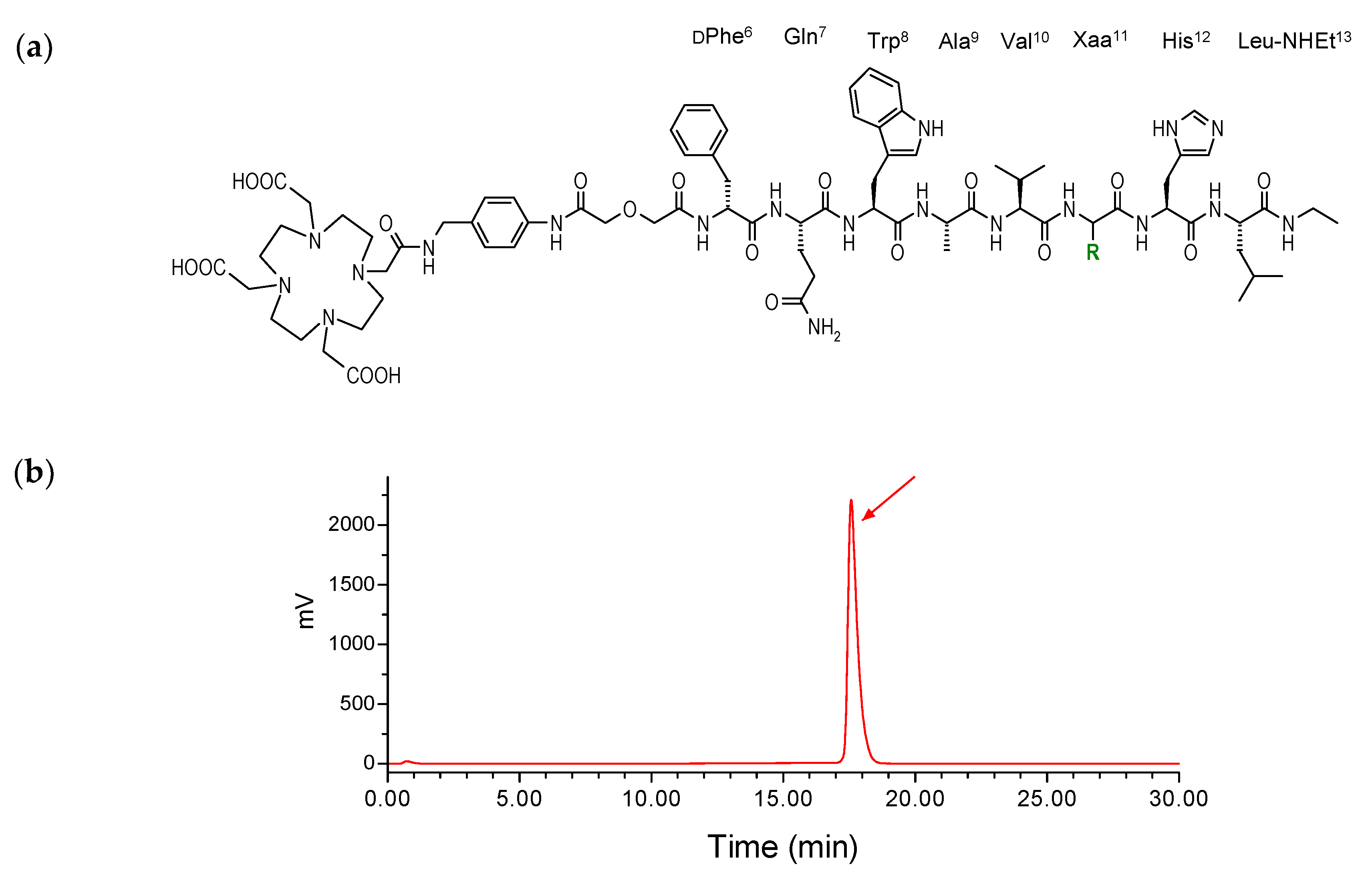
2.1. Peptides and Radioligands
2.2. In Vitro Assays in PC-3 Cells
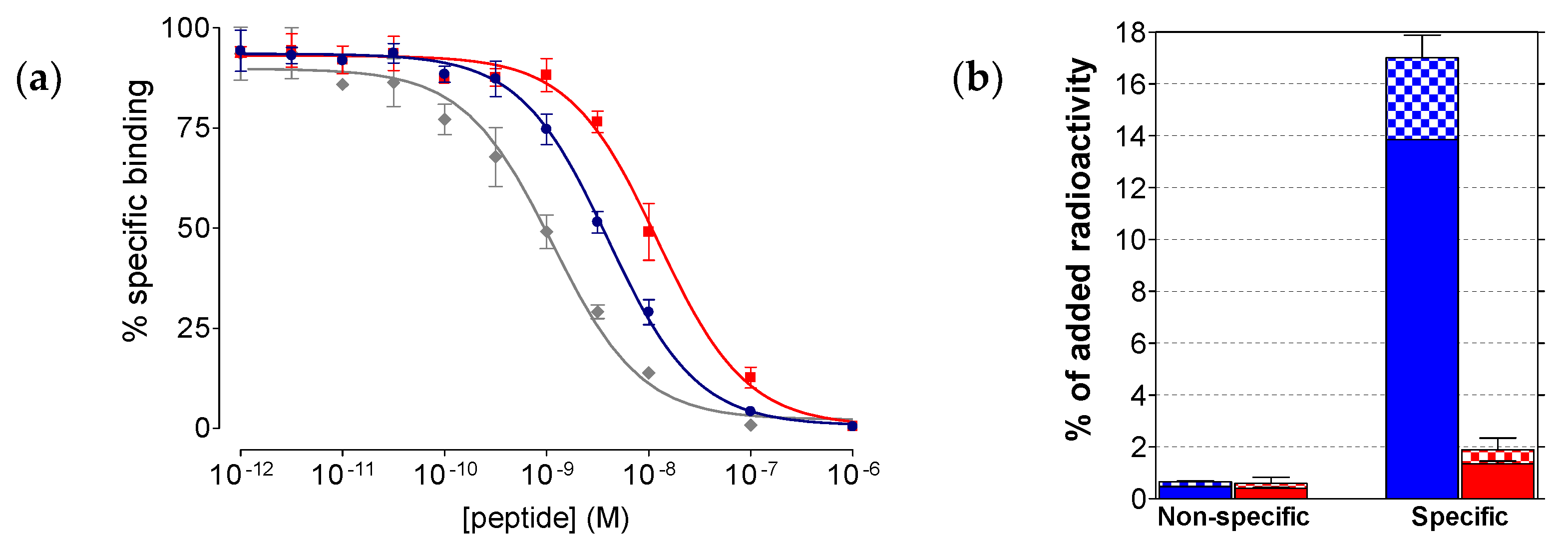
2.2.1. Affinity of SB3 and SB4 for the GRPR
2.2.2. Comparative Uptake of 111In-SB3 and 111In-SB4 by PC-3 Cells
2.3. In Vivo Comparison of 111In-SB3 and 111In-SB4
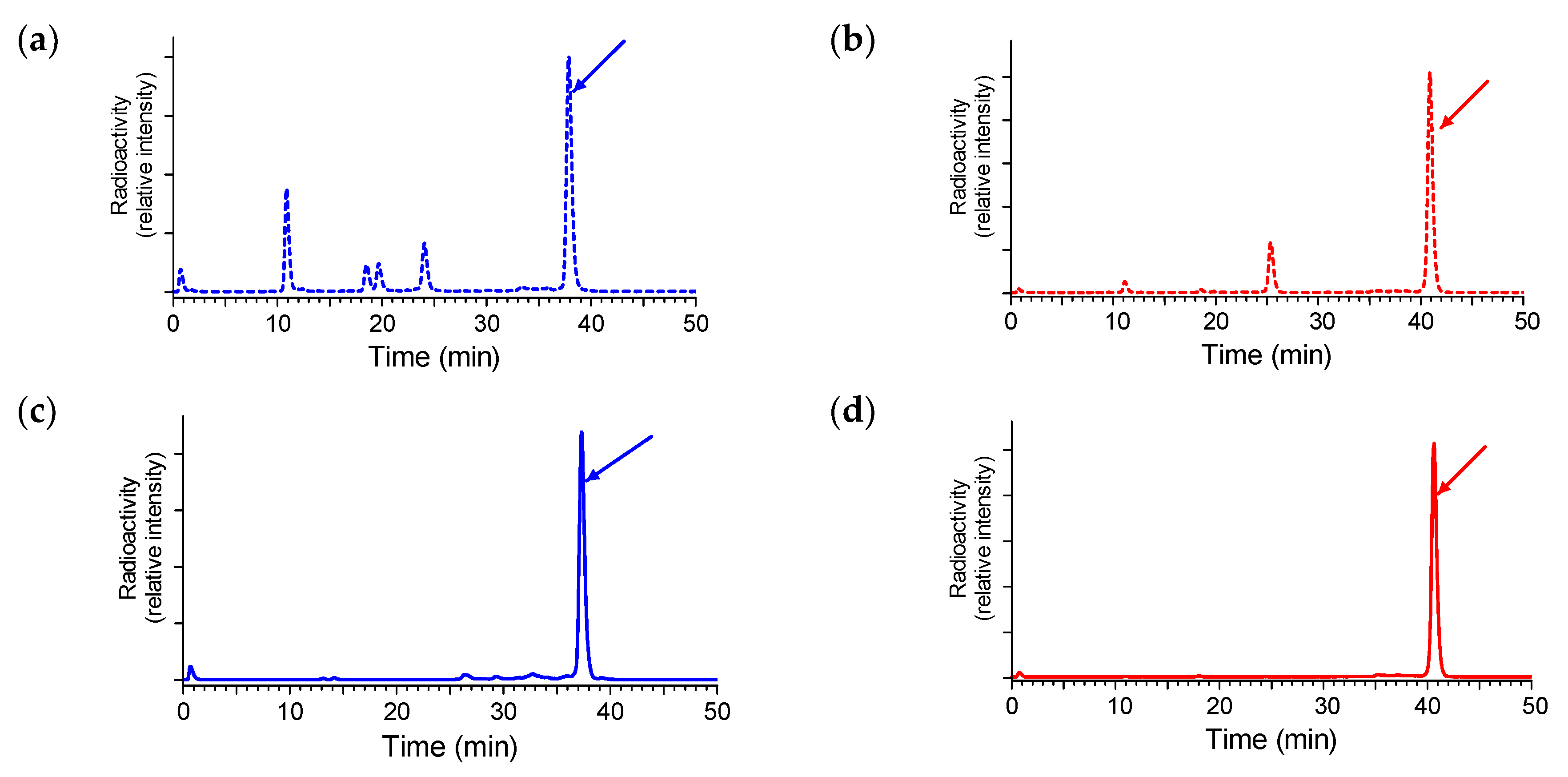
2.3.1. Stability of 111In-SB3 and 111In-SB4 in Healthy Mice
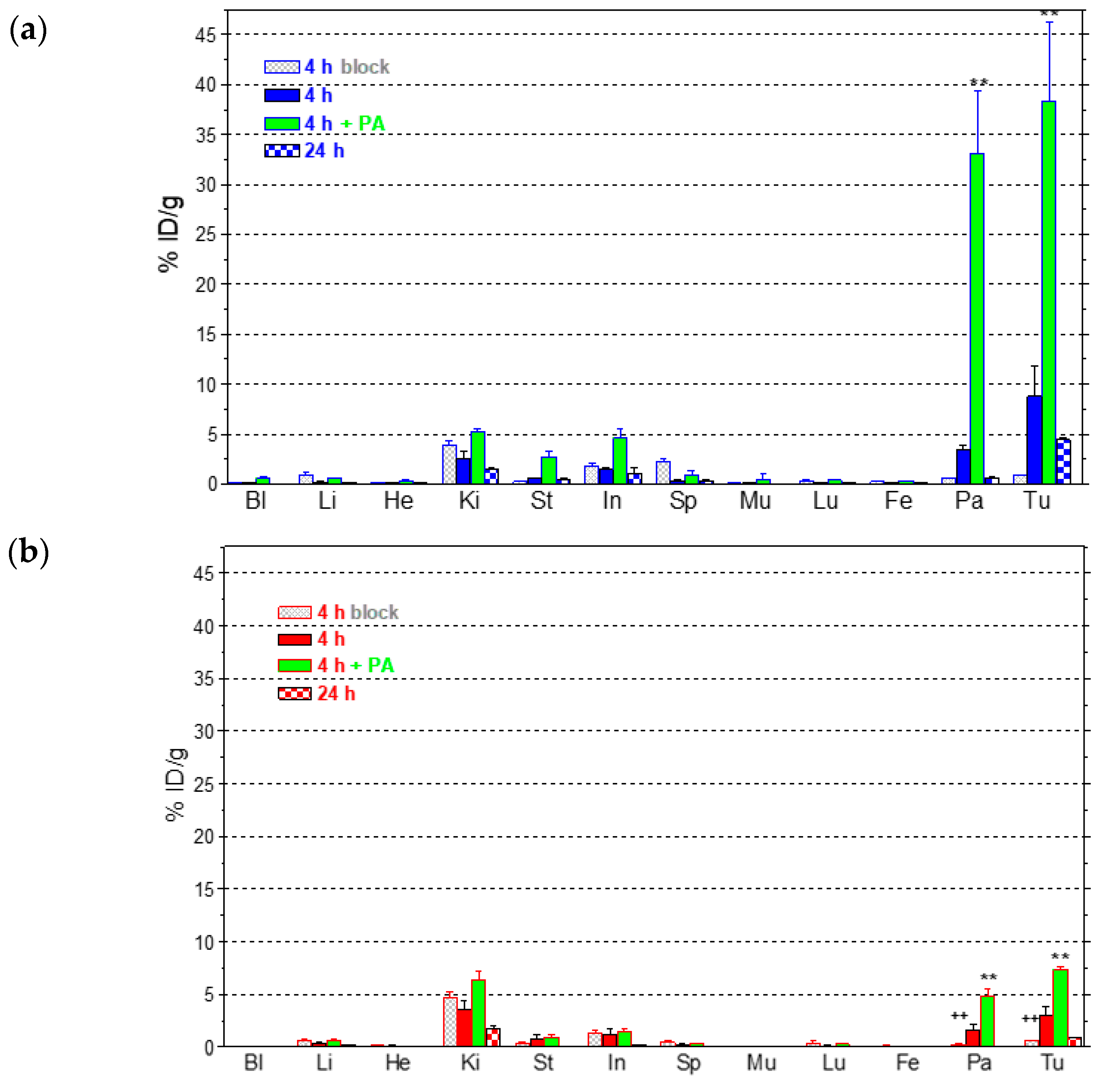
2.3.2. Comparative Biodistribution of 111In-SB3 and 111In-SB4 in SCID Mice Bearing PC-3 Xenografts
3. Discussion
4. Materials and Methods
4.1. Peptides and Radioligands
Preparation and Quality Control of 111In-SB3 and 111In-SB4
4.2. In Vitro Assays
4.2.1. Cell Lines and Culture
4.2.2. Competition Binding in PC-3 Cell-Membranes
4.2.3. Internalization Assay in PC-3 Cells
4.3. Animal Studies
4.3.1. In Vivo Stability Tests
4.3.2. Induction of PC-3 Xenografts in SCID Mice
4.3.3. Biodistribution in PC-3 Xenograft-Bearing SCID Mice
4.4. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Kroog, G.S.; Jensen, R.T.; Battey, J.F. Mammalian bombesin receptors. Med. Res. Rev. 1995, 15, 389–417. [Google Scholar] [CrossRef]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International union of pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert. Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Nock, B.A. From bench to bed: New gastrin-releasing peptide receptor-directed radioligands and their use in prostate cancer. PET Clin. 2017, 12, 205–217. [Google Scholar] [CrossRef]
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar]
- Körner, M.; Waser, B.; Rehmann, R.; Reubi, J.C. Early over-expression of grp receptors in prostatic carcinogenesis. Prostate 2014, 74, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Muntener, M.; Kristiansen, G. Profiling gastrin-releasing peptide receptor in prostate tissues: Clinical implications and molecular correlates. Prostate 2012, 72, 318–325. [Google Scholar] [CrossRef]
- Gugger, M.; Reubi, J.C. Gastrin-releasing peptide receptors in non-neoplastic and neoplastic human breast. Am. J. Pathol. 1999, 155, 2067–2076. [Google Scholar] [CrossRef]
- Halmos, G.; Wittliff, J.L.; Schally, A.V. Characterization of bombesin/gastrin-releasing peptide receptors in human breast cancer and their relationship to steroid receptor expression. Cancer Res. 1995, 55, 280–287. [Google Scholar] [PubMed]
- Mattei, J.; Achcar, R.D.; Cano, C.H.; Macedo, B.R.; Meurer, L.; Batlle, B.S.; Groshong, S.D.; Kulczynski, J.M.; Roesler, R.; Dal Lago, L.; Brunetto, A.T.; Schwartsmann, G. Gastrin-releasing peptide receptor expression in lung cancer. Arch. Pathol. Lab. Med. 2014, 138, 98–104. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A.; Kulkarni, H.; Singh, A.; Baum, R.P. Theranostic prospects of gastrin-releasing peptide receptor-radioantagonists in oncology. PET Clin. 2017, 12, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Coy, D.H.; Taylor, J.E.; Jiang, N.Y.; Kim, S.H.; Moreau, J.P.; Huang, S.C.; Mantey, S.A.; Frucht, H.; Jensen, R.T. Desmethionine alkylamide bombesin analogues: A new class of bombesin receptor antagonists with potent antisecretory activity in pancreatic acini and antimitotic activity in swiss 3T3 cells. Biochemistry 1990, 29, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Coy, D.H.; Taylor, J.E.; Jiang, N.Y.; Moreau, J.P.; Huang, S.C.; Frucht, H.; Haffar, B.M.; Jensen, R.T. Des.-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J. Biol. Chem. 1990, 265, 15695–15703. [Google Scholar] [PubMed]
- Nock, B.; Nikolopoulou, A.; Chiotellis, E.; Loudos, G.; Maintas, D.; Reubi, J.C.; Maina, T. [99mTc]Demobesin 1, a novel potent bombesin analogue for grp receptor-targeted tumour imaging. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Lymperis, E.; Kaloudi, A.; Sallegger, W.; Bakker, I.L.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Radiometal-dependent biological profile of the radiolabeled gastrin-releasing peptide receptor antagonist SB3 in cancer theranostics: Metabolic and biodistribution patterns defined by neprilysin. Bioconjug. Chem. 2018, 29, 1774–1784. [Google Scholar] [CrossRef]
- Bakker, I.L.; van Tiel, S.T.; Haeck, J.; Doeswijk, G.N.; de Blois, E.; Segbers, M.; Maina, T.; Nock, B.A.; de Jong, M.; Dalm, S.U. In vivo stabilized SB3, an attractive GRPR antagonist, for pre- and intra-operative imaging for prostate cancer. Mol. Imaging Biol. 2018, 20, 973–983. [Google Scholar] [CrossRef]
- Bakker, I.L.; van Leenders, G.J.L.H.; Segbers, M.; Fröberg, A.C.; Dalm, Y.K.; Veenland, J.; Konijnenberg, M.; Busstra, M.B.; Verzijlbergen, J.F.; Schoots, J.; et al. Correlation of clinical GRP receptor PET imaging of prostate cancer to receptor expression status. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, S147. [Google Scholar]
- de Castiglione, R.; Gozzini, L. Bombesin receptor antagonists. Crit. Rev. Oncol. Hematol. 1996, 24, 117–151. [Google Scholar] [CrossRef]
- Coy, D.H.; Mungan, Z.; Rossowski, W.J.; Cheng, B.L.; Lin, J.T.; Mrozinski, J.E., Jr.; Jensen, R.T. Development of a potent bombesin receptor antagonist with prolonged in vivo inhibitory activity on bombesin-stimulated amylase and protein release in the rat. Peptides 1992, 13, 775–781. [Google Scholar] [CrossRef]
- Nock, B.A.; Charalambidis, D.; Sallegger, W.; Waser, B.; Mansi, R.; Nicolas, G.P.; Ketani, E.; Nikolopoulou, A.; Fani, M.; Reubi, J.C.; Maina, T. New gastrin releasing peptide receptor-directed [99mTc]Demobesin 1 mimics: Synthesis and comparative evaluation. J. Med. Chem. 2018, 61, 3138–3150. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. "To serve and protect": Enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Reile, H.; Armatis, P.E.; Schally, A.V. Characterization of high-affinity receptors for bombesin/gastrin releasing peptide on the human prostate cancer cell lines PC-3 and DU-145: Internalization of receptor bound 125I-(Tyr4)bombesin by tumor cells. Prostate 1994, 25, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Nock, B.; Nikolopoulou, A.; Sotiriou, P.; Loudos, G.; Maintas, D.; Cordopatis, P.; Chiotellis, E. [99mTc]Demotate, a new 99mTc-based [Tyr3]octreotate analogue for the detection of somatostatin receptor-positive tumours: Synthesis and preclinical results. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 742–753. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. Comparing Gly11/dAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair. Molecules 2019, 24, 1015. https://doi.org/10.3390/molecules24061015
Lymperis E, Kaloudi A, Kanellopoulos P, de Jong M, Krenning EP, Nock BA, Maina T. Comparing Gly11/dAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair. Molecules. 2019; 24(6):1015. https://doi.org/10.3390/molecules24061015
Chicago/Turabian StyleLymperis, Emmanouil, Aikaterini Kaloudi, Panagiotis Kanellopoulos, Marion de Jong, Eric P. Krenning, Berthold A. Nock, and Theodosia Maina. 2019. "Comparing Gly11/dAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair" Molecules 24, no. 6: 1015. https://doi.org/10.3390/molecules24061015
APA StyleLymperis, E., Kaloudi, A., Kanellopoulos, P., de Jong, M., Krenning, E. P., Nock, B. A., & Maina, T. (2019). Comparing Gly11/dAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair. Molecules, 24(6), 1015. https://doi.org/10.3390/molecules24061015