Prenyleudesmanes and A Hexanorlanostane from the Roots of Lonicera macranthoides

 ,
,
Abstract
:1. Introduction
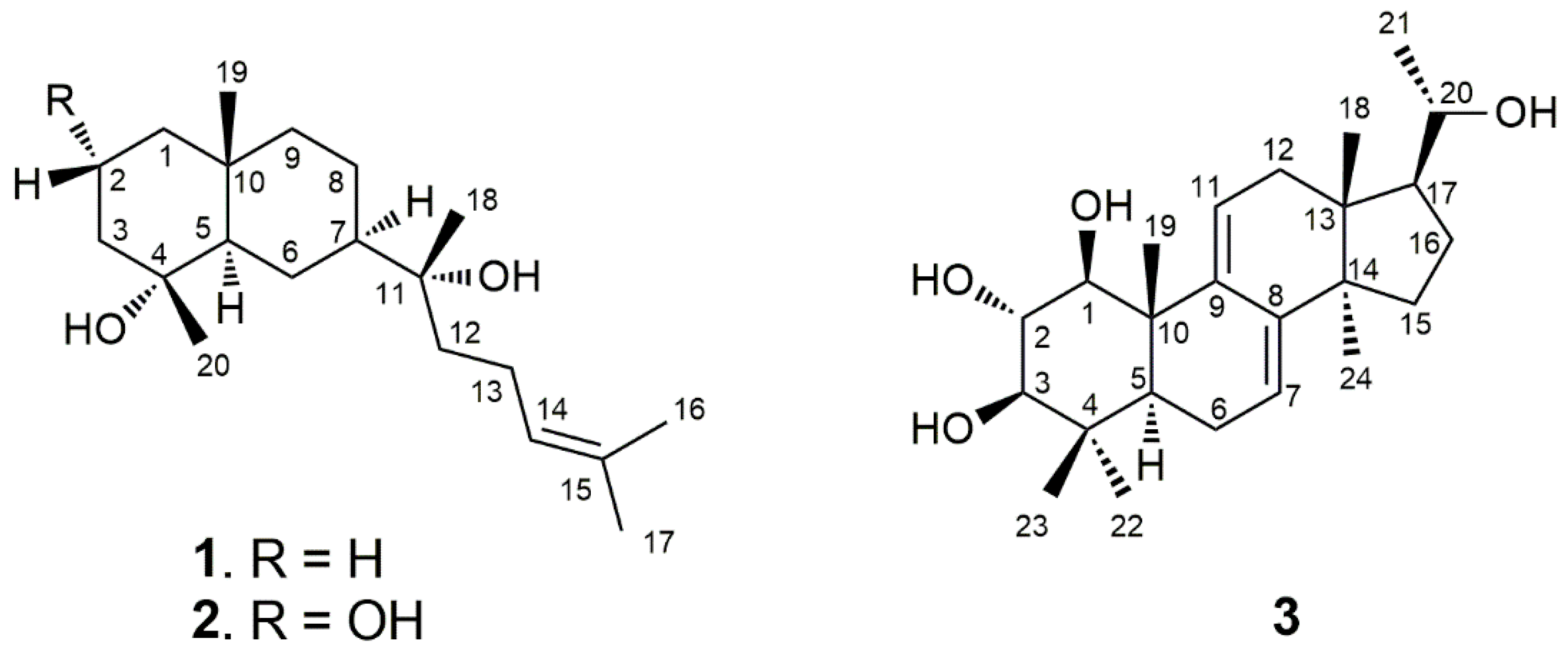
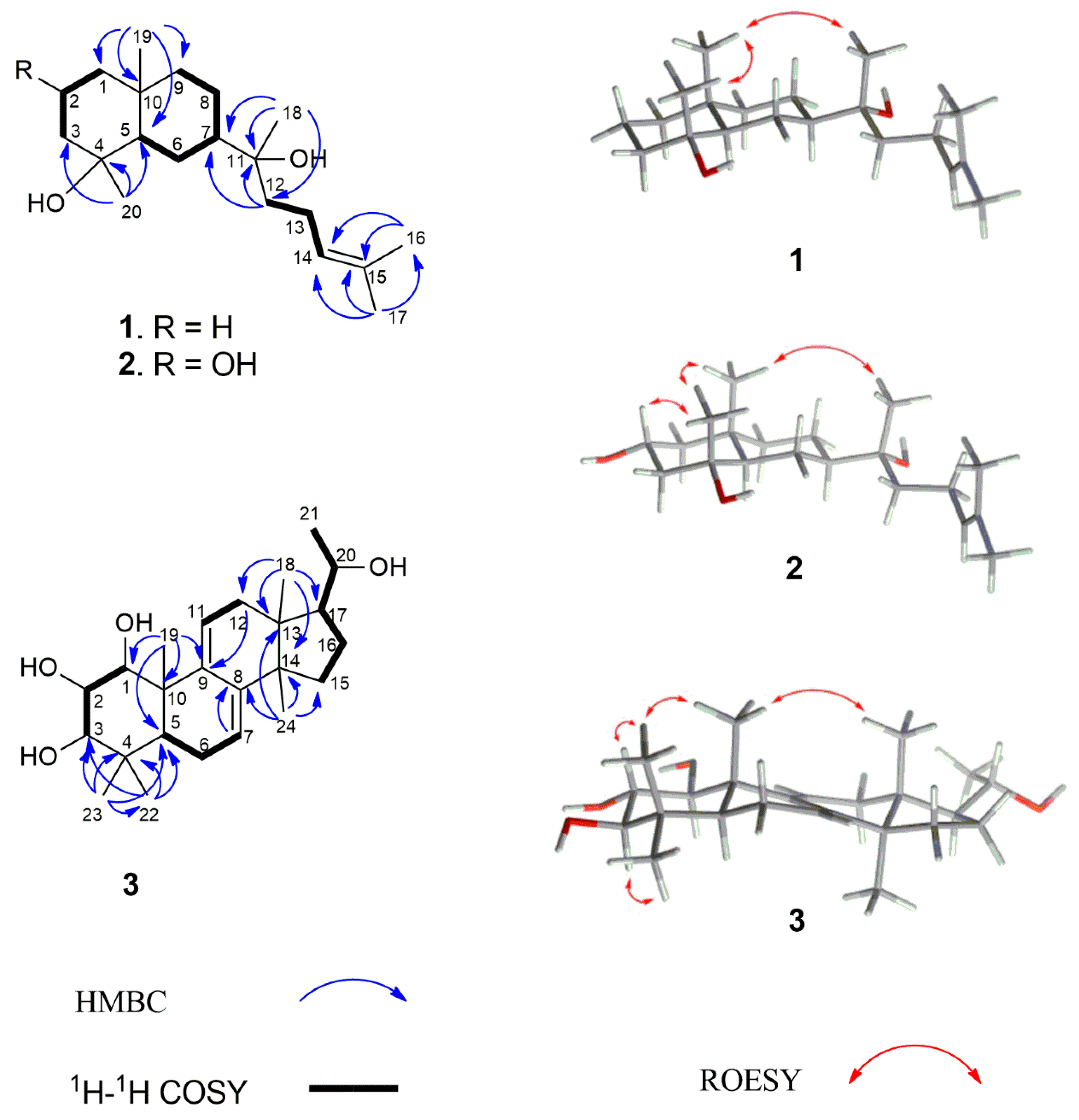
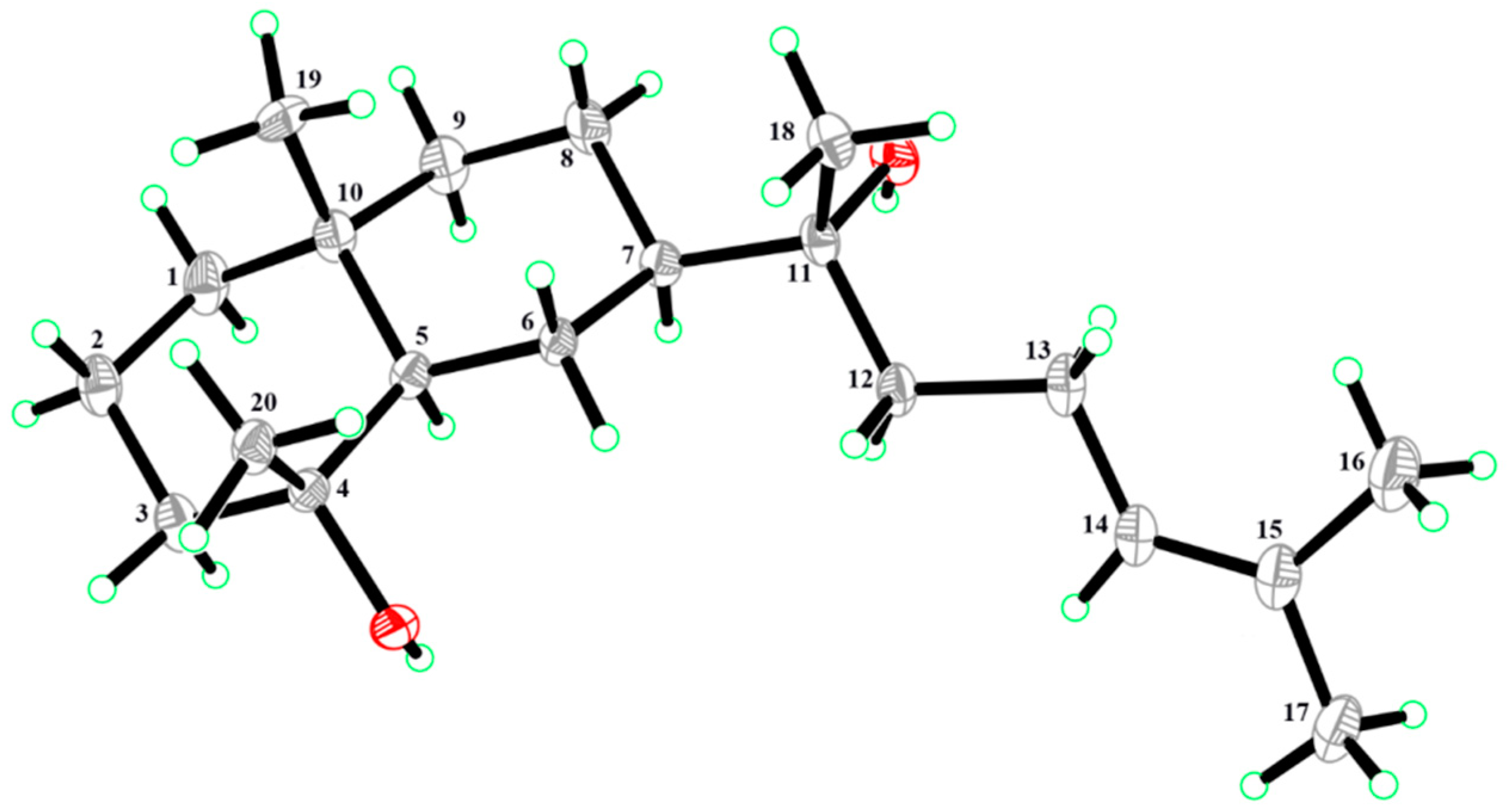
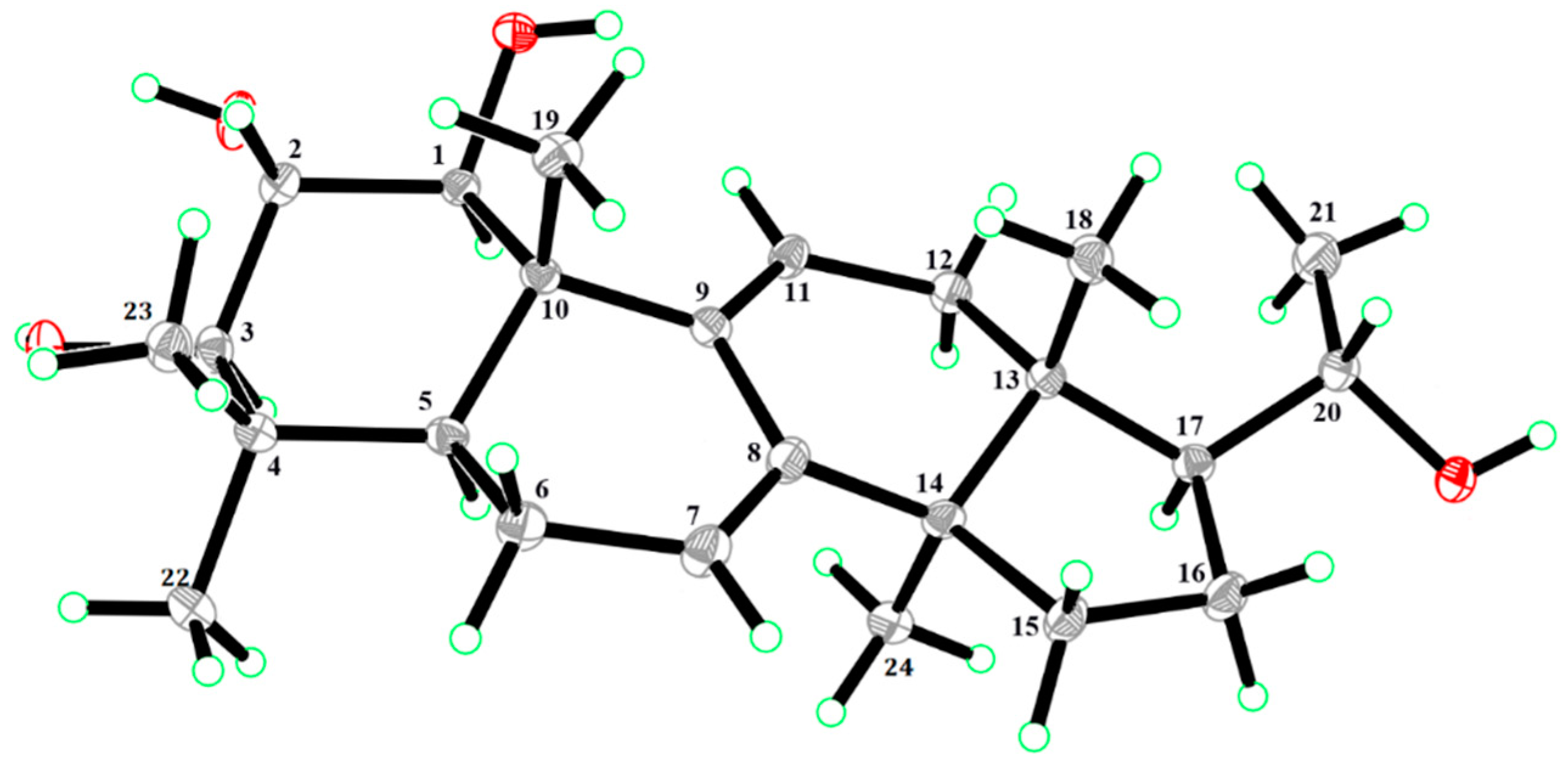
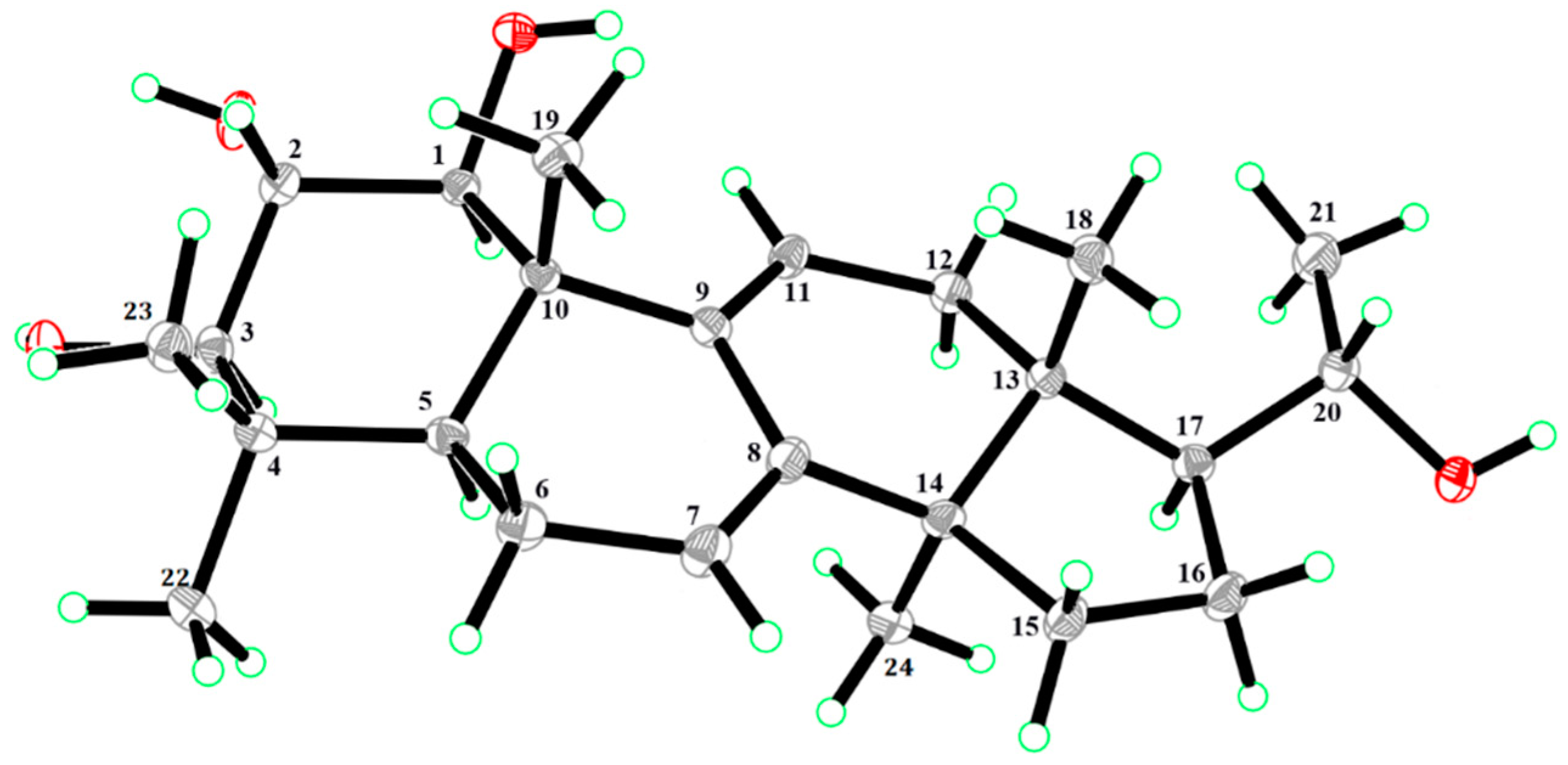
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Compound Characterization
3.5. Biological Assay
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- FOC (Flora of China) Flora of China Website. 1988. Available online: http://frps.iplant.cn/frps/Lonicera%20macranthoides (accessed on 15 October 2019).
- Chinese Pharmacopeia Commission. Pharmacopeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2015; Volume 1, p. 30. [Google Scholar]
- Chen, Y.; Zhao, Y.; Wang, M.; Wang, Q.; Shan, Y.; Guan, F.; Feng, X. A new lupane-type triterpenoid saponin from Lonicera macranthoides. Chem. Nat. Compd. 2014, 49, 1087–1090. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, X.; Jia, X.; Wang, M.; Liang, J.; Dong, Y. Triterpene glycosides from Lonicera. Isolation and structural determination of seven glycosides from flower buds of Lonicera macranthoides. Chem. Nat. Compd. 2008, 44, 39–43. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, X.; Wang, M.; Zhao, Y.Y.; Dong, Y.F. Triterpene glycosides from Lonicera. II. Isolation and structural determination of glycosides from flower buds of Lonicera macranthoides. Chem. Nat. Compd. 2009, 45, 514–518. [Google Scholar]
- Chen, Y.; Shan, Y.; Zhao, Y.Y.; Wang, Q.Z.; Wang, M.; Feng, X.; Liang, J.Y. Two new triterpenoid saponins from Lonicera macranthoides. Chin. Chem. Lett. 2012, 3, 325–328. [Google Scholar] [CrossRef]
- Sun, M.; Feng, X.; Yin, M.; Chen, Y.; Zhao, X.; Dong, Y. A biflavonoid from stems and leaves of Lonicera macranthoides. Chem. Nat. Compd. 2012, 48, 231–233. [Google Scholar] [CrossRef]
- Sun, M.; Feng, X.; Lin, X.H.; Yin, M.; Zhao, X.Z.; Chen, Y.; Shan, Y. Studies on the chemical constituents from stems and leaves of Lonicera macranthoides. J. Chin. Med. Mater. 2011, 34, 218–220. [Google Scholar]
- Liu, J.; Zhang, J.; Wang, F.; Chen, X.F. Chemical constituents from the buds of Lonicera macranthoides in Sichuan, China. Biochem. Syst. Ecol. 2014, 54, 68–70. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, Y.; Ma, X.; Zhao, Y.Y.; Feng, X. Study on chemical constituents from roots of Lonicera macranthoides. Chin. Med. Mat. 2014, 37, 2207–2209. [Google Scholar]
- Bai, B.; Chen, Y.; Liu, W.J.; Yin, M.; Wang, M.; Feng, X. Chemical constituents of petroleum ether fraction of Lonicera macranthoides roots. J. Chin. Med. Mater. 2015, 38, 518–520. [Google Scholar]
- Lyu, H.; Liu, W.J.; Xu, S.; Shan, Y.; Feng, X.; Chen, Y. Two 9, 10-syn-pimarane diterpenes from the roots of Lonicera macranthoides. Phytochem. Lett. 2018, 25, 175–179. [Google Scholar] [CrossRef]
- Fujioka, T.; Yamamoto, M.; Kashiwada, Y.; Fujii, H.; Mihashi, K.; Ikeshiro, Y.; Chen, I.S.; Lee, K.H. Novel cytotoxic diterpenes from the stem of Dysoxylum kuskusense. Bioorg. Med. Chem. Lett. 1998, 8, 3479–3482. [Google Scholar] [CrossRef]
- Flack, H.D. On enantiomorph-polarity estimation. Acta. Cryst. A Found. Crystallogr. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef]
- Avilov, S.A.; Kalinovsky, A.I.; Kalinin, V.I.; Stonik, V.A.; Riguera, R.; Jiménez CKoreoside, A. A new nonholostane triterpene glycoside from the sea cucumber Cucumaria koraiensis. J. Nat. Prod. 1997, 60, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Non-holostane aglycones of sea cucumber triterpene glycosides. Structure, biosynthesis, evolution. Steroids 2018, 147, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.H.; Waraszkiewicz, S.M.; Erickson, K.L.; Finer, J.; Clardy, J. Dictyoxepin and dictyolene, two new diterpenes from the marine alga Dictyota acutiloba (Phaeophyta). J. Am. Chem. Soc. 1977, 99, 3516–3517. [Google Scholar] [CrossRef]
- Takahashi, Y.; Suzuki, M.; Abe, T.; Masuda, M. Anhydroaplysiadiol from Laurencia japonensis. Phytochemistry 1998, 48, 987–990. [Google Scholar] [CrossRef]
- Ojika, M.; Yoshida, Y.; Okumura, M.; Ieda, S.; Yamada, K. Aplysiadiol, A new brominated diterpene from the marine mollusc Aplysia kurodai. J. Nat. Prod. 1990, 53, 1619–1622. [Google Scholar] [CrossRef]
- Matthée, G.F.; König, G.M.; Wright, A.D. Three new diterpenes from the marine soft coral Lobophytum crassum. J. Nat. Prod. 1998, 61, 237–240. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Chuang, C.T.; Wang, S.K.; Wen, Z.H.; Chiou, S.F.; Hsu, C.H.; Dai, C.F.; Duh, C.Y. Antiviral and anti-inflammatory diterpenoids from the soft coral Sinularia gyrosa. J. Nat. Prod. 2010, 73, 1184–1187. [Google Scholar] [CrossRef]
- Li, L.; Sheng, L.; Wang, C.Y.; Zhou, Y.B.; Huang, H.; Li, X.B.; Li, J.; Molllo, E.; Gavagnin, M.; Guo, Y.W. Diterpenes from the Hainan soft coral Lobophytum cristatum Tixier-Durivault. J. Nat. Prod. 2011, 74, 2089–2094. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhu, Z.D.; Gu, Y.C.; Li, J.; Zhu, W.L.; Guo, Y.W. Further new diterpenoids as PTP1B inhibitors from the Xisha soft coral Sinularia polydactyla. Mar. Drugs 2018, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Liang, B.W.; Li, X.F.; Liu, Q. Induced production of new diterpenoids in the fungus Penicillium funiculosum. Nat. Prod. Commun. 2014, 9, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Duh, C.Y.; Wang, S.K.; Chen, I.S. Cytotoxic prenyleudesmane diterpenes from the fruits of Dysoxylum kuskusense. J. Nat. Prod. 2000, 63, 1546–1547. [Google Scholar] [CrossRef]
- Gu, J.; Qian, S.Y.; Zhao, Y.L.; Cheng, G.G.; Hu, D.B.; Zhang, B.H.; Liu, Y.P.; Luo, X.D. Prenyleudesmanes, rare natural diterpenoids from Dysoxylum densiflorum. Tetrahedron 2014, 70, 1375–1382. [Google Scholar] [CrossRef]
- Zhang, P.; Lin, Y.; Wang, F.; Fang, D.; Zhang, G. Diterpenes from Dysoxylum lukii Merr. Phytochem. Lett. 2019, 29, 53–56. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1998, 48, 589–601. [Google Scholar]
Sample Availability: Samples of the compounds 1 and 3 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | 1 a, c | 2 a,c | 3 b,c | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 1α | 41.0 | 1.07 ddd 12.5/12.5/6.0 | 46.7 | 1.34 dd 14.0/3.2 | 78.4 | 3.57 d 10.0 |
| 1β | 1.38 d 12.5 | 1.67 m | ||||
| 2α | 20.1 | 1.58 m | 68.2 | 4.28 ddd 7.0/3.0/3.0 | 72.9 | 3.48 dd 10.0/10.0 |
| 2β | 1.56 m | |||||
| 3α | 43.5 | 1.35 ddd 12.5/12.5/5.5 | 48.5 | 1.66 m | 78.7 | 3.04 d 10.0 |
| 3β | 1.79 d 12.5 | 2.01 d 14.0 | ||||
| 4 | 72.3 | 71.6 | 38.1 | |||
| 5 | 55.0 | 1.19 d 12.5 | 54.4 | 1.29 dd 12.5/2.0 | 47.1 | 1.16 dd 5.0/12.0 |
| 6α | 21.4 | 1.86 d 12.5 | 21.2 | 1.87 d 12.5 | 22.4 | 2.20 dd 12.0/17.0 |
| 6β | 1.03 ddd 12.5/12.5/12.5 | 1.14 ddd 12.5/12.5/12.5 | 2.15 dd 5.0/5.0/17.0 | |||
| 7 | 48.3 | 1.41 dddd 12.5/12.5/3.0/3.0 | 48.5 | 1.42 dddd 12.5/12.5/3.0/3.0 | 119.5 | 5.49 d 5.0 |
| 8α | 21.8 | 1.59 d 12.5 | 21.2 | 1.56 d 12.5 | 142.1 | |
| 8β | 1.33 dddd 12.5/12.5/12.5/3.0 | 1.37 dddd 12.5/12.5/12.5/3.5 | ||||
| 9α | 44.6 | 1.45 ddd 12.5/3.0/3.0 | 44.9 | 1.49 ddd 12.5/3.0/3.0 | 144.0 | |
| 9β | 1.15 ddd 12.5/12.5/3.0 | 1.12 m | ||||
| 10 | 34.6 | 34.0 | 43.2 | |||
| 11 | 74.5 | 74.6 | 119.1 | 6.31 d 6.0 | ||
| 12α | 39.7 | 1.53 dd 8.2/8.2 | 39.5 | 1.52 dd 8.1/8.1 | 36.4 | 2.18 d 17.0 |
| 12β | 2.06 d 6.0/17.0 | |||||
| 13 | 22.3 | 2.06 m | 22.3 | 2.04 m | 42.0 | |
| 14 | 124.6 | 5.14 dd 7.0/7.0 | 124.5 | 5.13 dd 6.7/6.7 | 49.9 | |
| 15α | 131.6 | 131.6 | 31.1 | 1.68 ddd 7.5/11.5/11.5 | ||
| 15β | 1.46 dd 9.0/11.5 | |||||
| 16α | 17.6 | 1.63 s | 17.8 | 1.62 s | 25.8 | 2.07 ddd 9.0/14.0/17.0 |
| 16β | 1.60 dd 9.0/14.0 | |||||
| 17 | 25.7 | 1.69 s | 25.8 | 1.68 s | 53.0 | 1.78 dd 9.0/17.0 |
| 18 | 24.1 | 1.16 s | 24.2 | 1.16 s | 15.4 | 0.57 s |
| 19 | 18.7 | 0.86 s | 20.3 | 1.14 s | 15.9 | 1.06 s |
| 20 | 22.6 | 1.11 s | 25.0 | 1.33 s | 70.4 | 3.62 dd 6.2/9.0 |
| 21 | 22.4 | 1.21 d 6.2 | ||||
| 22 | 15.9 | 0.90 s | ||||
| 23 | 27.6 | 1.02 s | ||||
| 24 | 24.9 | 0.91 s | ||||
| Cell Line | 1 | 2 | 3 | Etoposide |
|---|---|---|---|---|
| HepG2 | 36.3 ± 2.1 | 64.9 ± 3.5 | 46.0 ± 2.4 | 25.4 ± 1.7 |
| HeLa | 13.8 ± 1.1 | 27.1 ± 1.4 | 12.5 ± 0.9 | 21.2 ± 1.3 |
| HASMC | >100 | >100 | >100 | 63.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, H.; Liu, W.; Bai, B.; Shan, Y.; Paetz, C.; Feng, X.; Chen, Y. Prenyleudesmanes and A Hexanorlanostane from the Roots of Lonicera macranthoides. Molecules 2019, 24, 4276. https://doi.org/10.3390/molecules24234276
Lyu H, Liu W, Bai B, Shan Y, Paetz C, Feng X, Chen Y. Prenyleudesmanes and A Hexanorlanostane from the Roots of Lonicera macranthoides. Molecules. 2019; 24(23):4276. https://doi.org/10.3390/molecules24234276
Chicago/Turabian StyleLyu, Hui, Wenjuan Liu, Bai Bai, Yu Shan, Christian Paetz, Xu Feng, and Yu Chen. 2019. "Prenyleudesmanes and A Hexanorlanostane from the Roots of Lonicera macranthoides" Molecules 24, no. 23: 4276. https://doi.org/10.3390/molecules24234276
APA StyleLyu, H., Liu, W., Bai, B., Shan, Y., Paetz, C., Feng, X., & Chen, Y. (2019). Prenyleudesmanes and A Hexanorlanostane from the Roots of Lonicera macranthoides. Molecules, 24(23), 4276. https://doi.org/10.3390/molecules24234276