5.2. Chemical Synthesis
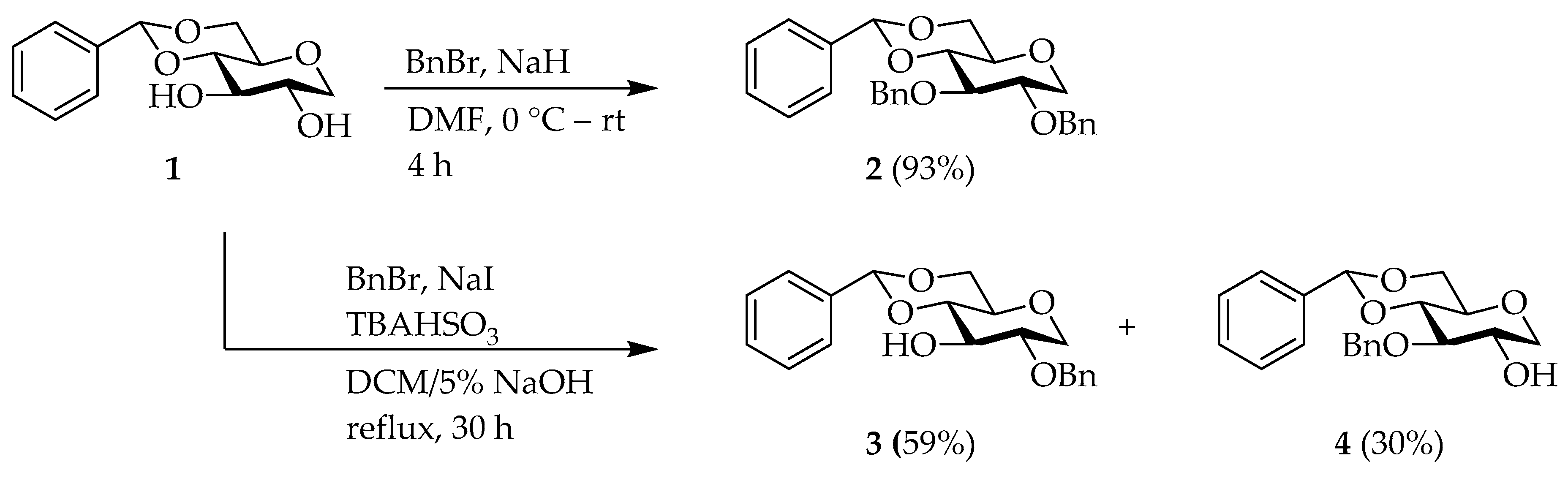
2-O-Benzyl-4,6-O-benzylidene-1,5-anhydro-d-glucitol (
3) [
35] and
3-O-Benzyl-4,6-O-benzylidene-1,5-anhydro-d-glucitol (
4) [
35]; Compound
1 (2.5 g, 10 mmol) and
(0.68 g, 2.0 mmol) in 160 mL of DCM and 14 mL of 5% NaOH was stirred at rt. Then, BnBr (2.1 mL, 17 mmol) was slowly added, and the mixture was refluxed for 30 h. After the addition 50 mL of water, the mixture was extracted with DCM (3 × 100). The combined organic layers were washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude product was separated by C.C (Hex/EtOAc = 4/1) to obtain 2-
O-Bn compound
3 (2.0 g, 59%) as colorless needles and 3-
O-Bn compound
4 (1.0 g, 30% yield) as a white solid. Compound
3: m.p. 163 °C;
= −3.16 (c 1.0,
);
NMR (
, 600 MHz) δ 7.49–7.47 (m, 2H), 7.38–7.29 (m, 8H), 5.50 (s, 1H), 4.76, 4.67 (ABq,
J = 11.7 Hz, 2H), 4.30 (dd,
J = 10.5, 5.0 Hz, 1H), 4.01 (dd,
J = 11.3, 5.5 Hz, 1H), 3.84 (m, 1H), 3.65 (t,
J = 10.3 Hz, 1H), 3.59–3.55 (m, 1H), 3.45 (t,
J = 9.3 Hz, 1H), 3.35 (m, 1H), 3.30 (t,
J = 11.0 Hz, 1H);
NMR (
, 150 MHz) δ 138.02, 137.05, 129.23, 128.56, 128.33, 128.03, 127.89, 126.29, 101.88, 81.05, 77.74, 74.81, 73.43, 70.91, 68.79, 68.45. Compound
4: m.p. 137 °C;
= +5.3 (c 1.0,
);
NMR (
600 MHz) δ 7.50–7.48 (m, 2H), 7.41–7.29 (m, 8H), 5.58 (s, 1H), 5.03, 4.72 (ABq,
J = 11.3 Hz, 2H), 4.34 (dd,
J = 10.5, 5.0 Hz, 1H), 4.06 (dd,
J = 11.2, 5.7 Hz, 1H), 3.80–3.76 (m, 1H), 3.72 (t,
J = 10.3 Hz, 1H), 3.66 (t,
J = 9.1 Hz, 1H), 3.58 (t,
J = 8.8 Hz, 1H), 3.44–3.40 (m, 1H), 3.34 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 138.30, 137.32, 129.00, 128.61, 128.30, 128.13, 128.01, 125.98, 101.21, 82.66, 82.13, 74.71, 71.53, 69.91, 69.79, 68.88.
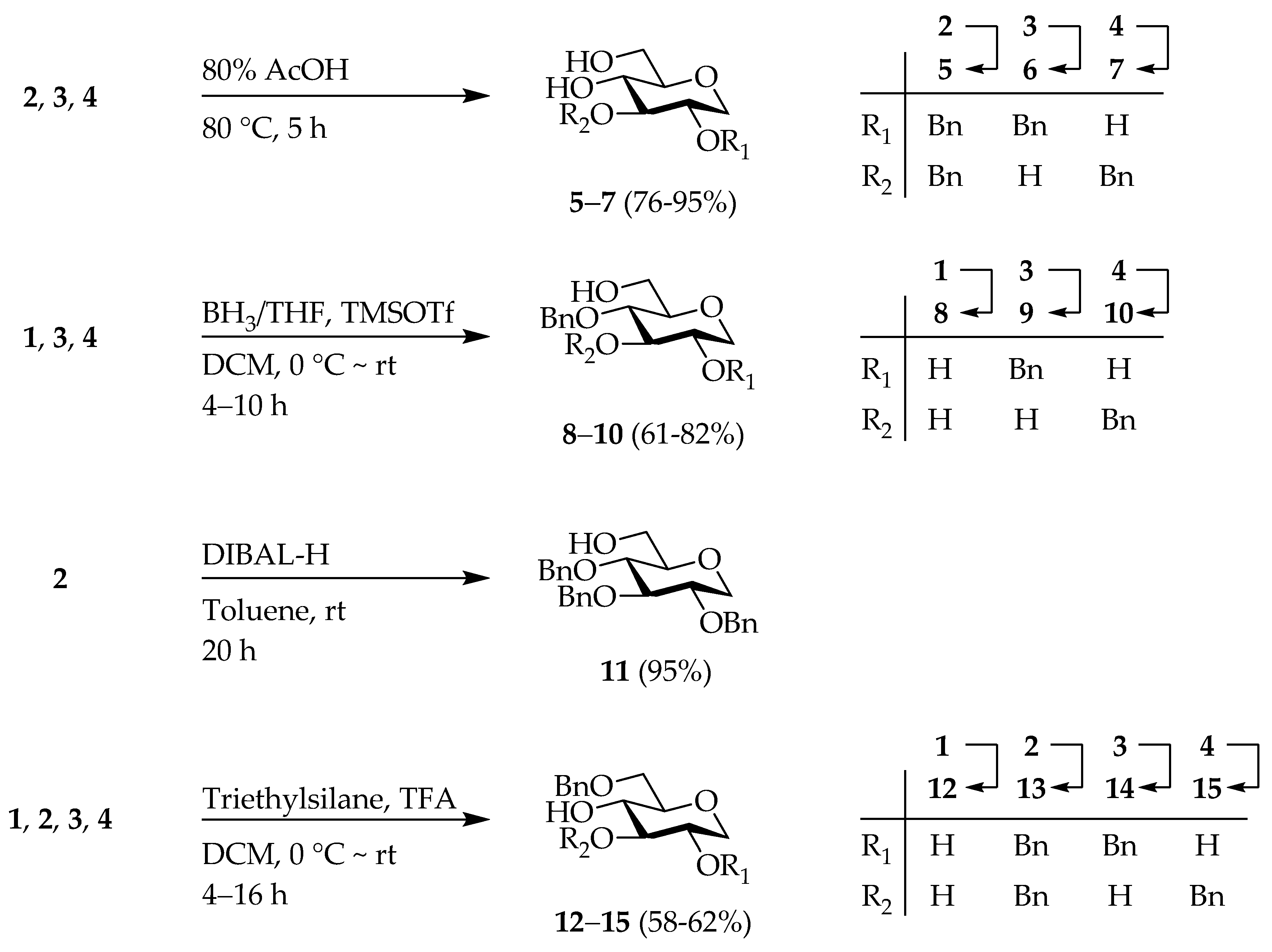
2,3-Di-O-benzyl-1,5-anhydro-d-glucitol (
5) [
42]; Compound
2 (0.86g, 2.0 mmol) in 10 mL of 80% AcOH/
solution was stirred at 80 °C for 5 h. After the addition 5 mL of saturated aq.
, the reaction solution was extracted by EtOAc (3 × 30). The organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by recrystallization (EtOAc/Hex) to obtain
5 (0.65 g, 95%) as colorless needles. m.p. 129 °C;
= −10.4 (c 1.00,
);
NMR (
, 600 MHz) δ 7.43–7.29 (m, 10H), 5.03, 4.70 (ABq,
J = 11.7 Hz, 2H), 4.64, 4.03 (ABq,
J = 11.3, 2H), 3.86–3.82 (m, 1H), 3.72–3.68 (m, 1H), 3.63–3.59 (m, 1H), 3.50–3.44 (m, 2H), 3.28–3.23 (m, 2H);
NMR (
, 150 MHz) δ 138.54, 137.97, 128.67, 128.53, 127.98, 127.93, 127.83, 85.30, 79.53, 78.29, 75.10, 73.07, 70.45, 67.94, 62.89.
1,5-Anhydro-2-O-benzyl-d-glucitol (
6) [
35]; Compound
3 (860 mg, 2.5 mmol) in 10 mL of 80% AcOH/
solution was stirred at 80 °C for 5 h. By the same procedure previously described for the preparation of compound
5, compound
6 (503 mg, 76%) was obtained as colorless needles. m.p. 131 °C;
= +8.4 (c 1.00, MeOH);
NMR (
, 600 MHz); δ 7.40–7.36 (m, 2H), 7.33–7.31 (m, 2H), 7.28–7.25 (m, 1H), 4.75, 4.64 (ABq,
J = 11.7 Hz, 2H), 3.96 (dd,
J = 11.0, 5.2 Hz, 1H), 3.81 (dd,
J = 11.9, 2.2 Hz, 1H), 3.59 (dd,
J = 11.9, 6.0 Hz, 1H), 3.43 (t,
J = 8.9 Hz, 1H), 3.39–3.35 (m, 1H), 3.26–3.22 (m, 1H), 3.15–3.11 (m, 2H);
NMR (
, 150 MHz); δ 140.09, 129.37, 129.05, 128.76, 82.42, 79.31, 79.27, 74.14, 71.92, 68.95, 63.07.
1,5-Anhydro-3-O-benzyl-d-glucitol (7); Compound 4 (680 mg, 2.0 mmol) in 10 mL of 80% AcOH/ solution was stirred at 80 °C for 5 h. By the same procedure previously described for the preparation of compound 5, compound 7 (410 mg, 80%) was obtained as colorless needles. m.p. 153 °C; = +28.6 (c 1.00, MeOH); NMR (, 600 MHz); δ 7.44 (m, 2H), 7.32–7.30 (m, 2H), 7.26–7.23 (m, 1H), 4.90 (overlap, 2H), 3.89 (dd, J = 11.3, 5.5 Hz, 1H), 3.83 (dd, J = 11.9, 2.2 Hz, 1H), 3.62–3.57 (m, 2H), 3.37 (dd, J = 18.0, 8.71 Hz, 1H), 3.31–3.28 (overlap, 1H), 3.22–3.16 (m, 2H); NMR (, 150 MHz); δ 140.53, 129.20, 129.09, 128.49, 88.20, 82.68, 76.10, 71.67, 71.54, 71.09, 63.05. HRMS (ESI, m/z): , calcd for : 277.1052; found 277.1052.
1,5-Anhydro-4-O-benzyl-d-glucitol (
8) [
35]; Compound
1 (0.76 g, 3.0 mmol) in 15 mL of DCM was stirred at 0 °C. Then, 15 mL of borane-THF (ca. 1M THF solution) and TMSOTf (0.11 mL, 0.60 mmol) were successively added to the mixture. The mixture was allowed to stir for 4 h and added MeOH carefully. After the addition 1 mL of saturated aq.
, the reaction solution was extracted with DCM (5 × 40 mL). The combined organic layer was dried over
, filtered and concentrated under reduced pressure. The crude product was purified by C.C.
/MeOH = 8/1) to obtain
8 (0.64 g, 74%) as a white solid.
= +27.7 (c 0.45,
);
NMR (
, 600 MHz) δ 7.38–7.25 (m, 5H), 4.94, 4.64 (ABq,
J = 11.0 Hz, 2H), 3.89 (dd,
J = 13.3, 5.4 Hz, 1H), 3.78 (dd,
J = 12.0, 2.1 Hz, 1H), 3.60 (dd, 1H,
J = 11.4, 5.73 Hz,), 3.49–3.46 (m, 2H), 3.33–3.30 (overlap, 1H), 3.20 (ddd,
J = 9.7, 5.2, 2.1 Hz, 1H), 3.14 (t,
J = 10.7, 1H);
NMR (
, 150 MHz); δ 140.09, 129.33, 129.14, 128.70, 81.75, 80.43, 79.50, 75.90, 71.77, 70.93, 62.72.
2,4-Di-O-benzyl-1,5-anhydro-d-glucitol (
9) [
46]; Compound
3 (1.0 g, 3.0 mmol) in 15 mL of DCM was successively added 15 mL of borane-THF (ca. 1M THF solution) and TMSOTf (0.11 mL, 0.6 mmol) at 0 °C. The mixture was allowed to stir at rt for 10 h and added 1 mL of MeOH carefully. After the addition 1 mL of saturated aq.
, the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by recrystallization (Hex/EtOAc) to obtain
9 (820 mg, 82%) as colorless needles. m.p. 112 °C;
= +34.6 (c 1.00,
);
NMR (
, 600 MHz) δ 7.37–7.28 (m, 10H), 4.86, 4.70 (ABq,
J = 11.3 Hz, 2H), 4.65 (s, 2H), 3.99 (dd,
J = 11.3, 5.2 Hz, 1H), 3.84 (ddd,
J = 11.8, 5.8, 2.7 Hz, 1H), 3.74 (td,
J = 8.9, 2.1 Hz, 1H), 3.68–3.64 (m, 1H), 3.45–3.41 (m, 1H), 3.41 (t,
J = 9.2 Hz, 1H), 3.26 (ddd,
J = 9.6, 4.5, 2.7 Hz, 1H), 3.19 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 138.18, 138.00, 128.60, 128.56, 128.09, 127.98, 127.85, 79.34, 78.18, 77.97, 77.39, 74.74, 73.05, 67.44, 62.27.
3,4-Di-O-benzyl-1,5-anhydro-d-glucitol (
10) [
47]; Compound
4 (850 g, 2.5 mmol) in 15 mL of DCM was successively added 12.5 mL of borane-THF (ca. 1M THF solution) and TMSOTf (90 μL, 0.5 mmol) at 0 °C. The mixture was allowed to stir at rt for 7 h and added 5 mL of MeOH carefully. After the addition 1 mL of saturated aq.
, the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by recrystallization (Hex/EtOAc) to obtain
10 (523 mg, 61%) as colorless needles. m.p. 99 °C;
= +48.3 (c 1.40,
);
NMR (
, 600 MHz) δ 7.38–7.30 (m, 10H), 4.97, 4.77 (ABq,
J = 11.3 Hz, 2H), 4.86, 4.68 (ABq,
J = 11.0 Hz, 2H), 3.98 (dd,
J = 11.2, 5.3 Hz, 1H), 3.84 (ddd,
J = 11.9, 5.5, 2.6 Hz, 1H), 3.71–3.65 (m, 2H), 3.52 (t,
J = 9.1 Hz, 1H), 3.47 (t,
J = 8.8 Hz, 1H), 3.32–3.29 (m, 1H), 3.23 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 138.45, 137.84, 128.71, 128.56, 128.02, 127.86, 86.73, 79.92, 77.79, 75.25, 74.95, 70.16, 69.30, 61.99.
2,3,4-Tri-O-benzyl-1,5-anhydro-d-glucitol (
11) [
46]; Compound
2 (860 mg, 2.0 mmol) in 10 mL of toluene was stirred at rt. The reaction solution was added DIBAL-H (ca. 1M toluene solution, 6 mL) and stirred at rt for 20 h. The reaction solution was slowly added 4.2 mL of MeOH and 7.2 mL of 30% Rochelle salt solution and stirred for 1 h. After the addition 20 mL of EtOAc, the mixture was extracted with 30% Rochelle salt solution (3 × 15 mL). The organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by C.C. (Hex/EtOAc = 4/1–2/1) to obtain
11 (760 mg, 90% yield) as colorless needles. m.p. 83 °C;
= +8.9 (c 0.90,
);
NMR (
, 600 MHz) δ 7.37–7.28 (m, 15H), 4.98–4.64 (m, 6H), 3.99 (dd,
J = 11.3, 5.2 Hz, 1H), 3.84–3.81 (m, 1H), 3.67–3.58 (m, 3H), 3.48 (t,
J = 9.3 Hz, 1H), 3.29–3.25 (m, 1H), 3.23 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 138.61, 138.09, 138.01, 128.48, 128.41, 128.05, 127.89, 127.83, 127.64, 86.17, 79.69, 78.55, 77.57, 75.55, 75.13, 73.34, 67.96, 62.25.
1,5-Anhydro-6-O-benzyl-d-glucitol (
12) [
35]; Compound
1 (760 mg, 3.0 mmol) in 15 mL of DCM was added triethylsilane (2.4 mL, 15 mmol) and trifluoracetic acid (1.2 mL, 15 mmol) at 0 °C. The reaction solution was stirred at rt for 8 h. After the addition 10 mL of saturated aq.
, the mixture was extracted with DCM (5 × 30 mL). The combined organic layer was dried over
, filtered and concentrated under reduced pressure. The crude was purified by C.C (
/MeOH = 100/1–10/1) to obtain
12 (471 mg, 62%) as a colorless oil.
= +8.9 (c 0.30,
);
NMR (
, 600 MHz) δ 7.35–7.25 (m, 5H), 4.55 (d,
J = 2.1 Hz, 2H), 3.88 (dd,
J = 11.0, 5.5 Hz, 1H), 3.77 (dd,
J = 10.8, 1.9 Hz, 1H), 3.60 (dd,
J = 10.8, 5.7 Hz, 1H), 3.48–3.42 (m, 1H), 3.32–3.24 (overlap, 3H), 3.15 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 139.57, 129.34, 128.90, 128.67, 81.38, 79.95, 74.48, 71.85, 71.31, 71.19, 70.95.
2,3,6-Tri-O-benzyl-1,5-anhydro-d-glucitol (
13) [
49]; Compound
2 (860 mg, 2.0 mmol) in 10 mL of DCM was added triethylsilane (1.6 mL, 10 mmol) and tifluoroacetic acid (0.8 mL, 10 mmol) at 0 °C. The reaction solution was stirred at rt for 15 h. After the addition 5 mL of saturated aq.
, the mixture was extracted with DCM (3 × 30 mL). The organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude product was purified by C.C. (Hex/EtOAc = 4/1) to obtain
13 (505 mg, 59%) as a colorless oil.
= −7.0 (c 1.00,
);
NMR (
, 600 MHz) δ 7.37–7.25 (m, 15H), 5.00, 4.76 (ABq,
J = 11.5 Hz, 2H), 4.69, 4.63 (ABq,
J = 11.7 Hz, 2H), 4.58, 4.54 (ABq,
J = 12.2 Hz, 2H), 4.04 (dd,
J = 11.3, 5.2 Hz, 1H), 3.70 (dd,
J = 10.5, 3.3 Hz, 1H), 3.64–3.60 (m, 2H), 3.54 (td,
J = 9.2, 2.1 Hz, 1H), 3.44 (t,
J = 8.9 Hz, 1H), 3.37–3.34 (m, 1H), 3.23 (t,
J = 11.0 Hz, 1H);
NMR (
, 150 MHz) δ 138.64, 138.05, 137.84, 128.58, 128.48, 128.39, 127.95, 127.90, 127.87, 127.83, 127.77, 127.70, 85.40, 78.75, 78.04, 75.10, 73.65, 73.05, 70.87, 69.98, 68.06.
2,6-Di-O-benzyl-1,5-anhydro-d-glucitol (14); Compound 3 (860 mg, 2.5 mmol) in 15 mL of DCM was added triethylsilane (2.0 mL, 12.5 mmol) and tifluoroacetic acid (1.0 mL, 12.5 mmol) at 0 °C. The reaction solution was stirred at rt for 8 h. After the addition 10 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. /MeOH = 8/1) to obtain 14 (500 mg, 58%) as a white solid. = +17.4 (c 1.00, ); NMR (, 600 MHz) δ 7.36–7.27 (m, 10H), 4.64 (s, 2H), 4.59, 4.54 (ABq, J = 12.0 Hz, 2H), 4.02 (dd, J = 11.2, 5.0 Hz, 1H), 3.69 (dd, J = 10.5, 3.6 Hz, 1H), 3.65 (dd, J = 10.5, 5.0 Hz, 1H), 3.55 (td, J = 8.8, 2.1 Hz, 1H), 3.50 (td, J = 9.0, 2.6 Hz, 1H), 3.47–3.43 (m, 1H), 3.37–3.34 (m, 1H), 3.19 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 137.96, 137.71, 128.59, 128.46, 128.07, 127.90, 127.81, 78.31, 77.74, 77.41, 73.68, 72.95, 71.34, 70.01, 67.70. HRMS (ESI, m/z): , calcd for : 367.1521; found 367.1521.
1,5-Anhydro-3,6-di-O-benzyl-d-glucitol (15); Compound 4 (680 mg, 2.0 mmol) in 15 mL of DCM was added triethylsilane (1.6 mL, 10 mmol) and trifluoroacetic acid (0.80 mL, 10 mmol) at 0 °C. The reaction solution was stirred at rt for 4 h. After the addition 10 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. /MeOH = 8/1) to obtain 15 (420 mg, 62%) as a white solid. = +22.7 (c 1.00, ); NMR (, 600 MHz) δ 7.39–7.27 (m, 10H), 4.91, 4.83 (ABq, J = 11.7 Hz, 2H), 4.59, 4.54 (ABq, J = 12.2 Hz, 2H), 3.96 (dd, J = 11.2, 5.3 Hz, 1H), 3.72–3.66 (m, 3H), 3.61 (dt, J = 9.1, 2.6 Hz, 1H), 3.39–3.36 (m, 1H), 3.31 (t, J = 8.8 Hz, 1H), 3.21 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.60, 137.61, 128.70, 128.47, 128.02, 127.93, 127.86, 127.82, 86.47, 78.47, 74.87, 73.74, 72.11, 70.38, 69.79, 69.57. HRMS (ESI, m/z): , calcd for : 367.1521; found 367.1522.
1,5-Anhydro-2-O-(3′,4′,5′-tribenzyloxybenzoyl)-3-O-benzyl-4,6-O-benzylidene-d-glucitol (
17); Compound
3 (340 mg, 1.0 mmol), compound
16 [
39] (650 mg, 1.5 mmol), 2-chloro-1-methylpyridinium iodide (380 mg, 1.5 mmol), DMAP (37 mg, 0.30 mmol), TEA (416 μL, 3.0 mmol) in 15 mL of DCM was stirred at rt for 20 h. After the addition 100 mL of saturated aq.
, the reaction solution was extracted with DCM (3 × 60 mL). The combined organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by C.C. (DCM/MeOH = 500/1 − 200/1) to obtain
17 (350 mg, 41%) as a colorless amorphous oil.
= +8.4 (c 1.04,
);
NMR (
, 600 MHz) δ 7.53–7.11 (m, 29H), 5.61 (s, 1H), 5.27–5.20 (m, 1H), 5.15 (s, 2H), 5.09 (s, 4H), 4.86, 4.71 (ABq,
J = 12.0 Hz, 2H), 4.36 (dd,
J = 10.3, 4.8 Hz, 1H), 4.19 (dd,
J = 11.0, 5.8 Hz, 1H), 3.87 (t,
J = 9.1 Hz, 1H), 3.79–3.74 (m, 2H), 3.45 (td,
J = 9.7, 4.9 Hz, 1H), 3.36 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 165.06, 152.55, 142.74, 138.10, 137.31, 137.28, 136.56, 129.03, 128.57, 128.30, 128.25, 128.22, 128.07, 128.02, 127.89, 127.58, 127.47, 126.02, 124.50, 109.41, 101.32, 81.96, 79.12, 75.14, 74.33, 71.50, 71.30, 68.76, 67.68; HRMS (ESI,
m/z):
, calcd for
: 787.2883; found 787.2881.
1,5-Anhydro-2-O-benzyl-3-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-O-benzylidene-d-glucitol (18); Compound 2 (0.51 g, 1.5 mmol), compound 16 (1.0 g, 2.3 mmol), 2-chloro-1-methylpyridinium iodide (0.59 g, 2.3 mmol), DMAP (28 mg, 0.23 mmol), TEA (0.62 mL, 4.5 mmol) in 20 mL of DCM was stirred at rt for 20 h. By the same procedure previously described for the preparation of compound 17, compound 18 (0.87 g, 68%) was obtained as a colorless amorphous oil. = −45.6 (c 0.95, ); NMR (, 600 MHz) δ 7.24–7.44 (m, 24H), 7.20–7.14 (m, 5H), 5.53 (t, J = 9.3 Hz, 1H), 5.46 (s, 1H), 5.15–5.10 (m, 6H), 4.55, 4.46 (ABq, J = 12.4 Hz, 2H), 4.35 (dd, J = 10.5, 5.0 Hz, 1H), 4.13 (dd, J = 11.3, 5.5 Hz, 1H), 3.76–3.72 (m, 1H), 3.70 (t, J = 10.3 Hz, 1H), 3.65 (t, J = 9.5 Hz, 1H), 3.51 (td, J = 9.7, 4.8 Hz, 1H), 3.48 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.16, 152.47, 142.58, 137.53, 137.39, 136.69, 128.95, 128.54, 128.38, 128.21, 128.16, 128.02, 127.98, 127.87, 127.53, 126.14, 125.05, 109.55, 101.35, 79.18, 75.62, 75.15, 74.94, 72.97, 71.48, 71.33, 68.80, 68.74; HRMS (ESI, m/z): , calcd for : 787.2883; found 787.2882.
1,5-Anhydro-2,3,6-tris-O-benzyl-4-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (19); Compound 13 (340 mg, 0.78 mmol), compound 16 (530 mg, 1.2 mmol), 2-chloro-1-methylpyridinium iodide (307 mg, 1.2 mmol), DMAP (95 g, 0.78 mmol), TEA (315 μL, 2.3 mmol) in 15 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 19 (607 mg, 91%) was obtained as a colorless amorphous oil. = −32.0 (c 0.95, ); NMR (, 600 MHz) δ 7.44–7.26 (m, 20H), 7.23–7.15 (m, 7H), 7.11–7.03 (m, 5H), 5.18–5.15 (m, 3H), 5.13–5.07 (m, 4H), 4.76, 4.54 (ABq, J = 11.3 Hz, 2H), 4.74, 4.65 (ABq, J = 11.7 Hz, 1H), 4.45 (s, 2H), 4.08 (dd, J = 11.3, 5.2 Hz, 1H), 3.75–3.71 (m, 1H), 3.65 (t, J = 9.1 Hz, 1H), 3.59–3.56 (m, 1H), 3.50 (dd, J = 10.8, 2.6 Hz, 1H), 3.45 (dd, J = 10.7, 5.8 Hz, 1H), 3.29 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 164.83, 152.41, 142.58, 138.03, 137.58, 137.37, 136.62, 128.57, 128.50, 128.23, 128.14, 128.06, 128.01, 127.94, 127.86, 127.83, 127.58, 127.49, 127.45, 124.67, 109.34, 83.08, 78.18, 78.08, 75.13, 74.94, 73.67, 73.40, 71.24, 69.47, 68.23; HRMS (ESI, m/z): , calcd for : 879.3509; found 879.3511.
1,5-Anhydro-2,3,4-tris-O-benzyl-6-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (20); Compound 11 (0.43 g, 1.0 mmol), compound 16 (0.66 g, 1.5 mmol), 2-chloro-1-methylpyridinium iodide (0.38 g, 1.5 mmol), DMAP (0.18 g, 1.5 mmol), TEA (0.42 mL, 3.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 20 (0.50 g, 58%) was obtained as a colorless amorphous oil. = +27.9 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.18 (m, 32H), 5.14–5.10 (m, 6H), 5.03, 4.88 (ABq, J = 10.7 Hz, 2H), 4,85, 4.53 (ABq, J = 11.8 Hz, 2H), 4.74, 4.67 (ABq, J = 11.7 Hz, 2H), 4.52 (dd, J = 12.0, 2.1 Hz, 1H), 4.37 (dd, J = 12.0, 4.5 Hz, 1H), 4.03 (dd, J = 11.3, 5.2 Hz, 1H), 3.69 (t, J = 8.8 Hz, 1H), 3.66–3.62 (m, 1H), 3.52–3.49 (m, 1H), 3.46 (t, J = 8.9 Hz, 1H), 3.23 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.82, 152.41, 142.46, 138.55, 138.04, 137.69, 137.37, 136.65, 128.59, 128.53, 128.48, 128.46, 128.20, 128.08, 128.01, 127.96, 127.93, 127.85, 127.76, 127.45, 124.91, 109.30, 86.33, 78.48, 77.83, 77.57, 75.73, 75.23, 75.09, 73.29, 71.17, 68.12, 63.86; HRMS (ESI, m/z): , calcd for : 879.3509; found 879.3509.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-O-benzylidene-d-glucitol (21); Compound 1 (380 mg, 1.5 mmol), compound 16 (2.0 g, 4.5 mmol), 2-chloro-1-methylpyridinium iodide (1.2 g, 4.5 mmol), DMAP (0.55 g, 4.5 mmol), TEA (1.3 mL, 9.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 21 (1.6 g, 88%) was obtained as a colorless amorphous oil. = +46.4 (c 1.00, ); NMR ( , 600 MHz) δ 7.44–7.42 (m, 6H), 7.34–7.31 (m, 26H), 7.25–7.22 (m, 4H), 5.80 (t, J = 9.5 Hz, 1H), 5.56 (s, 1H), 5.27–5.23 (m, 1H), 5.11–4.93 (m, 12H), 4.45–4.41 (m, 2H), 3.87 (t, J = 16.0 Hz, 1H), 3.82 (t, J = 17.0 Hz, 1H), 3.65–3.62 (m, 1H), 3.55 (t, J = 10.5 Hz, 1H); NMR (, 150 MHz) δ 165.6, 165.4, 152.6, 142.9, 142.8, 137.4, 136.8, 136.5, 129.1, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.5, 126.2, 109.3, 109.1, 101.6, 78.8, 75.1, 73.0, 71.9, 71.2, 71.2, 71.1, 68.7, 67.7; HRMS (ESI, m/z): , calcd for : 1119.3932; found 1119.3901.
1,5-Anhydro-2,4-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-3,6-bis-O-benzyl-d-glucitol (22); Compound 15 (0.30 mg, 0.87 mmol), compound 16 (1.3 g, 3.0 mmol), 2-chloro-1-methylpyridinium iodide (0.77 g, 3.0 mmol), DMAP (0.37 g, 3.0 mmol), TEA (0.83 mL, 6.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 22 (0.93 g, 77%) was obtained as a colorless amorphous oil. = +7.87 (c 1.00, ); NMR NMR (, 600 MHz) δ 7.58–7.14 (m, 45H), 7.08–6.95 (m, 5H), 5.32–5.25 (m, 2H), 5.19–5.08 (m, 13H), 4.61–4.45 (m, 4H), 4.29 (dd, J = 11.3, 5.5 Hz, 1H), 3.94 (t, J = 9.1 Hz, 1H), 3.72–3.68 (m, 1H), 3.58–3.53 (m, 2H), 3.39 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 164.87, 164.71, 152.61, 152.51, 142.88, 142.79, 137.54, 137.52, 137.32, 137.27, 136.56, 136.49, 128.59, 128.57, 128.55, 128.28, 128.24, 128.22, 128.16, 128.10, 128.05, 128.02, 127.85, 127.76, 127.65, 127.53, 127.49, 124.51, 109.43, 80.72, 78.29, 75.16, 74.16, 73.72, 72.12, 71.35, 71.32, 69.44, 67.03; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4554.
1,5-Anhydro-2,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-3,4-bis-O-benzyl-d-glucitol (23); Compound 10 (0.45 g, 1.3 mmol), compound 16 (1.8 g, 4.0 mmol), 2-chloro-1-methylpyridinium iodide (1.0 g, 4.0 mmol), DMAP (0.49 g, 4.0 mmol), TEA (1.1 mL, 8.0 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 23 (1.4 g, 92%) was obtained as a colorless amorphous oil. = +51.2 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.18 (m, 44H), 5.27–5.21 (m, 1H), 5.17–5.07 (m, 12H), 4.82, 4.52 (ABq, J = 11.0 Hz, 2H), 4.77, 4.70 (ABq, J = 11.3 Hz, 2H), 4.56 (m, 1H), 4.42–4.39 (m, 1H), 4.19 (dd, J = 11.2, 5.7 Hz, 1H), 3.85–3.82 (m, 1H), 3.60–3.56 (m, 2H), 3.31 (t, J = 10.7 Hz, 1H); NMR (, 150 MHz) δ 165.80, 165.05, 152.56, 152.43, 142.76, 142.47, 137.88, 137.49, 137.37, 137.28, 136.66, 136.53, 128.62, 128.60, 128.57, 128.51, 128.39, 128.21, 128.12, 128.06, 128.01, 127.88, 127.80, 127.40, 127.37, 124.80, 124.49, 109.32, 109.25, 84.32, 77.91, 77.42, 75.45, 75.26, 75.13, 75.09, 72.22, 71.26, 71.15, 67.11, 63.52; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4550.
1,5-Anhydro-2,6-bis-O-benzyl-3,4-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (24); Compound 14 (380 mg, 1.1 mmol), compound 16 (1.5 g, 3.3 mmol), 2-chloro-1-methylpyridinium iodide (0.84 g, 3.3 mmol), DMAP (0.91 g, 3.3 mmol), TEA (0.91 mL, 6.6 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 24 (1.2 g, 92%) was obtained as a colorless amorphous oil. = −80.8 (c 1.00, ); NMR (, 600 MHz) δ 7.41–7.14 (m, 44H), 5.58 (t, J = 9.5 Hz, 1H), 5.31 (t, J = 9.8 Hz, 1H), 5.09–5.00 (m, 12H), 4.54, 4.48 (ABq, J = 12.2 Hz, 2H), 4.52, 4.47 (ABq, J = 12.0 Hz, 2H), 4.19 (dd, J = 11.5, 5.3 Hz, 1H), 3.79 (td, J = 9.9, 5.2 Hz, 1H), 3.73 (m, 1H), 3.59 (dd, J = 10.7, 2.4 Hz, 1H), 3.51 (dd, J = 10.8, 5.3 Hz, 1H), 3.46 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.62, 165.18, 152.43, 142.67, 142.53, 137.56, 137.48, 137.43, 137.39, 136.57, 136.56, 128.49, 128.41, 128.38, 128.36, 128.27, 128.16, 127.98, 127.89, 127.87, 127.84, 127.65, 127.55, 124.67, 124.20, 109.15, 77.99, 76.35, 75.22, 75.10, 75.08, 73.70, 72.96, 71.12, 70.07, 69.06, 68.30; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4558.
1,5-Anhydro-2,4-bis-O-benzyl-3,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (25); Compound 9 (0.52 mg, 1.5 mmol), compound 16 (2.0 g, 4.5 mmol), 2-chloro-1-methylpyridinium iodide (1.2 g, 4.5 mmol), DMAP (0.55 g, 4.5 mmol), TEA (1.2 mL, 9.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 25 (1.6 g, 91%) was obtained as a colorless amorphous oil. = +16.1 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.24 (m, 34H), 7.18–7.05 (m, 10H), 5.50 (t, J = 9.3 Hz, 1H), 5.19–5.08 (m, 12H), 4.56, 4.43 (ABq, J = 12.4 Hz, 2H), 4.55 (dd, J = 12.0 Hz, 2.0 Hz, 1H), 4.45 (dd, J = 12.0, 5.5 Hz, 1H) 4.41 (s, 2H), 4.11 (dd, J = 11.3, 5.2 Hz, 1H), 3.67–3.62 (m, 2H), 3.54 (t, J = 9.5 Hz, 1H), 3.36 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.22, 152.49, 142.66, 137.65, 137.31, 136.99, 136.59, 128.56, 128.36, 128.22, 128.16, 128.05, 127.94, 127.83, 127.52, 127.47, 124.96, 124.88, 109.54, 109.51, 78.19, 77.87, 76.42, 75.26, 75.13, 74.68, 72.63, 71.32, 67.97, 63.91; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4557.
1,5-Anhydro-2,3-bis-O-benzyl-4,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (26); Compound 5 (0.34 g, 1.0 mmol), compound 16 (1.3 g, 3.0 mmol), 2-chloro-1-methylpyridinium iodide (0.77 g, 3.0 mmol), DMAP (0.37 g, 3.0 mmol), TEA (0.83 mL, 6.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 26 (1.0 g, 87%) was obtained as a colorless amorphous oil. = +13.7 (c 1.00, ); NMR (, 600 MHz) δ 7.45–7.21 (m, 39H), 7.12–7.03 (m, 5H), 5.32 (t, J = 9.5 Hz, 1H), 5.15–5.08 (m, 8H), 5.03–5.01 (m, 4H), 4.79, 4.58 (ABq, J = 11.5, 2H), 4.76, 4.66 (ABq, J = 11.5 Hz, 2H), 4.63 (dd, J = 12.0, 2.8 Hz, 1H), 4.12 (dd, J = 12.2, 5.3 Hz, 1H), 4.07 (dd, J = 11.5, 5.0 Hz, 1H), 3.70–3.68 (m, 2H), 3.70 (t, J = 8.9 Hz, 1H), 3.30 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.80, 164.76, 152.48, 152.44, 142.77, 142.42, 137.98, 137.91, 137.47, 137.37, 136.73, 136.56, 128.53, 128.50, 128.47, 128.20, 128.17, 128.07, 128.03, 128.01, 127.94, 127.84, 127.60, 127.53, 124.75, 124.54, 109.40, 109.14, 82.95, 78.22, 76.56, 75.15, 75.08, 73.47, 71.24, 71.06, 70.96, 68.32, 63.44; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4553.
1,5-Anhydro-2,3,4-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-6-O-benzyl-d-glucitol (27); Compound 12 (180 mg, 0.70 mmol), compound 16 (1.4 g, 3.2 mmol), 2-chloro-1-methylpyridinium iodide (0.82 g, 3.2 mmol), DMAP (0.39 g, 3.2 mmol), TEA (0.89 mL, 6.4 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 27 (0.94 g, 91%) was obtained as a colorless amorphous oil. = −4.9 (c 0.65, ); NMR (, 600 MHz) δ 7.43–7.16 (m, 56H), 5.82 (t, J = 9.6 Hz, 1H), 5.55 (t, J = 10.0 Hz, 1H), 5.29 (td, J = 10.0, 5.5 Hz, 1H), 5.13–4.96 (m, 14H), 4.90 (s, 4H), 4.58, 4.53 (ABq, J = 12.0, 2H), (dd, J = 10.2, 5.6 Hz, 1H), 3.88–3.84 (m, 1H), 3.67 (dd, J = 11.0, 2.4 Hz, 1H), 3.61 (dd, J = 10.8, 5.3 Hz, 1H), 3.57 (t, J = 10.8 Hz, 1H) NMR (, 150 MHz) δ 165.92, 165.17, 165.02, 152.54, 152.50, 142.88, 142.82, 142.71, 137.44, 137.34, 136.49, 136.45, 136.36, 128.55, 128.51, 128.39, 128.32, 128.27, 128.17, 128.15, 128.10, 128.06, 128.02, 127.95, 127.92, 127.89, 127.81, 127.72, 127.56, 127.52, 124.08, 124.03, 109.20, 109.11, 109.02, 78.36, 75.12, 75.09, 75.07, 74.65, 73.77, 71.18, 71.10, 71.02, 70.75, 69.71, 69.00, 67.26; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5601.
1,5-Anhydro-2,3,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-4-O-benzyl-d-glucitol (28); Compound 8 (0.38 mg, 1.5 mmol), compound 16 (3.0 g, mmol), 2-chloro-1-methylpyridinium iodide (1.8 g, 7.0 mmol), DMAP (0.12 g, 1.0 mmol), TEA (1.9 mL, 14 mmol) in 30 mL of DCM was stirred at rt for 1 d. By the same procedure previously described for the preparation of compound 17, compound 28 (1.0 g, 45% yield) was obtained as a colorless amorphous oil. = +47.2 (c 0.40, ); NMR (, 600 MHz) δ 7.47–7.19 (m, 51H), 7.11–7.05 (m, 5H), 5.75 (t, J = 9.1 Hz, 1H), 5.19–5.03 (m, 15H), 5.00, 4.92 (ABq, J = 11.7 Hz, 4H), 4.60–4.53 (m, 2H), 4.45 (s, 2H), 4.41 (dd, J = 11.3, 5.5 Hz, 1H), 3.75–3.70 (m, 2H), 3.46 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.51, 165.41, 152.59, 152.54, 142.94, 142.75, 142.65, 137.35, 137.32, 137.27, 136.86, 136.62, 136.51, 136.39, 128.60, 128.54, 128.46, 128.44, 128.38, 128.22, 128.18, 128.14, 128.08, 128.06, 128.01, 127.97, 127.90, 127.61, 127.50, 127.43, 124.81, 124.40, 124.05, 109.54, 109.29, 109.03, 78.11, 76.54, 76.04, 75.14, 75.11, 75.09, 74.96, 71.34, 71.20, 71.06, 70.81, 67.07, 63.58; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5604.
1,5-Anhydro-2,4,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-3-O-benzyl-d-glucitol (29); Compound 7 (0.33 mg, 1.3 mmol), compound 16 (2.6 g, 6.0 mmol), 2-chloro-1-methylpyridinium iodide (1.5 g, 6.0 mmol), DMAP (0.73 g, 6.0 mmol), TEA (1.7 mL, 12 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 29 (1.3 g, 65% yield) was obtained as a colorless amorphous oil. = +26.7 (c 0.95, ); NMR (, 600 MHz) δ 7.46–7.21 (m, 49H), 7.07–6.96 (m, 5H), 5.48 (t, J = 9.5 Hz, 1H), 5.34–5.29 (m, 1H), 5.17–5.07 (m, 14H), 5.04 (s, 4H), 4.68 (dd, J = 12.0, 3.1 Hz, 1H), 4.62, 4.54 (ABq, J = 11.7 Hz, 2H), 4.30 (dd, J = 11.2, 5.7 Hz, 1H), 4.22 (dd, J = 12.2, 5.0 Hz, 1H), 3.99 (t, J = 9.1 Hz, 1H), 3.90–3.87 (m, 1H), 3.42 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.81, 164.83, 164.61, 152.63, 152.55, 152.46, 142.94, 142.49, 137.46, 137.41, 137.33, 137.24, 136.67, 136.50, 136.46, 128.57, 128.53, 128.51, 128.46, 128.21, 128.19, 128.14, 128.10, 128.06, 128.02, 127.95, 127.85, 127.80, 127.62, 127.56, 127.47, 124.64, 124.41, 124.38, 109.47, 109.44, 109.13, 80.62, 76.53, 75.16, 75.08, 74.34, 72.02, 71.35, 71.30, 71.05, 70.98, 67.05, 63.33; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5599.
1,5-Anhydro-2-O-benzyl-3,4,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (30); Compound 6 (0.38 g, 1.5 mmol), compound 16 (3.0 g, 6.8 mmol), 2-chloro-1-methylpyridinium iodide (1.7 g, 6.8 mmol), DMAP (0.83 g, 6.8 mmol), TEA (2.0 mL, 14 mmol) in 30 mL of DCM was stirred at rt for 2 days. By the same procedure previously described for the preparation of compound 17, compound 30 (2.0 g, 89% yield) was obtained as a colorless amorphous oil. = −36.2 (c 1.00, ); NMR (, 600 MHz) δ 7.42–7.14 (m, 54H), 5.67 (t, J = 9.6 Hz, 1H), 5.41 (t, J = 9.8 Hz, 1H), 5.13–4.94 (m, 18H), 4.69 (dd, J = 12.2, 2.9 Hz, 1H), 4.59, 4.51 (ABq, J = 12.4 Hz, 2H), 4.23 (dd, J = 12.4, 5.5 Hz, 1H), 4.19 (dd, J = 11.5, 5.3 Hz, 1H) 3.92 (m, 1H), 3.83 (td, J = 9.8, 5.3 Hz, 1H), 3.48 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.68, 165.65, 165.29, 152.51, 152.48, 152.40, 142.87, 142.66, 142.53, 137.54, 137.43, 137.38, 136.63, 136.46, 128.51, 128.46, 128.43, 128.38, 128.29, 128.15, 128.11, 127.98, 127.92, 127.89, 127.87, 127.80, 127.78, 127.57, 127.52, 124.65, 124.53, 123.97, 109.18, 76.69, 76.16, 75.17, 75.08, 72.95, 71.13, 71.03, 70.20, 68.38, 63.54; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5607.
1,5-Anhydro-2-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
31) [
31];
on C (20 wt.%, 20 mg) was added to a solution of
17 (270 mg, 0.35 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. After the replaced argon atmosphere to hydrogen gas, the suspension was stirred at rt for 6 h. The reaction mixture was filtered and concentrated under reduced pressure to obtain purple amorphous oil. The purple amorphous oil was dissolved by 2 mL acetone and filtered through a whatman™ puradisc 0.1 μM TF and concentrated under reduced pressure. In addition, the purple amorphous oil dissolved with 2 mL of MeOH and added acidic resin until becoming a clear solution. After the filtered through the whatman™ puradisc 0.1 μm TF, the solution was concentrated under reduced pressure to give
31 (106 mg, 96%) as pale yellow amorphous oil.
= +58.5 (c 0.70, MeOH);
NMR (
, 600 MHz) δ 7.08 (d,
J = 5.5 Hz, 2H), 4.87–4.83 (overlap, 1H), 4.08 (dd,
J = 10.5, 5.5 Hz, 1H), 3.86 (dd,
J = 11.9, 2.2 Hz, 1H), 3.66–3.63 (m, 2H), 3.36 (t,
J = 9.5 Hz, 1H), 3.29 (t,
J = 10.7 Hz, 1H), 3.24 (ddd,
J = 9.7, 5.9, 2.3 Hz, 1H).
NMR (
, 150 MHz) δ 167.80, 146.42, 139.92, 121.14, 110.25, 82.56, 77.06, 73.28, 71.97, 67.84, 62.93; HRMS (
,
m/
z):
, calcd for
: 315.0716; found 315.0718. Please find
NMR,
NMR spectra of compounds
31–
44,
54–
58,
61 and
67 in the
Supplementary Materials.
1,5-Anhydro-3-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
32) [
29];
on C (20 wt.%, 20 mg) was added to a solution of compound
18 (270 mg, 0.35 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
32 (110 mg, quant.) was obtained as a colorless amorphous oil.
= +24.8 (c 1.0 MeOH);
NMR
, 600 MHz) δ 7.13 (s, 2H), 5.04 (t,
J = 9.3 Hz, 1H), 3.97 (dd,
J = 11.3, 5.5 Hz, 1H), 3.85 (dd,
J = 11.9, 2.2 Hz, 1H), 3.73–3.69 (m, 1H), 3.66 (dd,
J = 12.0, 5.8 Hz, 1H), 3.51 (t,
J = 9.5 Hz, 1H), 3.31–3.27 (overlap, 2H).
NMR (
, 150 MHz) δ 168.50, 146.39, 139.64, 121.88, 110.34, 82.51, 81.20, 70.95, 69.98, 69.89, 62.75; HRMS (
,
m/
z):
, calcd for
: 315.0716; found 315.0721.
1,5-Anhydro-4-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
33) [
29];
on C (20 wt.%, 20 mg) was added to a solution of compound
19 (280 mg, 0.33 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
33 (100 mg, 96%) was obtained as a colorless amorphous oil.
= −5.7 (c 0.90, MeOH);
NMR (
600 MHz) δ 7.14 (s, 2H), 4.88 (t,
J = 9.3 Hz, 1H), 3.94 (dd,
J = 11.2, 5.3 Hz, 1H), 3.68 (t,
J = 8.9 Hz, 1H), 3.64–3.60 (m, 1H), 3.57 (dd,
J = 12.0, 2.1 Hz, 1H), 3.51–3.44 (m, 2H), 3.25 (t,
J = 10.8 Hz, 1H);
δ 166.59, 145.90, 138.86, 121.46, 110.07, 80.56, 77.44, 72.70, 71.38, 70.45, 62.54; HRMS (
,
m/
z):
, calcd for
: 315.0716; found 315.0718.
1,5-Anhydro-6-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
34) [
31];
on C (20 wt.%, 20 mg) was added to a solution of compound
20 (210 mg, 0.25 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
55 (74 mg, 96%) was obtained as a colorless amorphous oil.
= +30.3 (c 1.00, MeOH);
NMR (
, 600 MHz) δ 7.09 (s, 2H), 4.54 (dd,
J = 11.9, 1.9 Hz, 1H), 4.35 (dd,
J = 12.0, 5.5 Hz, 1H), 3.93 (dd,
J = 11.2, 5.3 Hz, 1H), 3.51–3.55 (m, 1H), 3.48–3.45 (m, 1H), 3.40 (t,
J = 8.6 Hz, 1H), 3.37 (t,
J = 8.6 Hz, 1H), 3.23 (t,
J = 10.8 Hz, 1H).
NMR (
, 150 MHz) δ 168.32, 146.36, 139.71, 121.26, 110.09, 79.91, 79.63, 71.59, 71.20, 70.86, 65.02; HRMS (
,
m/
z):
, calcd for
: 315.0716; found 315.0718.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
35) [
29];
on C (20 wt.%, 50 mg) was added to a solution of compound
21 (210 mg, 0.25 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
35 (313 mg, quant.) was obtained as a colorless amorphous oil.
= +139.7 (c 1.00, MeOH);
NMR (
, 600 MHz) δ 7.05 (s, 2H), 6.96 (s, 2H), 5.41 (t,
J = 9.5 Hz, 1H), 5.08 (td,
J = 10.1, 5.3 Hz, 1H), 4.20 (dd,
J = 11.0, 5.5 Hz, 1H), 3.90 (dd,
J = 11.9, 2.2 Hz, 1H), 3.74–3.69 (m, 2H), 3.45 (t,
J = 10.8 Hz, 1H), 3.40 (ddd,
J = 9.6, 5.5, 2.1 Hz, 1H);
NMR (
, 150 MHz) δ 168.18, 167.38, 146.38, 146.34, 140.08, 139.85, 121.29, 120.55, 110.30, 110.25, 82.65, 77.91, 71.39, 69.83, 67.81, 62.57; HRMS (
,
m/
z):
, calcd for
: 467.0826; found 467.0831.
1,5-Anhydro-2,4-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
36) [
29];
on C (20 wt.%, 50 mg) was added to a solution of compound
22 (680 mg, 0.57 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
36 (267 mg, quant.) was obtained as a colorless amorphous oil.
= +11.3 (c 0.70, MeOH);
NMR (
600 MHz) δ 7.17 (s, 2H), 7.14 (dd,
J = 9.8, 4.0 Hz, 2H), 5.04 (t,
J = 9.5 Hz, 1H), 5.01–4.97 (m, 1H), 4.15–4.11 (m, 2H), 3.63–3.54 (m, 3H), 3.41 (t,
J = 10.8 Hz, 1H);
δ 166.39, 166.21, 146.00, 145.98, 138.99, 138.94, 121.43, 121.38, 110.15, 110.09, 80.72, 74.30, 73.19, 72.73, 67.38, 62.49; HRMS (
,
m/
z):
, calcd for
: 467.0826; found 467.0827.
1,5-Anhydro-2,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
37) [
26];
on C (20 wt.%, 50 mg) was added to a solution of compound
23 (440 mg, 0.37 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
37 (170 mg, quant.) was obtained as a yellow amorphous oil.
= +19.4 (c 1.00 in MeOH);
NMR (
600 MHz) δ 7.16 (s, 2H), 7.14 (s, 2H), 4.92–4.88 (m, 1H), 4.57 (d,
J = 10.7 Hz, 1H), 4.41–4.35 (m, 1H), 4.07 (dd,
J = 10.8, 5.3 Hz, 1H), 3.83–3.78 (m, 1H), 3.61–3.57 (m, 2H), 3.37 (t,
J = 10.7 Hz, 1H);
δ 166.63, 166.30, 146.02, 145.96, 138.89, 138.77, 121.74, 121.46, 110.07, 109.85, 79.47, 76.57, 72.94, 71.60, 67.45, 64.54; HRMS (
,
m/
z):
, calcd for
: 467.0826; found 467.0831.
1,5-Anhydro-3,4-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (38); on C (20 wt.%, 50 mg) was added to a solution of compound 24 (710 mg, 0.60 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 38 (280 mg, quant.) was obtained as a yellow amorphous oil. = −78.2 (c 1.00, MeOH); NMR (600 MHz) δ 7.06 (s, 2H), 7.04 (s, 2H), 5.40 (t, J = 9.5 Hz, 1H), 5.16 (t, J = 9.6 Hz, 1H), 4.06 (dd, J = 11.3, 5.5 Hz, 1H), 3.98–3.93 (m, 1H), 3.71–3.64 (m, 2H), 3.58 (dd, J = 12.5, 5.7 Hz, 1H), 3.45 (t, J = 10.8 Hz, 1H); δ 166.41, 166.12, 145.83, 145.72, 139.04, 138.66, 121.52, 120.78, 110.05, 110.01, 80.29, 78.04, 70.50, 70.17, 69.50, 62.25; HRMS (, m/z): , calcd for : 467.0826; found 467.0829.
1,5-Anhydro-3,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
39) [
35];
on C (20 wt.%, 50 mg) was added to a solution of compound
25 (800 mg, 0.67 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
39 (310 mg, quant.) was obtained as a yellow amorphous oil.
= +28.7 (c 0.60, MeOH);
NMR (
600 MHz) δ 7.17 (s, 2H), 7.16 (s, 2H), 5.11 (t,
J = 9.1 Hz, 1H), 4.57 (dd,
J = 11.9, 1.9 Hz, 1H), 4.40 (dd,
J = 11.9, 5.3 Hz, 1H), 3.99 (dd,
J = 11.0, 5.5 Hz, 1H), 3.84–3.79 (m, 1H), 3.72 (t,
J = 9.5 Hz, 1H), 3.65–3.63 (m, 1H), 3.38 (t,
J = 10.8 Hz, 1H);
δ 166.90, 166.63, 145.97, 145.82, 138.71, 138.52, 122.16, 121.76, 110.10, 109.85, 81.10, 79.61, 70.73, 69.66, 69.46, 64.54; HRMS (
,
m/
z):
, calcd for
: 467.0826; found 467.0828.
1,5-Anhydro-4,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
40) [
35];
on C (20 wt.%, 50 mg) was added to a solution of compound
26 (430 mg, 0.36 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
40 (160 mg, 95%) was obtained as a yellow amorphous oil.
= +41.7 (c 0.60, MeOH);
NMR (
600 MHz) δ 7.15 (s, 2H), 7.14 (s, 2H), 5.09 (t,
J = 9.5 Hz, 1H), 4.41 (dd,
J = 12.0, 2.1 Hz, 1H), 4.13 (dd,
J = 12.0, 5.8 Hz, 1H), 3.97 (dd,
J = 11.2, 5.3 Hz, 1H), 3.82–3.79 (m, 1H), 3.75 (t,
J = 9.1 Hz, 1H), 3.71–3.66 (m, 1H), 3.33 (t,
J = 10.8 Hz, 1H);
δ 166.47, 166.13, 145.92, 138.90, 138.77, 121.53, 121.38, 110.12, 109.91, 77.56, 77.42, 72.24, 71.28, 70.51, 69.79, 63.98, 55.32; HRMS (
,
m/
z):
, calcd for
: 467.0826; found 467.0829.
1,5-Anhydro-2,3,4-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
41) [
35];
on C (20 wt.%, 50 mg) was added to a solution of compound
27 (520 mg, 0.34 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
41 (210 mg, quant.) was obtained as a yellow amorphous oil.
= −4.8 (c 1.00, MeOH);
NMR (
600 MHz) δ 7.07 (d,
J = 3.8 Hz, 4H), 7.01 (d,
J = 3.8 Hz, 2H), 5.77 (t,
J = 9.6 Hz, 1H), 5.36 (t,
J = 9.8 Hz, 1H), 5.23 (td,
J = 10.1, 5.4 Hz, 1H), 4.28 (dd,
J = 11.2, 5.7 Hz, 1H), 3.83–3.80 (m, 1H), 3.72 (dd,
J = 12.4, 2.1 Hz, 1H), 3.62–3.65 (m, 2H);
δ 166.12, 165.91, 165.89, 145.80, 145.65, 139.00, 138.76, 120.93, 120.70, 120.62, 110.03, 109.96, 109.91, 80.37, 74.46, 70.83, 69.96, 67.27, 61.99; HRMS (
,
m/
z):
, calcd for
: 619.0935; found 619.0934.
1,5-Anhydro-2,3,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
42) [
30];
on C (20 wt.%, 50 mg) was added to a solution of compound
28 (410 mg, 0.27 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
42 (170 mg, quant.) was obtained as a yellow amorphous oil.
= +80.9 (c 0.70, MeOH);
NMR (
600 MHz) δ 7.19 (s, 2H), 7.10 (s, 2H), 7.04 (s, 2H), 5.50 (t,
J = 9.5 Hz, 1H), 5.13–5.09 (m, 1H), 4.60 (dd,
J = 11.9, 1.9 Hz, 1H), 4.47 (dd,
J = 12.0, 4.8 Hz, 1H), 4.20 (dd,
J = 11.0, 5.5 Hz, 1H), 3.93 (t,
J = 9.5 Hz, 1H), 3.79–3.77 (m, 1H), 3.56 (t,
J = 10.8 Hz, 1H)
δ 166.60, 166.39, 166.03, 146.05, 145.96, 145.87, 139.15, 138.86, 138.81, 121.64, 121.56, 120.75, 110.06, 110.00, 109.89, 79.67, 77.07, 70.84, 69.60, 67.44, 64.23; HRMS (
,
m/
z):
, calcd for
: 619.0935; found 619.0931.
1,5-Anhydro-2,4,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
43) [
29];
on C (20 wt.%, 50 mg) was added to a solution of compound
29 (480 mg, 0.32 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. B By the same procedure previously described for the preparation of compound
31, desired compound
37 (200 mg, quant.) was obtained as a yellow amorphous oil.
= +36.3 (c 1.00, MeOH);
NMR (
600 MHz) δ 7.17 (s, 2H), 7.16 (s, 2H), 7.15 (s, 2H), 5.25 (dd,
J = 10.0, 9.3 Hz, 1H), 5.07–5.03 (m, 1H), 4.46 (dd,
J = 12.2, 1.9 Hz, 1H), 4.20–4.16 (m, 3H), 3.95–3.92 (m, 1H), 3.50 (t,
J = 10.8 Hz, 1H);
δ 166.44, 166.19, 165.95, 145.98, 138.98, 138.83, 121.56, 121.35, 121.31, 110.20, 110.11, 109.96, 77.63, 74.27, 72.96, 72.22, 67.45, 63.76; HRMS (
,
m/
z):
, calcd for
: 619.0935; found 619.0934.
1,5-Anhydro-3,4,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (
44) [
35];
on C (20 wt.%, 50 mg) was added to a solution of compound
30 (765 mg, 0.50 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
37 (310 mg, quant.) was obtained as a colorless amorphous oil.
= −6.12 (c 0.80, MeOH);
NMR (
600 MHz) δ 7.20 (s, 2H), 7.07 (s, 2H), 7.06 (s, 2H), 5.46 (t,
J = 9.5 Hz, 1H), 5.34 (t,
J = 9.8 Hz, 1H), 4.47 (dd,
J = 12.4, 2.1 Hz, 1H), 4.27 (dd,
J = 12.2, 5.3 Hz, 1H), 4.12 (dd,
J = 11.3, 5.5 Hz, 1H), 4.04–4.00 (m, 2H), 3.55 (t,
J = 10.8 Hz, 1H)
δ 166.39, 166.35, 165.72, 145.83, 145.73, 145.66, 138.98, 138.74, 138.64, 121.44, 121.38, 120.67, 110.05, 109.97, 109.94, 77.86, 77.41, 70.54, 69.73, 69.41, 63.52; HRMS (
,
m/
z):
, calcd for
: 619.0935; found 619.0941.
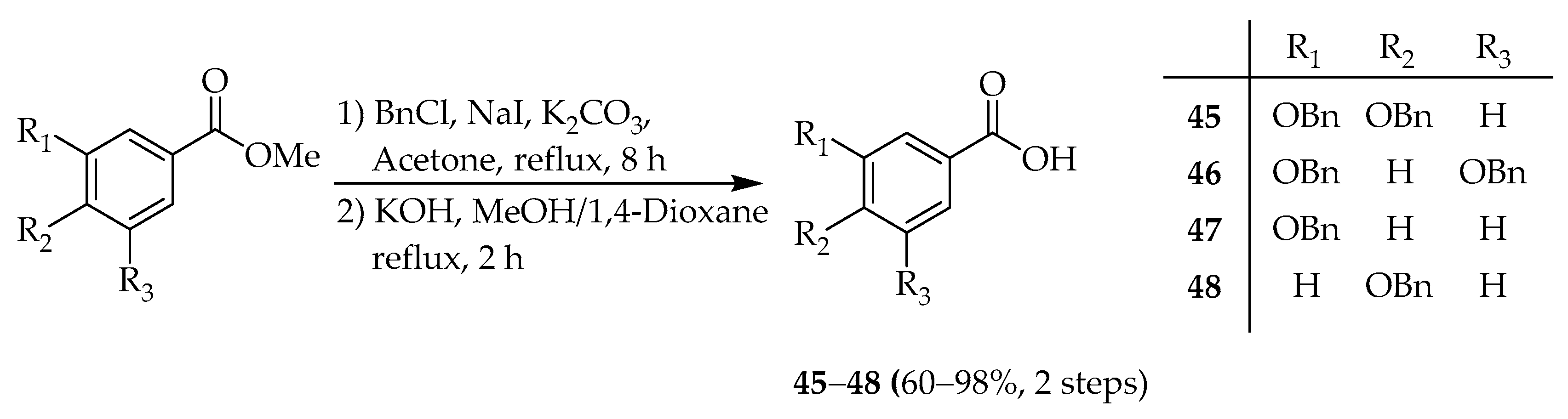
3,4-bis(Benzyloxy)benzoic acid (
45) [
51]; Methyl 3,4-dihydroxybenzoate (2.5 g, 15 mmol) and
(8.2 g, 60 mmol) and KI (2.0 g, 12 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (4.2 mL, 36 mmol) and refluxed for 7 h. TLC indicated full conversion of the start material, added MeOH and stirred 1 h. The reaction mixture was filtered by celite and filtrate was evaporated under reduced pressure. The residue was purified by recrystallization with hexane, and the mother liquid was purified by C.C (Hex/EtOAc = 100/1–4/1) to afford methyl ester of
45 (total 5.0 g, 97%) as a white solid. Further on, methyl ester of
45 (3.0 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. TLC indicated full conversion of the start material, the reaction mixture was cooled to 0 °C and slowly added 6 M HCl until pH = 1. The precipitating muddy suspension was filtrated, and the white residue was washed by water and MeOH until pH = 7. The white solid was dried
in vacuo, purified by recrystallizing with MeOH to obtain desired compound
45 (2.4 g, 79%) as colorless needles. m.p. 211 °C;
NMR (DMSO-
d6, 600 MHz) δ 7.56–7.15 (m, 13H), 5.22 (s, 2H), 5.18 (s, 2H);
NMR (
, 150 MHz) δ 166.88, 151.94, 147.50, 136.93, 136.62, 128.36, 128.30, 127.82, 127.71, 127.46, 127.34, 123.36, 123.22, 114.43, 112.97, 69.87, 69.73.
3,5-bis(Benzyloxy)benzoic acid (
46) [
53]; Methyl 3,5-dihydroxybenzoate (2.5 g, 15 mmol) and
(8.2 g, 60 mmol) and KI (2.0 g, 12 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (4.2 mL, 36 mmol) and refluxed for 9 h. By the same procedure previously described for the preparation of compound
45, methyl ester of
46 (5.2 g, quant.) was obtained as a white solid. Further on, methyl ester of
46 (3.0 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound
45, desire compound
46 (2.5 g, 81%) was obtained as colorless needles. m.p. 185 °C;
NMR (
, 600 MHz) δ 7.46–7.30 (m, 11H), 7.19–7.16 (m, 2H), 6.93–6.93 (m, 1H), 5.15 (s, 4H);
NMR (
, 150 MHz) δ 166.80, 159.30, 136.64, 132.80, 128.36, 127.79, 127.58, 107.87, 106.43, 69.38.
3-(Benzyloxy)benzoic acid (
47) [
52]; Methyl 3-hydroxybenzoate (2.3 g, 15 mmol) and
(4.2 g, 30 mmol) and KI (1.0 g, 6.0 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (2.1 mL, 18 mmol) and refluxed for 10 h. By the same procedure previously described for the preparation of compound
45, methyl ester of
47 (3.7 g, 95%) was obtained as a white solid. Further on, methyl ester of
47 (1.9 g, 7.4 mmol), KOH (4.2 g, 74 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound procedure described for previously
45 preparation, desire compound
47 (1.4 g, 85%) was obtained as colorless needles. m.p. 136 °C;
NMR (600 MHz, DMSO-
d6) δ 7.57–7.53 (m, 2H), 7.48–7.39 (m, 5H), 7.35–7.32 (m, 1H), 7.28–7.26 (m, 1H), 5.17 (s, 2H);
-NMR (150 MHz, DMSO-
d6) δ 167.12, 158.34, 136.84, 132.23, 129.79, 128.50, 127.92, 127.70, 121.82, 119.77, 114.89, 69.35.
4-(Benzyloxy)benzoic acid (
48) [
51]; Methyl 4-hydroxybenzoate (2.3 g, 15 mmol) and
(4.2 g, 30 mmol) and KI (1.0 g, 6.0 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (2.1 mL, 18 mmol) and refluxed for 9 h. By the same procedure previously described for the preparation of compound
45, methyl ester of
48 (3.6 g, quant.) was obtained as a white solid. Further on, methyl ester of
48 (2.3 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound procedure described for previously
45 preparation, desired compound
48 (1.4 g, 85%) was obtained as colorless needles. m.p. 192 °C;
NMR (600 MHz, DMSO-
d6) δ 7.95–7.90 (m, 2H), 7.48–7.46 (m, 2H), 7.42–7.40 (m, 2H), 7.36–7.34 (m, 1H), 7.12–7.09 (m, 2H), 5.19 (s, 2H);
-NMR (150 MHz, DMSO-
d6) δ 166.90, 161.83, 136.43, 131.27, 128.40, 127.92, 127.73, 123.09, 114.50, 69.35.
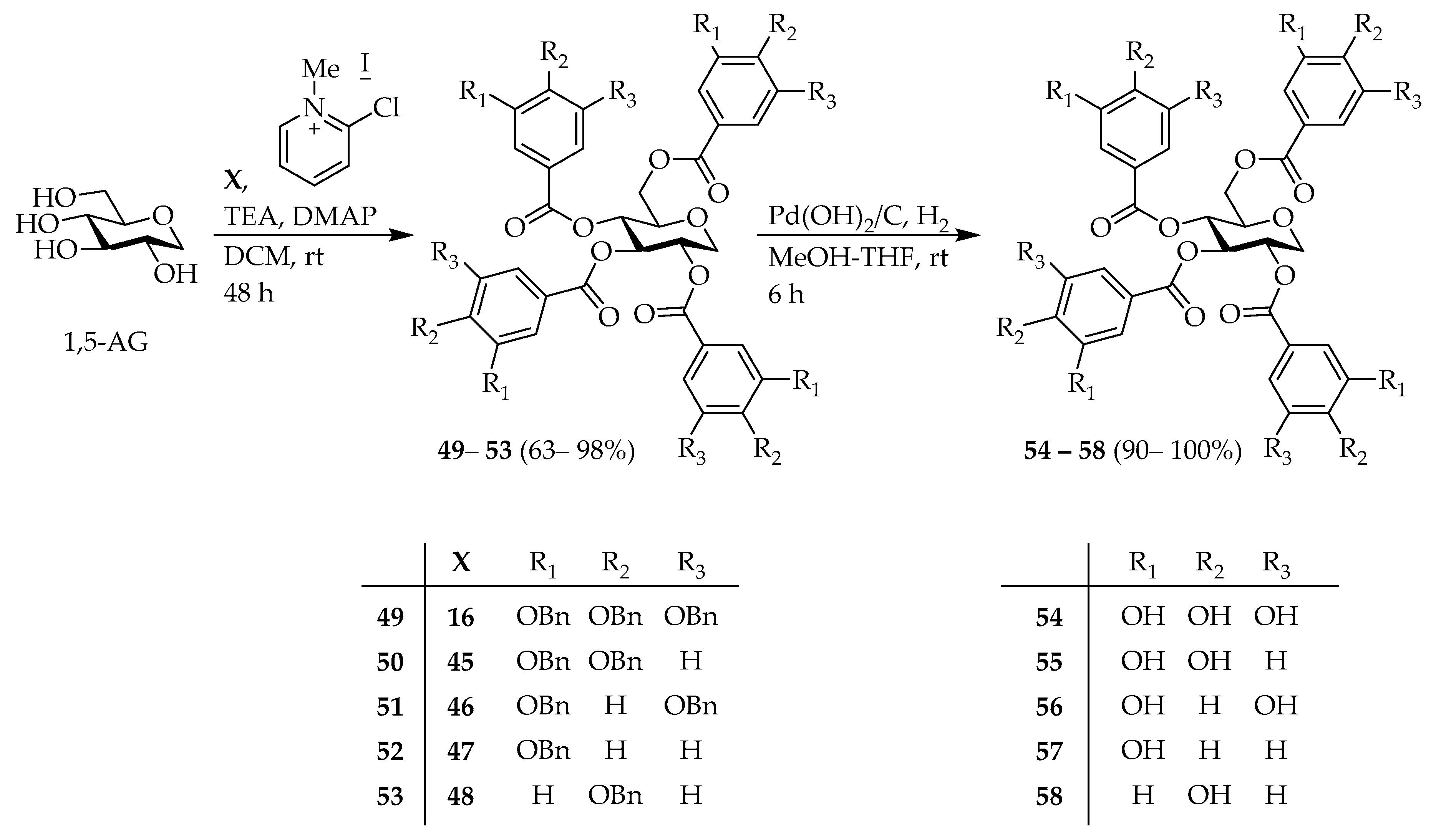
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (
49) [
50]; 1,5-AG (82 mg, 0.50 mmol), compound
16 (1.1 g, 2.4 mmol), 2-chloro-1-methylpyridinium iodide (0.61 g, 2.4 mmol), DMAP (0.29 g, 2.4 mmol), TEA (0.67 mL, 4.8 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound
17, desire compound
49 (0.86 g, 92%) was obtained as a colorless amorphous oil.
= +7.0 (c 1.00,
);
NMR (600 MHz, CHLOROFORM-D) δ 7.56–7.07 (m, 68H), 5.89 (t,
J = 9.8 Hz, 1H), 5.63 (t,
J = 9.8 Hz, 1H), 5.35–5.31 (m, 1H), 5.18–4.94 (m, 22H), 4.88–4.84 (m, 4H), 4.76 (dd,
J = 12.2, 2.9 Hz, 1H), 4.52 (dd,
J = 11.3, 5.5 Hz, 1H), 4.30 (dd,
J = 12.4, 5.2 Hz, 1H), 4.06–4.03 (m, 1H), 3.60 (t,
J = 10.8 Hz, 1H);
NMR (
, 150 MHz) δ 165.91, 165.71, 165.14, 165.10, 152.54, 152.43, 143.06, 142.93, 142.79, 142.62, 137.41, 137.31, 136.59, 136.44, 136.35, 136.25, 128.54, 128.45, 128.44, 128.37, 128.27, 128.23, 128.14, 128.11, 128.08, 128.02, 128.00, 127.95, 127.90, 127.87, 127.81, 127.55, 127.52, 124.56, 123.95, 123.79, 109.22, 109.14, 109.03, 75.10, 75.06, 74.53, 71.18, 71.11, 71.05, 70.51, 69.78, 67.31, 63.36 HRMS (ESI,
m/z):
, calcd for
: 1875.6644; found 1875.6653.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′-dibenzyloxybenzoyl)-d-glucitol (50); 1,5-AG (0.12 g, 0.7 mmol), compound 45 (1.4 g, 4.2 mmol), 2-chloro-1-methylpyridinium iodide (1.1 g, 4.2 mmol), DMAP (0.52 g, 4.2 mmol), TEA (1.1 mL, 8.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desired compound 50 (0.61 g, 72%) was obtained as a colorless amorphous oil. = +18.8 (c 0.90, ); NMR (, 600 MHz) δ 7.65–7.23 (m, 48H), 6.89–6.86 (m, 2H), 6.82–6.80 (m, 1H), 6.77–6.75 (m, 1H), 5.81 (t, J = 9.8 Hz, 1H), 5.56 (t, J = 9.6 Hz, 1H), 5.29 (td, J = 10.1, 5.5 Hz, 1H), 5.22–5.03 (m, 14H), 4.99 (s, 2H), 4.63 (dd, J = 12.4, 2.7 Hz, 1H), 4.43 (dd, J = 11.3, 5.5 Hz, 1H), 4.29 (dd, J = 12.4, 5.2 Hz, 1H), 3.97–3.94 (m, 1H), 3.53 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.83, 165.69, 165.17, 164.91, 153.26, 153.14, 152.96, 148.27, 148.17, 136.87, 136.68, 136.51, 136.38, 128.57, 128.53, 128.46, 128.40, 127.94, 127.86, 127.46, 127.41, 127.09, 127.01, 126.98, 124.36, 124.25, 121.79, 115.37, 115.20, 113.01, 76.91, 73.90, 71.07, 71.04, 70.95, 70.70, 70.64, 70.16, 69.38, 67.29, 63.16; HRMS (ESI, m/z): , calcd for : 1451.4980; found 1451.4977.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,5′-dibenzyloxybenzoyl)-d-glucitol (51); 1,5-AG (0.15 g, 0.9 mmol), compound 46 (1.8 g, 5.4 mmol), 2-chloro-1-methylpyridinium iodide (1.3 g, 5.4 mmol), DMAP (0.66 g, 5.4 mmol), TEA (1.5 mL, 10.8 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desired compound 51 (0.69 g, 63%) was obtained as a colorless amorphous oil. = +8.3 (c 0.90, ); NMR (, 600 MHz) δ 7.45–7.18 (m, 48H), 6.79–6.76 (m, 2H), 6.72–6.69 (m, 2H), 5.89 (t, J = 9.6 Hz, 1H), 5.62 (t, J = 9.8 Hz, 1H), 5.37 (td, J = 10.1, 5.6 Hz, 1H), 5.06–4.98 (m, 8H), 4.95–4.84 (m, 8H), 4.66 (dd, J = 12.2, 2.9 Hz, 1H), 4.49 (dd, J = 11.3, 5.5 Hz, 1H), 4.40 (dd, J = 12.2, 5.3 Hz, 1H), 4.04–4.01 (m, 1H), 3.58 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.80, 165.20, 165.06, 159.76, 159.75, 159.70, 136.44, 136.31, 136.26, 136.22, 131.45, 130.91, 130.87, 130.71, 128.60, 128.57, 128.51, 128.09, 128.06, 127.59, 127.57, 108.53, 108.49, 108.41, 108.11, 107.74, 107.70, 107.51, 74.28, 70.41, 70.23, 70.19, 69.73, 67.21, 63.51; HRMS (ESI, m/z): , calcd for : 1451.4980; found 1451.4980.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′-benzyloxybenzoyl)-d-glucitol (52); 1,5-AG (0.10 g, 0.6 mmol), compound 47 (0.82 g, 3.6 mmol), 2-chloro-1-methylpyridinium iodide (0.81 g, 3.6 mmol), DMAP (0.44 g, 3.6 mmol), TEA (1.0 mL, 7.2 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desire compound 52 (0.56 g, 92%) was obtained as a colorless amorphous oil. = +22.7 (c 1.00, ); NMR (, 600 MHz) δ 7.68–7.05 (m, 36H), 5.90 (t, J = 9.6 Hz, 1H), 5.65 (t, J = 9.8 Hz, 1H), 5.40 (td, J = 10.1, 5.6 Hz, 1H), 5.02–5.13 (m, 4H), 5.00 (s, 2H), 4.96 (s, 2H), 4.65 (dd, J = 12.0, 2.7 Hz, 1H), 4.48 (dd, J = 11.2, 5.7 Hz, 1H), 4.42 (dd, J = 12.2, 5.3 Hz, 1H), 4.02–4.05 (m, 1H), 3.59 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 166.03, 165.85, 165.34, 165.11, 158.66, 136.57, 136.37, 130.91, 130.30, 130.12, 129.56, 129.49, 128.61, 128.56, 128.13, 127.62, 127.58, 122.52, 121.12, 120.85, 120.71, 120.53, 115.16, 115.12, 115.00, 114.84, 76.89, 74.07, 70.27, 70.12, 69.50, 67.21, 63.31; HRMS (ESI, m/z): , calcd for : 1027.3306; found 1027.3301.
1,5-Anhydro-2,3,4,6-tetrakis-O-(4′-benzyloxybenzoyl)-d-glucitol (53); 1,5-AG (0.16 g, 1.0 mmol), compound 48 (1.4 g, 6.1 mmol), 2-chloro-1-methylpyridinium iodide (1.4 g, 6.0 mmol), DMAP (0.73 g, 6.0 mmol), TEA (1.7 mL, 12 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desire compound 53 (0.99 g, 98%) was obtained as a colorless amorphous oil. = +38.2 (c 0.90, ); NMR (, 600 MHz) δ 8.06–7.96 (m, 2H), 7.92–7.86 (m, 6H), 7.44–7.17 (m, 20H), 6.93–6.78 (m, 8H), 5.89 (t, J = 9.5 Hz, 1H), 5.63 (t, J = 9.8 Hz, 1H), 5.40 (td, J = 10.0, 5.7 Hz, 1H), 5.02–4.85 (m, 8H), 4.61–4.59 (m, 1H), 4.41 (td, J = 12.5, 5.4 Hz, 2H), 3.99–3.96 (m, 1H), 3.54 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.83, 165.55, 165.15, 164.86, 162.78, 162.62, 162.57, 136.22, 136.07, 131.93, 131.86, 128.63, 128.61, 128.17, 127.46, 127.41, 127.19, 122.30, 121.65, 121.59, 121.46, 114.52, 114.47, 114.43, 114.38, 73.69, 69.96, 69.90, 69.24, 67.23, 63.05; HRMS (ESI, m/z): , calcd for : 1027.3306; found 1027.3304.
2,3,4,6-tetrakis-O-(3′,4′,5′-Trihydroxybenzoyl)-d-glucitol (
54) [
50];
on C (20 wt.%, 50 mg) was added to a solution of compound
50 (860 mg, 0.46 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desired compound
54 (321 mg, 90%) was obtained as a yellow amorphous oil.
= +58.0 (c 1.04, MeOH);
NMR (
600 MHz) δ 7.19 (s, 2H), 7.06 (s, 2H) × 2, 6.99 (s, 2H), 5.81 (t,
J = 9.6 Hz, 1H), 5.50 (t,
J = 9.8 Hz, 1H), 5.28–5.24 (m, 1H), 4.49 (dd,
J = 12.4, 2.1 Hz, 1H), 4.33–4.29 (m, 2H), 4.17–4.15 (m, 1H), 3.72 (t,
J = 10.8 Hz, 1H);
δ 166.36, 166.05, 165.90, 165.63, 146.00, 145.97, 145.92, 145.80, 139.22, 138.97, 138.90, 121.50, 120.90, 120.63, 110.17, 110.08, 110.01, 109.98, 77.57, 74.31, 70.74, 69.61, 67.44, 63.32; HRMS-ESI (
m/z):
, calcd for
: 795.1021; found 795.1019.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′-dihydroxybenzoyl)-d-glucitol (55); on C (20 wt.%, 50 mg) was added to a solution of compound 50 (365 mg, 0.30 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 55 (196 mg, 92%) was obtained as a colorless amorphous oil. = +48.3 (c 0.65, MeOH); NMR (600 MHz); δ 7.56 (d, J = 2.1 Hz, 1H), 7.49 (dd, J = 8.4, 1.9 Hz, 1H), 7.43–7.33 (m, 6H), 6.90 (d, J = 8.2 Hz, 1H), 6.84 (dd, J = 11.3, 8.2 Hz, 2H), 6.76 (d, J = 8.2 Hz, 1H), 5.85 (t, J = 9.6 Hz, 1H), 5.55 (t, J = 9.8 Hz, 1H), 5.30 (td, J = 10.1, 5.4 Hz, 1H), 4.52 (dd, J = 12.0, 2.4 Hz, 1H), 4.37–4.32 (m, 2H), 4.20–4.17 (m, 1H), 3.75 (t, J = 10.8 Hz, 1H); δ 166.22, 166.00, 165.76, 165.56, 151.21, 151.01, 145.55, 145.39, 123.78, 123.71, 123.62, 122.53, 121.96, 121.74, 117.41, 117.29, 117.19, 117.09, 115.74, 115.68, 77.53, 74.40, 70.78, 69.87, 67.48, 63.52; HRMS (, m/z): , calcd for : 707.1248; found 707.1255.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3,5-dihydroxybenzoyl)-d-glucitol (56); on C (20 wt.%, 50 mg) was added to a solution of compound 51 (365 mg, 0.30 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 56 (210 mg, quant.) was obtained as a colorless amorphous oil. = +39.1 (c 0.50, MeOH); NMR (600 MHz); δ 7.09 (s, 2H), 6.95 (d, J = 1.4 Hz, 4H), 6.89 (d, J = 1.7 Hz, 2H), 6.63–6.49 (m, 4H), 5.90 (t, J = 9.6 Hz, 1H), 5.59 (t, J = 9.6 Hz, 1H), 5.37 (td, J = 10.0, 5.6 Hz, 1H), 4.55 (d, J = 12.4 Hz, 1H), 4.43 (dd, J = 12.2, 4.6 Hz, 1H), 4.37 (dd, J = 11.3, 5.5 Hz, 1H), 4.25 (dd, J = 9.8, 4.3 Hz, 1H), 3.80 (t, J = 10.8 Hz, 1H) δ 166.33, 166.13, 165.82, 165.67, 159.36, 159.31, 159.24, 132.73, 132.10, 131.91, 129.67, 128.96, 108.81, 108.74, 108.65, 108.39, 108.24, 108.08, 77.25, 74.65, 70.85, 69.79, 67.25, 63.45; HRMS (, m/z): , calcd for : 707.1248; found 707.1255.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3-hydroxybenzoyl)-d-glucitol (57); on C (20 wt.%, 50 mg) was added to a solution of compound 52 (300 mg, 0.30 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 57 (181 mg, 94%) was obtained as a colorless amorphous oil. = +36.4 (c 0.70, MeOH); NMR 600 MHz); δ 7.55–6.97 (m, 20H), 5.94 (t, J = 9.5 Hz, 1H), 5.65 (t, J = 9.6 Hz, 1H), 5.41 (td, J = 10.1, 5.4 Hz, 1H), 4.57 (dd, J = 12.2, 2.6 Hz, 1H), 4.47 (dd, J = 12.4, 4.8 Hz, 1H), 4.39 (dd, J = 11.2, 5.7 Hz, 1H), 4.29–4.26 (m, 1H), 3.83 (t, J = 10.8 Hz, 1H); δ 166.38, 166.23, 165.90, 165.82, 158.46, 158.25, 132.14, 131.57, 131.43, 130.49, 130.44, 130.40, 129.72, 129.00, 126.08, 121.43, 121.39, 121.21, 121.08, 117.04, 116.93, 116.88, 116.73, 77.23, 74.83, 70.96, 70.23, 67.33, 63.83; HRMS (, m/z): , calcd for : 643.1452; found 643.1459.
1,5-Anhydro-2,3,4,6-tetrakis-O-(4-hydroxybenzoyl)-d-glucitol (58); on C (20 wt.%, 50 mg) was added to a solution of compound 54 (288 mg, 0.29 mmol) in 15 mL of MeOH and 15 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 58 (190 mg, quant.) was obtained as a colorless amorphous oil. = +45.3 (c 0.50, MeOH); NMR (600 MHz); δ 7.93–7.76 (m, 8H), 6.92–6.77 (m, 8H), 5.89 (t, J = 9.6 Hz, 1H), 5.61 (t, J = 9.8 Hz, 1H), 5.34 (td, J = 10.0, 5.3 Hz, 1H), 4.55 (dd, J = 12.4, 2.7 Hz, 1H), 4.41–4.35 (m, 2H), 4.23–4.20 (m, 1H), 3.77 (t, J = 10.8 Hz, 1H); δ 166.17, 165.99, 165.69, 165.54, 163.17, 163.13, 162.88, 162.82, 132.75, 132.68, 132.61, 132.58, 121.99, 121.44, 121.21, 121.19, 116.09, 116.05, 116.00, 115.94, 77.45, 74.47, 70.76, 70.05, 67.50, 63.65; HRMS (, m/z): , calcd for : 643.1452; found 643.1459.
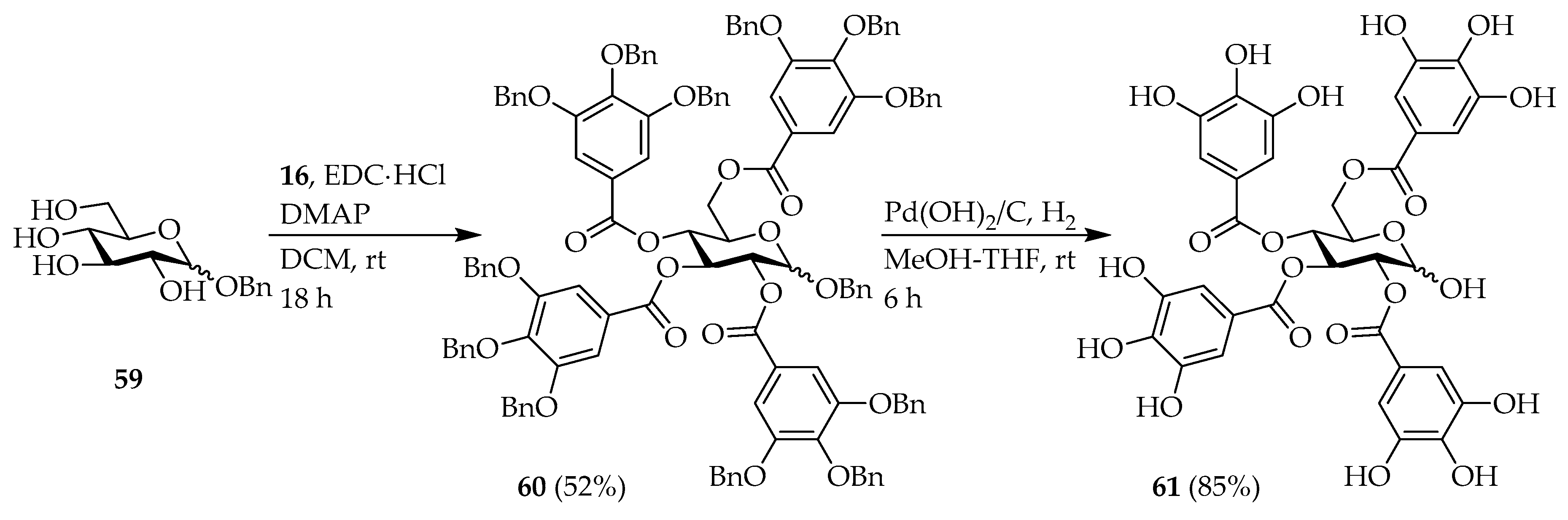
Benzyl 2,3,4,6-tetrakis-O-(3′,4′,5′-tribenzyloxybenzoyl)-α,β-d-glucopyranoside (
60); Compound
59 [
43] (211 mg, 0.78 mmol), EDC·HCl (1.2 g, 6.3 mmol), compound
16 (2.06 g, 4.7 mmol), DMAP (47.7 mg, 0.39 mmol) in 30 mL of DCM was stirred at rt for overnight. After the addition 20 mL of water, the reaction mixture was extracted with DCM (20 mL). The combined organic layer was washed with water (30 mL × 4) and brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by C.C (
/MeOH = 200/1) to obtain desired compound
60 (791 mg, 52%) as a colorless amorphous oil.
= +18.1 (c 1.00,
);
NMR (
, 600 MHz) (assigned for the major anomer; α) δ 7.42–7.16 (m, 80H), 6.21 (t,
J = 10.0, 1H), 5.69 (t,
J = 10.0 Hz, 1H), 5.45 (d,
J = 3.8 Hz, 1H), 5.12–4.95 (m, 16H), 4.73 (dd,
J = 3.1, 12.4 Hz, 1H), 4.54–4.51 (m, 1H), 4.73 (dd,
J = 3.1, 12.4 Hz, 1H);
NMR (
, 150 MHz) (assigned for the major anomer; α) δ 165.68, 165.16, 152.55, 152.52, 152.49, 143.00, 142.86, 142.69, 142.64, 137.41, 137.36, 137.32, 136.56, 136.53, 136.48, 136.40, 136.29, 128.54, 128.46, 128.38, 128.29, 128.24, 128.16, 128.12, 128.09, 128.05, 127.98, 127.94, 127.92, 127.88, 127.81, 127.58, 127.54, 127.52, 124.64, 124.15, 124.02, 123.85, 109.23, 109.16, 109.01, 95.27, 75.11, 75.09, 75.06, 72.17, 71.18, 71.15, 71.05, 70.16, 69.96, 67.96, 63.17. HRMS (ESI,
m/z):
, calcd for
: 1981.7073; found 1981.7067.
2,3,4,6-tetrakis-O-(3′,4′,5′-Trihydroxybenzoyl)-α,β-d-glucopyranose (
61) [
55];
on C (20 wt.%, 20 mg) was added to a solution of compound
54 (100 mg, 0.51 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound
31, desire compound
58 (34 mg, 85%) was obtained as a colorless amorphous oil.
= +54.4 (c 0.30, MeOH);
NMR (
, 600 MHz) δ [α-form] 7.20 (s, 2H), 7.09 (s, 2H), 7.07 (s, 2H), 7.00 (s, 2H), 6.11 (t,
J = 9.6 Hz, 1H), 5.62 (d,
J = 3.4 Hz, 1H), 5.56 (t,
J = 10.2 Hz, 1H), 5.16 (dd,
J = 10.3, 3.5 Hz, 1H), 4.47 (dd,
J = 2.4, 11.4 Hz, 1H), 4.35 (dd,
J = 12.6, 4.8 Hz, 1H), 4.32–4.29 (m, 1H); [β-form] 7.19 (s, 2H), 7.08 (s, 2H), 7.05 (s, 2H), 6.95 (s, 2H), 5.80 (t,
J = 9.6 Hz, 1H), 5.51 (t,
J = 9.6 Hz, 1H), 5.29 (t,
J = 9.0 Hz, 1H), 5.24 (d,
J = 9.0 Hz, 1H), 4.50 (d,
J = 1.7, 11.4 Hz, 1H), 4.31 (m, 2H);
(assigned for the major anomer; α) δ 166.48, 166.22, 165.99, 165.73, 146.04, 145.99, 145.84, 139.27, 139.24, 139.00, 138.93, 121.61, 121.02, 120.78, 120.67, 110.21, 110.17, 110.06, 110.01, 91.04, 73.02, 70.70, 69.88, 68.55, 63.27; HRMS (ESI,
m/z):
, calcd for
: 811.0970; found 811.0970.
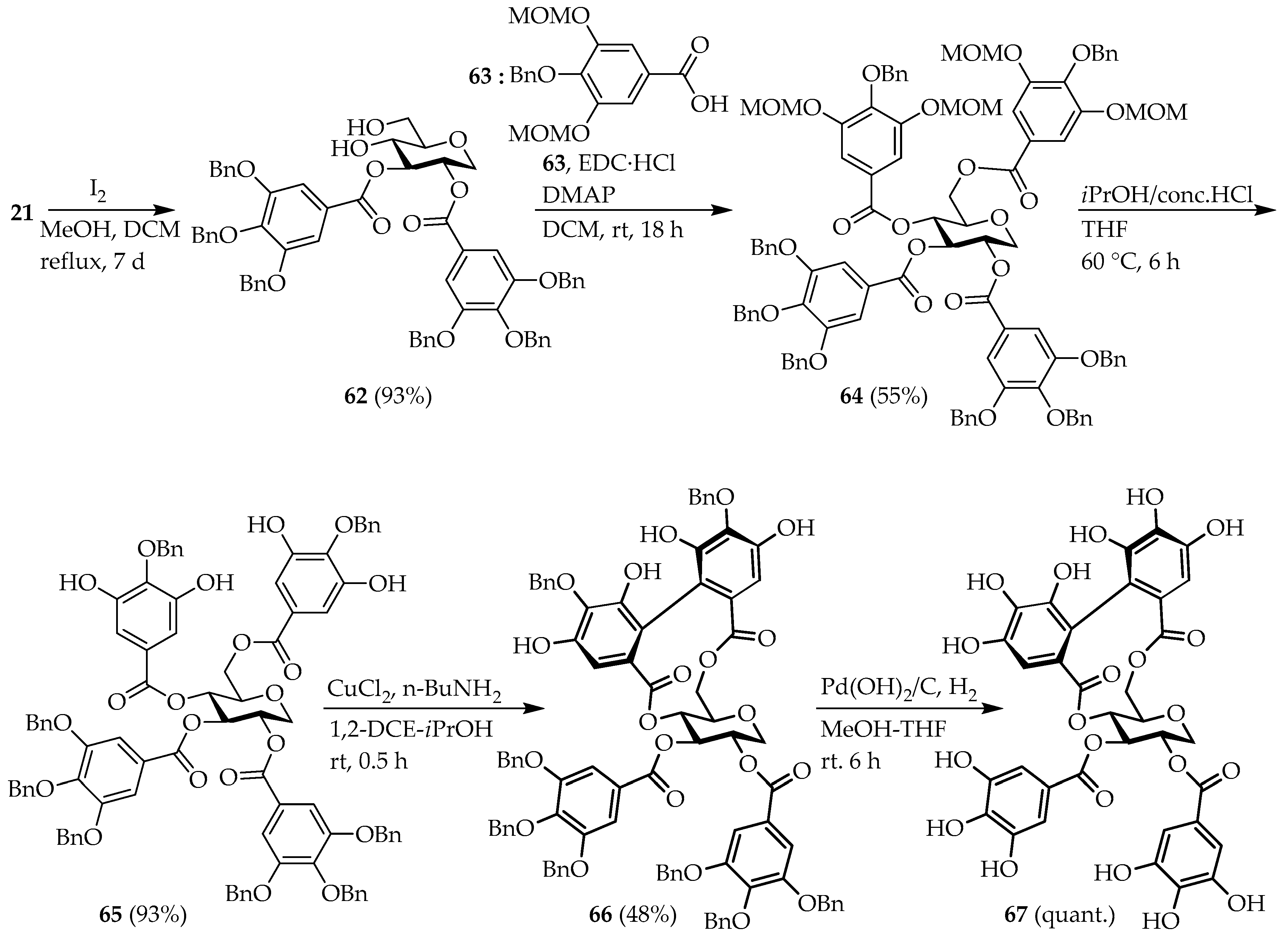
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (62); Compound 21 (3.5 g, 3.2 mmol), iodine (0.71 g, 5.6 mmol) in 100 mL of DCM and 50 mL of MeOH was stirred at 70 °C for 7 days. The reaction mixture was washed with sodium thiosulfate solution and brine. The crude product was purified by C.C (DCM/MeOH = 4/1) to obtain desired compound 62 (3.0 g, 93%) as a white solid. = +80.3 (c 1.06, ); NMR (, 600 MHz) δ 7.38–7.21 (m, 32H), 5.34 (t, J = 9.0 Hz, 1H), 5.28–5.26 (m, 1H), 5.07–4.97 (m, 12H), 4.35–4.32 (m, 1H), 4.00–3.99 (m, 1H), 3.91–3.88 (m, 2H), 3.51–3.48 (m, 2H), 3.07 (d, J = 4.0 Hz, 1H), 2.01 (t, J = 6.0 Hz, 1H); NMR (, 150 MHz) δ 167.2, 165.3, 152.6, 143.1, 142.9, 137.3, 136.5, 136.4, 128.5, 128.4, 128.2, 128.1, 128.0, 127.7, 127.5, 109.3, 109.1, 80.4, 78.5, 75.1, 71.1, 67.0, 69.8, 66.9, 62.4; HRMS-ESI (m/z): , calcd for : 1031.3612; found 1031.3619.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-bis-O-(3′,5′-dimetoxymetoxy-4′-benzyloxybenzoyl)-d-glucitol (
64); Compound
62 (2.5 g, 2.5 mmol),
63 [
46] (2.5 g, 6.2 mmol), EDC·HCl (1.4 g, 7.2 mmol), DMAP (0.15 g, 1.2 mmol) in 15 mL of DCM was stirred at rt for 18 h. After the addition 30 mL of water, the reaction mixture was extracted with EtOAc (3 × 50 mL). The combined organic layer was washed with brine, dried over
, filtered and concentrated under reduced pressure. The crude was purified by C.C (
/acetone = 100/1) to obtain
64 (2.3 g, 55% yield) as a colorless amorphous oil.
= +24.7 (c 2.15 in
);
NMR (
, 600 MHz) δ 7.58 (s, 2H), 7.46–7.25 (m, 50H), 5.85 (t,
J = 10Hz, 1H), 5.61 (t,
J = 10.0Hz, 1H), 5.28–5.26 (m, 1H), 5.22–4.93 (m, 24H), 4.73–4.71 (m, 1H), 4.50–4.47 (m, 1H), 4.35–4.32 (m, 1H), 4.06–4.03 (m, 1H), 3.56 (t,
J = 11.0 Hz, 1H), 3.48 (s, 6H), 3.42(s, 6H);
NMR (
, 150 MHz) δ 165.7, 165.5, 165.1, 164.8, 152.5, 150.9, 150.8, 143.7, 143.4, 142.8, 142.7, 137.4, 137.3, 137.2, 136.5, 136.4, 132.4, 130.8, 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 125.1, 124.2, 124.1, 124.0, 112.4, 112.3, 109.1, 109.0, 95.4, 75.2, 75.1, 75.0, 74.4, 71.1, 71.0, 70.6, 69.3, 68.1, 67.1, 63.3, 56.4; HRMS-ESI (
m/z):
, calcd for
: 1691.5824; found 1691.5825.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-bis-O-(3′,5′-dihydroxy-4′-benzyloxybenzoyl)-d-glucitol (65); Compound 64 (2.0 g, 1.2 mmol) in 8 mL of THF solution was added 8.8 mL of 2-propanol and 0.2 mL of conc. HCl solution, and the mixture was stirred at 60 °C for 6 h. After the addition 3 mL of , the mixture was extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (/MeOH = 1000/1–500/1) to obtain 65 (1.7 g, 93% yield) as a white amorphous oil. = +26.8 (c 2.36, ); NMR (, 600 MHz) δ 7.43–7.14 (m, 48H), 6.00 (s, 4H), 5.82 (t, J = 9.5 Hz, 1H), 5.59 (t, J = 9.5 Hz, 1H), 5.37–5.30 (m, 1H), 5.10–4.82 (m, 16H), 4.70–4.68 (m, 1H), 4.54–4.52 (m, 1H), 4.45–4.42 (m, 1H), 3.97–3.95 (m, 1H), 3.57 (t, J = 10.5 Hz, 1H); NMR (, 150 MHz) δ 166.2, 165.8, 165.5, 165.2, 152.5, 149.0, 148.9, 142.8, 142.7, 138.2, 137.8, 137.3, 137.2, 136.6, 136.4, 136.2,128.8, 128.7, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.7, 127.5, 124.5, 123.9, 123.8, 109.9, 109.8, 109.1, 108.9, 77.6, 75.2, 75.1, 75.0, 74.3, 71.1, 71.0, 70.7, 69.7, 68.6, 67.3, 62.6; HRMS-ESI (m/z): , calcd for : 1515.4775; found 1515.4777.
Benzyl protected 1,5-AG-tellimagrandin analog (66); To a solution of (135 mg, 1.0 mmol) in 10 mL of MeOH was added n-butylamine (400 μL, 4.0 mmol). After the stirred at rt for 1.5 h, the mixture was added to solution of compound 65 (500 mg, 0.34 mmol) in 20 mL of 1,2-dichloroethane (DCE) and stirred at rt for 30 min. The reaction mixture was diluted with 50 mL of diethyl ether, and 50 mL of 5M aq. HCl and 50 mL of diethyl ether were added. The separating organic layer washed with water, and brine. The organic layer was dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (DCM/MeOH = 250/1) and HPLC (column, FNED01048, 250 × 20 mm, SG80–5 μm, eluant DCM/MeOH = 200/1) to afford 66 (237 mg, 48%) as a peal yellow amorphous oil. = +69.5 (c 1.43, ); NMR (, 600 MHz) δ 7.43–7.23 (m, 50H), 6.75 (s, 1H), 6.67 (s, 1H), 5.67 (t, J = 10.0 Hz, 1H), 5.37–5.34 (m, 1H), 5.29 (t, J = 10.0 Hz, 1H), 5.25–5.24 (m, 1H), 5.13–4.85 (m, 16H), 4.47–4.45 (m, 1H), 3.98 (d, J = 12.5 Hz, 1H), 3.96–3.94 (m. 1H), 3.46 (t, J = 11.0 Hz, 1H); δ 167.1, 166.6, 165.8, 165.2, 152.5, 149.0, 147.0, 142.8, 137.4, 137.3, 136.5, 136.4, 136.3, 136.2, 135.7, 135.5, 130.3, 129.6, 129.0, 128.9, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 123.9, 113.7, 113.2, 109.2, 108.4, 107.9, 75.6, 75.1, 75.0, 74.4, 71.1, 71.0, 70.8, 70.1, 67.7, 63.6; HRMS-ESI (m/z): , calcd for ; found 1513.4620.
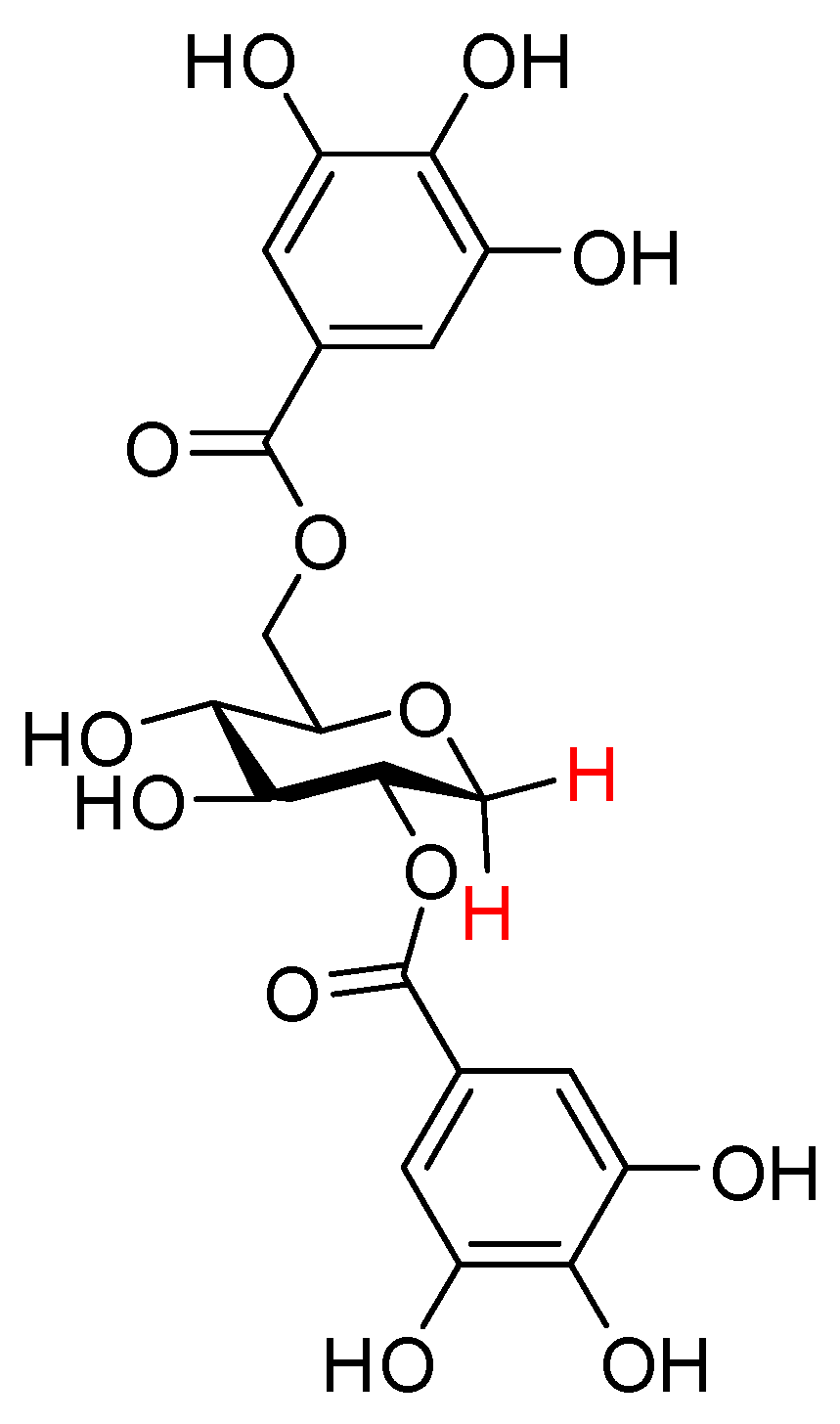
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-trihydroxybenzoyl)-4,6-O-hexahydroxydiphenyl-d-glucitol (67); on C (20 wt.%, 20 mg) was added to a solution of compound 66 (60 mg, 0.40 mmol) in 2 mL of MeOH and 2 mL of THF under the argon. By the same procedure previously described for the preparation of compound procedure described for previously 31 preparation, desired compound 67 (32 mg, quant.) was obtained as a peal yellow amorphous oil. = +113.3 (c 0.29, ); NMR (600 MHz) δ 7.00 (s, 2H), 6.95 (s, 2H), 6.60 (s, 1H), 6.40 (s, 1H), 5.61 (t, J = 9.5 Hz, 1H), 5.27–5.19 (m, 2H), 5.03 (t, J = 10.0 Hz, 1H), 4.25–4.22 (m, 1H), 4.08–4.07 (m, 1H), 3.80 (t, J = 13.0 Hz, 1H), 3.54 (t, J = 11.0 Hz, 1H); NMR ( 150 MHz); 171.1, 170.7, 168.8, 168.3, 150.6, 147.6, 147.1, 144.6, 143.5, 141.8, 139.7, 126.8, 126.7, 126.3, 120.7, 120.3, 120.1, 119.4, 111.1, 110.8, 108.3, 107.7, 78.7, 78.5, 76.2, 72.1, 72.0, 71.8, 71.4, 71.3, 69.4, 68.7, 64.7; HRMS (ESI, m/z): , calcd for : 793.0864; found 793.0865.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
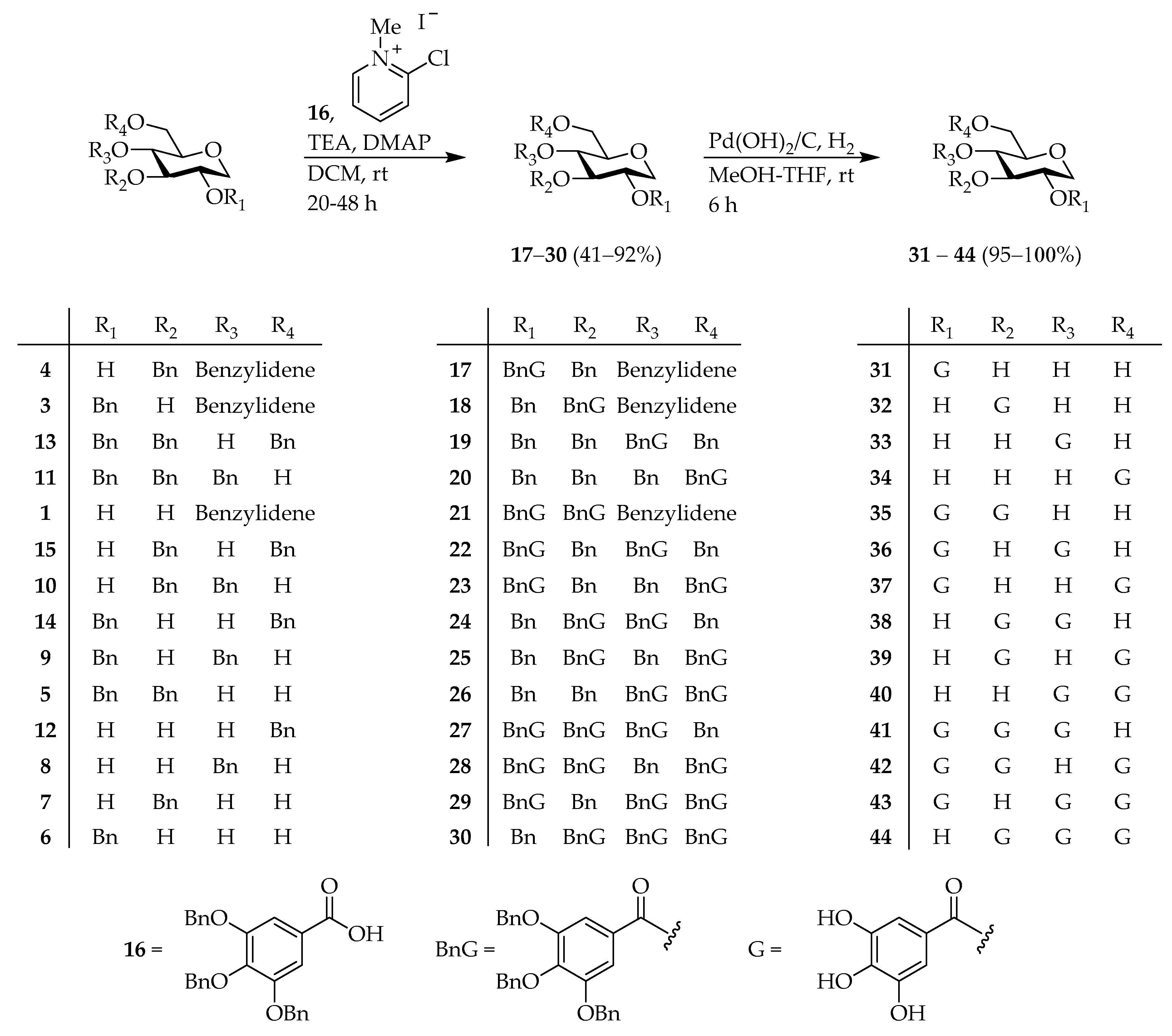{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}