
Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions


 , ,
, , 
Abstract
1. Introduction
2. Kynurenines and Their Role in Schizophrenia
2.1. Kynurenines and Associated Elements
2.1.1. The Kynurenine Pathway
2.1.2. KYNA and Its Target Receptors
2.2. The KYNA Hypothesis of Schizophrenia
3. The Endocannabinoid System and Its Role in Schizophrenia
3.1. Overview of the Endocannabinoid System
3.2. The Cannabinoid Hypothesis of Schizophrenia
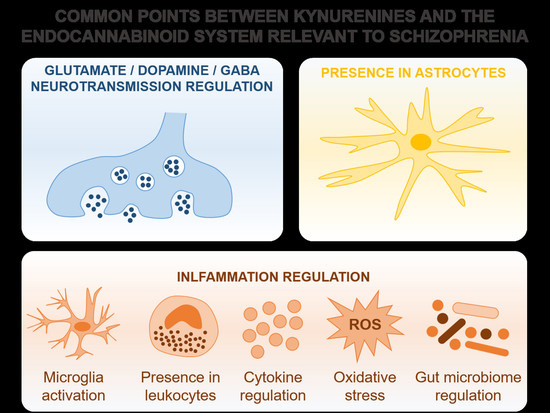
4. Common Points and Potential Interactions between the Endocannabinoid System and Kynurenines Relevant to Schizophrenia
4.1. Overview
4.2. Glutamatergic, Dopaminergic, and GABAergic Transmission Regulation by Kynurenines and the Endocannabinoid System in Schizophrenia
4.2.1. The Basics of the Dopaminergic, Glutamatergic, and GABAergic Hypothesis of Schizophrenia
4.2.2. KYNA and Dopaminergic/Glutamatergic/GABAergic Interactions in Schizophrenia
4.2.3. The Endocannabinoid System and Dopaminergic/Glutamatergic/GABAergic Interactions in Schizophrenia
4.3. Astrocytes as a Potential Stage for the Endocannabinoid System and Kynurenine Interaction in Schizophrenia
4.3.1. Overview of Astrocytes and Their Role in Schizophrenia
4.3.2. CB1Rs, KYNA Production, and Target Receptors of KYNA in Astrocytes
4.3.3. The Role of Astrocytic CB1Rs, α7nAChRs, and KYNA in Glutamate Neurotransmission and Its Significance in Schizophrenia
4.4. The Involvement of Kynurenines and the Endocannabinoid System in the Inflammatory Component of Schizophrenia
4.4.1. The Inflammatory Hypothesis of Schizophrenia
4.4.2. Neuroinflammation, Cytokines, and Microglia Activation
4.4.3. ROS and Oxidative Stress
4.4.4. Gastrointestinal Inflammation and Gut Microbiome
5. Therapeutic Potentials
5.1. Overview
5.2. Currently Available Medications
Non-Dopaminergic Agents in Clinical Studies Based on the Glutamatergic and GABAergic Hypothesis of Schizophrenia
5.3. Targeting the KP
5.4. Targeting the Endocannabinoid System
6. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet (Lond. Engl.) 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Erhardt, S.; Schwieler, L.; Imbeault, S.; Engberg, G. The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 2017, 112, 297–306. [Google Scholar] [CrossRef]
- Manseau, M.W.; Goff, D.C. Cannabinoids and Schizophrenia: Risks and Therapeutic Potential. Neurotherapeutics 2015, 12, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Schwieler, L.; Nilsson, L.; Linderholm, K.; Engberg, G. The kynurenic acid hypothesis of schizophrenia. Physiol. Behav. 2007, 92, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. Int. J. Mol. Sci. 2017, 18, 1617. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Aguilera, G.; Santamaría, A. Cannabinoids: Glutamatergic Transmission and Kynurenines. In Advances in Neurobiology; Springer: Cham, Switzerland, 2016; Volume 12, pp. 173–198. [Google Scholar]
- Myint, A.-M.; Kim, Y.-K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef]
- Plitman, E.; Iwata, Y.; Caravaggio, F.; Nakajima, S.; Chung, J.K.; Gerretsen, P.; Kim, J.; Takeuchi, H.; Chakravarty, M.M.; Remington, G.; et al. Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis. Schizophr. Bull. 2017, 43, 764–777. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Hernández, M.; Ramos, J.A. Cannabinoid-dopamine interaction in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 2010, 16, e72–e91. [Google Scholar] [CrossRef]
- Müller-Vahl, K.R.; Emrich, H.M. Cannabis and schizophrenia: Towards a cannabinoid hypothesis of schizophrenia. Expert Rev. Neurother. 2008, 8, 1037–1048. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Notarangelo, F.M.; Wu, H.-Q.; Bruno, J.P.; Schwarcz, R. Astrocytes as Pharmacological Targets in the Treatment of Schizophrenia: Focus on Kynurenic Acid. In Handbook of Behavioral Neuroscience; Elsevier: Amsterdam, The Netherlands, 2016; Volume 23, pp. 423–443. ISBN 9780128009819. [Google Scholar]
- Navarrete, M.; Díez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.P.; Turska, M.; Paluszkiewicz, P.; Parada-Turska, J.; Oxenkrug, G.F. Kynurenic Acid in the digestive system-new facts, new challenges. Int. J. Tryptophan Res. 2013, 6, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef] [PubMed]
- Pérez-De La Cruz, V.; Carrillo-Mora, P.; Santamaría, A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, 1–8. [Google Scholar] [PubMed]
- Bryleva, E.Y.; Brundin, L. Kynurenine pathway metabolites and suicidality. Neuropharmacology 2017, 112, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef]
- Lipina, C.; Hundal, H.S. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol. 2016, 6, 150276. [Google Scholar] [CrossRef]
- Gallelli, C.A.; Calcagnini, S.; Romano, A.; Koczwara, J.B.; de Ceglia, M.; Dante, D.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues. Antioxidants 2018, 7, 93. [Google Scholar] [CrossRef]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Evrard, E.; Gan, X.; Kim, J.-Y.; McAllister, L.A.; Pandit, J.; Rong, S.; Salafia, M.A.; et al. PF-04859989 as a template for structure-based drug design: Identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg. Med. Chem. Lett. 2013, 23, 1961–1966. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major Developments in the Design of Inhibitors along the Kynurenine Pathway. Curr. Med. Chem. 2017, 24, 2471–2495. [Google Scholar] [CrossRef] [PubMed]
- Jayawickrama, G.S.; Nematollahi, A.; Sun, G.; Gorrell, M.D.; Church, W.B. Inhibition of human kynurenine aminotransferase isozymes by estrogen and its derivatives. Sci. Rep. 2017, 7, 17559. [Google Scholar] [CrossRef] [PubMed]
- Jayawickrama, G.S.; Nematollahi, A.; Sun, G.; Church, W.B. Improvement of kynurenine aminotransferase-II inhibitors guided by mimicking sulfate esters. PLoS ONE 2018, 13, e0196404. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Myint, A.-M.; J. Schwarz, M. Kynurenine Pathway in Schizophrenia: Pathophysiological and Therapeutic Aspects. Curr. Pharm. Des. 2011, 17, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Church, W.B. Kynurenine Aminotransferase Isozyme Inhibitors: A Review. Int. J. Mol. Sci. 2016, 17, 946. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.; Weizman, A.; Weinstein, A. Positive and Negative Effects of Cannabis and Cannabinoids on Health. Clin. Pharmacol. Ther. 2019, 105, 1139–1147. [Google Scholar] [CrossRef]
- Rohleder, C.; Müller, J.K.; Lange, B.; Leweke, F.M. Cannabidiol as a Potential New Type of an Antipsychotic. A Critical Review of the Evidence. Front. Pharmacol. 2016, 7, 422. [Google Scholar] [CrossRef]
- Wyrofsky, R.; McGonigle, P.; Van Bockstaele, E.J. Drug discovery strategies that focus on the endocannabinoid signaling system in psychiatric disease. Expert Opin. Drug Discov. 2015, 10, 17–36. [Google Scholar] [CrossRef]
- Behan, W.M.H.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef]
- Rios, C.; Santamaria, A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 1991, 16, 1139–1143. [Google Scholar] [CrossRef]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.; Guidetti, P.; Okuno, E.; Schwarcz, R. Characterization of human brain kynurenine aminotransferases using [3H]kynurenine as a substrate. Neuroscience 1993, 55, 177–184. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, G.J. Expression of The Kynurenine Pathway Enzymes in Human Microglia and Macrophages. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2003; Volume 527, pp. 105–112. [Google Scholar]
- Beadle, G.W.; Mitchell, H.K.; Nyc, J.F. Kynurenine as an Intermediate in the Formation of Nicotinic Acid from Tryptophane by Neurospora. Proc. Natl. Acad. Sci. USA 1947, 33, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug. Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Franco, N.F.; Ng, M.L.; Pai, S.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Current Evidence for a Role of the Kynurenine Pathway of Tryptophan Metabolism in Multiple Sclerosis. Front. Immunol. 2016, 7, 246. [Google Scholar] [CrossRef]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Nicoletti, F. Kynurenine pathway metabolites in migraine. J. Headache Pain 2015, 16, A1. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur. J. Pharmacol. 1988, 154, 85–87. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Dobelis, P.; Staley, K.J.; Cooper, D.C. Lack of modulation of nicotinic acetylcholine alpha-7 receptor currents by kynurenic acid in adult hippocampal interneurons. PLoS ONE 2012, 7, e41108. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Schwarcz, R. Kynurenic acid as an antagonist of α7 nicotinic acetylcholine receptors in the brain: Facts and challenges. Biochem. Pharmacol. 2013, 85, 1027–1032. [Google Scholar] [CrossRef]
- Berlinguer-Palmini, R.; Masi, A.; Narducci, R.; Cavone, L.; Maratea, D.; Cozzi, A.; Sili, M.; Moroni, F.; Mannaioni, G. GPR35 activation reduces Ca2+ transients and contributes to the kynurenic acid-dependent reduction of synaptic activity at CA3-CA1 synapses. PLoS ONE 2013, 8, e82180. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic Acid as a Ligand for Orphan G Protein-coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand that Synergistically Induces Interleukin-6 in the Presence of Inflammatory Signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef]
- Füvesi, J.; Somlai, C.; Németh, H.; Varga, H.; Kis, Z.; Farkas, T.; Károly, N.; Dobszay, M.; Penke, Z.; Penke, B.; et al. Comparative study on the effects of kynurenic acid and glucosamine–kynurenic acid. Pharmacol. Biochem. Behav. 2004, 77, 95–102. [Google Scholar] [CrossRef]
- Robotka, H.; Németh, H.; Somlai, C.; Vécsei, L.; Toldi, J. Systemically administered glucosamine-kynurenic acid, but not pure kynurenic acid, is effective in decreasing the evoked activity in area CA1 of the rat hippocampus. Eur. J. Pharmacol. 2005, 513, 75–80. [Google Scholar] [CrossRef]
- Rózsa, É.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Wu, H.-Q.; Okuyama, M.; Kajii, Y.; Pocivavsek, A.; Bruno, J.P.; Schwarcz, R. Targeting kynurenine aminotransferase II in psychiatric diseases: Promising effects of an orally active enzyme inhibitor. Schizophr. Bull. 2014, 40 (Suppl. 2), S152–S158. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Wonodi, I.; Schwarcz, R. Cortical Kynurenine Pathway Metabolism: A Novel Target for Cognitive Enhancement in Schizophrenia. Schizophr. Bull. 2010, 36, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M. Kynurenines: From the perspective of major psychiatric disorders. FEBS J. 2012, 279, 1375–1385. [Google Scholar] [CrossRef]
- Olsson, S.K.; Sellgren, C.; Engberg, G.; Landén, M.; Erhardt, S. Cerebrospinal fluid kynurenic acid is associated with manic and psychotic features in patients with bipolar I disorder. Bipolar Disord. 2012, 14, 719–726. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.-Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Erhardt, S.; Blennow, K.; Nordin, C.; Skogh, E.; Lindström, L.H.; Engberg, G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci. Lett. 2001, 313, 96–98. [Google Scholar] [CrossRef]
- Beggiato, S.; Tanganelli, S.; Fuxe, K.; Antonelli, T.; Schwarcz, R.; Ferraro, L. Endogenous kynurenic acid regulates extracellular GABA levels in the rat prefrontal cortex. Neuropharmacology 2014, 82, 11–18. [Google Scholar] [CrossRef]
- Beggiato, S.; Antonelli, T.; Tomasini, M.C.; Tanganelli, S.; Fuxe, K.; Schwarcz, R.; Ferraro, L. Kynurenic acid, by targeting α7 nicotinic acetylcholine receptors, modulates extracellular GABA levels in the rat striatum in vivo. Eur. J. Neurosci. 2013, 37, 1470–1477. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Wu, H.-Q.; Potter, M.C.; Elmer, G.I.; Pellicciari, R.; Schwarcz, R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology 2011, 36, 2357–2367. [Google Scholar] [CrossRef]
- Varga, D.; Herédi, J.; Kánvási, Z.; Ruszka, M.; Kis, Z.; Ono, E.; Iwamori, N.; Iwamori, T.; Takakuwa, H.; Vécsei, L.; et al. Systemic L-Kynurenine sulfate administration disrupts object recognition memory, alters open field behavior and decreases c-Fos immunopositivity in C57Bl/6 mice. Front. Behav. Neurosci. 2015, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Alexander, K.S.; Wu, H.-Q.; Schwarcz, R.; Bruno, J.P. Acute elevations of brain kynurenic acid impair cognitive flexibility: Normalization by the alpha7 positive modulator galantamine. Psychopharmacology 2012, 220, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Chess, A.C.; Simoni, M.K.; Alling, T.E.; Bucci, D.J. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr. Bull. 2007, 33, 797–804. [Google Scholar] [CrossRef]
- Pershing, M.L.; Bortz, D.M.; Pocivavsek, A.; Fredericks, P.J.; Jørgensen, C.V.; Vunck, S.A.; Leuner, B.; Schwarcz, R.; Bruno, J.P. Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: Implications for schizophrenia. Neuropharmacology 2015, 90, 33–41. [Google Scholar] [CrossRef]
- DeAngeli, N.E.; Todd, T.P.; Chang, S.E.; Yeh, H.H.; Yeh, P.W.; Bucci, D.J. Exposure to Kynurenic Acid during Adolescence Increases Sign-Tracking and Impairs Long-Term Potentiation in Adulthood. Front. Behav. Neurosci. 2015, 8, 451. [Google Scholar] [CrossRef] [PubMed]
- Wonodi, I.; McMahon, R.P.; Krishna, N.; Mitchell, B.D.; Liu, J.; Glassman, M.; Hong, L.E.; Gold, J.M. Influence of kynurenine 3-monooxygenase (KMO) gene polymorphism on cognitive function in schizophrenia. Schizophr. Res. 2014, 160, 80. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.; Campbell, B.M.; Strick, C.A.; Horner, W.; Hoffmann, W.E.; Kiss, T.; Chapin, D.S.; McGinnis, D.; Abbott, A.L.; Roberts, B.M.; et al. Reduction of brain kynurenic acid improves cognitive function. J. Neurosci. 2014, 34, 10592–10602. [Google Scholar] [CrossRef]
- Linderholm, K.R.; Skogh, E.; Olsson, S.K.; Dahl, M.-L.; Holtze, M.; Engberg, G.; Samuelsson, M.; Erhardt, S. Increased Levels of Kynurenine and Kynurenic Acid in the CSF of Patients With Schizophrenia. Schizophr. Bull. 2012, 38, 426–432. [Google Scholar] [CrossRef]
- Miller, C.L.; Llenos, I.C.; Dulay, J.R.; Weis, S. Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res. 2006, 1073, 25–37. [Google Scholar] [CrossRef]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Wonodi, I.; Roberts, R.C.; Rassoulpour, A.; McMahon, R.P.; Schwarcz, R. Impaired Kynurenine Pathway Metabolism in The Prefrontal Cortex of Individuals with Schizophrenia. Schizophr. Bull. 2011, 37, 1147–1156. [Google Scholar] [CrossRef]
- Kegel, M.E.; Bhat, M.; Skogh, E.; Samuelsson, M.; Lundberg, K.; Dahl, M.-L.; Sellgren, C.; Schwieler, L.; Engberg, G.; Schuppe-Koistinen, I.; et al. Imbalanced Kynurenine Pathway in Schizophrenia. Int. J. Tryptophan Res. 2014, 7, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; MacDonald, M.L.; Elswick, D.E.; Sweet, R.A. The glutamate hypothesis of schizophrenia: Evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.Z.; Zhang, X.; Blennow, K.; Nordberg, A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport 1999, 10, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Geyer, M.A. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem. Pharmacol. 2013, 86, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez de Fonseca, F.; Del Arco, I.; Bermudez-Silva, F.J.; Bilbao, A.; Cippitelli, A.; Navarro, M. The endocannabinoid system: Physiology and pharmacology. Alcohol Alcohol. 2005, 40, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Endocannabinoids and Synaptic Function in the CNS. Neurosci 2007, 13, 127–137. [Google Scholar] [CrossRef]
- Lovinger, D.M. Presynaptic modulation by endocannabinoids. In Pharmacology of Neurotransmitter Release; Springer: Berlin/Heidelberg, Germany, 2008; Volume 184, pp. 435–477. [Google Scholar]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2009, 147, S163–S171. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. In Cannabinoids; Springer: Berlin/Heidelberg, Germany, 2005; pp. 299–325. [Google Scholar]
- Howlett, A.C.; Bidaut-Russell, M.; Devane, W.A.; Melvin, L.S.; Johnson, M.R.; Herkenham, M. The cannabinoid receptor: Biochemical, anatomical and behavioral characterization. Trends Neurosci. 1990, 13, 420–423. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Malan, T.P.; Ibrahim, M.M.; Deng, H.; Liu, Q.; Mata, H.P.; Vanderah, T.; Porreca, F.; Makriyannis, A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain 2001, 93, 239–245. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Ibarra-Lecue, I.; Pilar-Cuéllar, F.; Muguruza, C.; Florensa-Zanuy, E.; Díaz, Á.; Urigüen, L.; Castro, E.; Pazos, A.; Callado, L.F. The endocannabinoid system in mental disorders: Evidence from human brain studies. Biochem. Pharmacol. 2018, 157, 97–107. [Google Scholar] [CrossRef]
- Leweke, F.M.; Giuffrida, A.; Wurster, U.; Emrich, H.M.; Piomelli, D. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999, 10, 1665–1669. [Google Scholar] [CrossRef]
- Giuffrida, A.; Leweke, F.M.; Gerth, C.W.; Schreiber, D.; Koethe, D.; Faulhaber, J.; Klosterkötter, J.; Piomelli, D. Cerebrospinal Anandamide Levels are Elevated in Acute Schizophrenia and are Inversely Correlated with Psychotic Symptoms. Neuropsychopharmacology 2004, 29, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Ferretjans, R.; Moreira, F.A.; Teixeira, A.L.; Salgado, J.V. The Endocannabinoid System and its Role in Schizophrenia: A Systematic Review of the Literature. Rev. Bras. Psiquiatr. 2012, 34, 163–193. [Google Scholar] [CrossRef]
- Altintas, M.; Inanc, L.; Oruc, G.A.; Arpacioglu, S.; Gulec, H. Clinical characteristics of synthetic cannabinoid-induced psychosis in relation to schizophrenia: A single-center cross-sectional analysis of concurrently hospitalized patients. Neuropsychiatr. Dis. Treat. 2016, 12, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht, M.; Häfner, H. Cannabis, Vulnerability, and the Onset of Schizophrenia: An Epidemiological Perspective. Aust. N. Z. J. Psychiatry 2000, 34, 468–475. [Google Scholar] [CrossRef]
- Kuepper, R.; Morrison, P.D.; Van Os, J.; Murray, R.M.; Kenis, G.; Henquet, C. Does dopamine mediate the psychosis-inducing effects of cannabis? A review and integration of findings across disciplines. Psiquiatr. Biol. 2012, 19, 49–58. [Google Scholar] [CrossRef]
- Koethe, D.; Hoyer, C.; Leweke, F.M. The endocannabinoid system as a target for modelling psychosis. Psychopharmacology 2009, 206, 551–561. [Google Scholar] [CrossRef]
- Boggs, D.L.; Surti, T.; Gupta, A.; Gupta, S.; Niciu, M.; Pittman, B.; Schnakenberg Martin, A.M.; Thurnauer, H.; Davies, A.; D’Souza, D.C.; et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology 2018, 235, 1923–1932. [Google Scholar] [CrossRef]
- Moore, T.H.; Zammit, S.; Lingford-Hughes, A.; Barnes, T.R.; Jones, P.B.; Burke, M.; Lewis, G. Cannabis use and risk of psychotic or affective mental health outcomes: A systematic review. Lancet 2007, 370, 319–328. [Google Scholar] [CrossRef]
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Rampino, A.; Marakhovskaia, A.; Soares-Silva, T.; Torretta, S.; Veneziani, F.; Beaulieu, J.M. Antipsychotic Drug Responsiveness and Dopamine Receptor Signaling; Old Players and New Prospects. Front. Psychiatry 2019, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, J.C.; Vinkers, C.H.; Hulshoff Pol, H.E.; Marsman, A. GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front. Psychiatry 2017, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Gil, R.; Krystal, J.; Baldwin, R.M.; Seibyl, J.P.; Bowers, M.; van Dyck, C.H.; Charney, D.S.; Innis, R.B.; Laruelle, M. Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am. J. Psychiatry 1998, 155, 761–767. [Google Scholar] [PubMed]
- Williams, G.V.; Castner, S.A. Under the curve: Critical issues for elucidating D1 receptor function in working memory. Neuroscience 2006, 139, 263–276. [Google Scholar] [CrossRef]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Review: Glutamate and dopamine dysregulation in schizophrenia—A synthesis and selective review. J. Psychopharmacol. 2007, 21, 440–452. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef]
- Javitt, D.C.; Schoepp, D.; Kalivas, P.W.; Volkow, N.D.; Zarate, C.; Merchant, K.; Bear, M.F.; Umbricht, D.; Hajos, M.; Potter, W.Z.; et al. Translating glutamate: From pathophysiology to treatment. Sci. Transl. Med. 2011, 3, 102mr2. [Google Scholar] [CrossRef]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased glutamic acid decarboxylase67 mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J. Psychiatr. Res. 2009, 43, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Orhan, F.; Fatouros-Bergman, H.; Goiny, M.; Malmqvist, A.; Piehl, F.; Karolinska Schizophrenia Project (KaSP) Consortium; Cervenka, S.; Collste, K.; Victorsson, P.; Sellgren, C.M.; et al. CSF GABA is reduced in first-episode psychosis and associates to symptom severity. Mol. Psychiatry 2018, 23, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Modinos, G.; Ferrera, D.; McGuire, P. Neuroimaging studies of GABA in schizophrenia: A systematic review with meta-analysis. Transl. Psychiatry 2017, 7, e1147. [Google Scholar] [CrossRef] [PubMed]
- Konopaske, G.T.; Sweet, R.A.; Wu, Q.; Sampson, A.; Lewis, D.A. Regional specificity of chandelier neuron axon terminal alterations in schizophrenia. Neuroscience 2006, 138, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Tufvesson-Alm, M.; Schwieler, L.; Schwarcz, R.; Goiny, M.; Erhardt, S.; Engberg, G. Importance of kynurenine 3-monooxygenase for spontaneous firing and pharmacological responses of midbrain dopamine neurons: Relevance for schizophrenia. Neuropharmacology 2018, 138, 130–139. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Mö Ller, T.; Stella, N. Astrocytes in Culture Produce Anandamide and Other Acylethanolamides. J. Biol. Chem. 2002, 227, 20869–20876. [Google Scholar] [CrossRef]
- Walter, L.; Stella, N. Endothelin-1 increases 2-arachidonoyl glycerol (2-AG) production in astrocytes. Glia 2003, 44, 85–90. [Google Scholar] [CrossRef]
- Starowicz, K.; Maione, S.; Cristino, L.; Palazzo, E.; Marabese, I.; Rossi, F.; de Novellis, V.; Di Marzo, V. Tonic Endovanilloid Facilitation of Glutamate Release in Brainstem Descending Antinociceptive Pathways. J. Neurosci. 2007, 27, 13739–13749. [Google Scholar] [CrossRef]
- Melis, M.; Pistis, M. Hub and switches: Endocannabinoid signalling in midbrain dopamine neurons. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3276–3285. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed]
- Laviolette, S.R.; Grace, A.A. The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: Implications for schizophrenia and addiction. Cell. Mol. Life Sci. 2006, 63, 1597–1613. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.; Labruyere, J.; Wang, G.; Wozniak, D.; Price, M.; Sesma, M. NMDA antagonist neurotoxicity: Mechanism and prevention. Science 1991, 254, 1515–1518. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.M. Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learn. Mem. 2000, 7, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Steffens, M.; Engler, C.; Zentner, J.; Feuerstein, T.J. Cannabinoid CB1 receptor-mediated modulation of evoked dopamine release and of adenylyl cyclase activity in the human neocortex. Br. J. Pharmacol. 2004, 141, 1193–1203. [Google Scholar] [CrossRef][Green Version]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef]
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull. 2018, 136, 101–108. [Google Scholar] [CrossRef]
- Dityatev, A.; Frischknecht, R.; Seidenbecher, C.I. Extracellular matrix and synaptic functions. Results Probl. Cell Differ. 2006, 43, 69–97. [Google Scholar]
- Dityatev, A.; Rusakov, D.A. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 2011, 21, 353–359. [Google Scholar] [CrossRef]
- Richard, A.D.; Lu, X.-H. “Teaching old dogs new tricks”: Targeting neural extracellular matrix for normal and pathological aging-related cognitive decline. Neural Regen. Res. 2019, 14, 578–581. [Google Scholar]
- Chelini, G.; Pantazopoulos, H.; Durning, P.; Berretta, S. The tetrapartite synapse: A key concept in the pathophysiology of schizophrenia. Eur. Psychiatry 2018, 50, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Okuno, E.; Schwarcz, R. Characterization of rat brain kynurenine aminotransferases I and II. J. Neurosci. Res. 1997, 50, 457–465. [Google Scholar] [CrossRef]
- Shen, J.; Yakel, J.L. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J. Mol. Neurosci. 2012, 48, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, Z.; Oláh, T.; Kőszeghy, Á.; Piscitelli, F.; Holló, K.; Pál, B.; Csernoch, L.; Di Marzo, V.; Antal, M. CB1 receptor activation induces intracellular Ca2+ mobilization and 2-arachidonoylglycerol release in rodent spinal cord astrocytes. Sci. Rep. 2018, 8, 10562. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Dinh, T.; Stella, N. ATP induces a rapid and pronounced increase in 2-arachidonoylglycerol production by astrocytes, a response limited by monoacylglycerol lipase. J. Neurosci. 2004, 24, 8068–8074. [Google Scholar] [CrossRef]
- Metna-Laurent, M.; Marsicano, G. Rising stars: Modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia 2015, 63, 353–364. [Google Scholar] [CrossRef]
- Secci, M.E.; Mascia, P.; Sagheddu, C.; Beggiato, S.; Melis, M.; Borelli, A.C.; Tomasini, M.C.; Panlilio, L.V.; Schindler, C.W.; Tanda, G.; et al. Astrocytic Mechanisms Involving Kynurenic Acid Control Δ9-Tetrahydrocannabinol-Induced Increases in Glutamate Release in Brain Reward-Processing Areas. Mol. Neurobiol. 2019, 56, 3563–3575. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids Potentiate Synaptic Transmission through Stimulation of Astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, D.N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute Cannabinoids Impair Working Memory through Astroglial CB1 Receptor Modulation of Hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef]
- Guidetti, P.; Hoffman, G.E.; Melendez-Ferro, M.; Albuquerque, E.X.; Schwarcz, R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia 2007, 55, 78–92. [Google Scholar] [CrossRef]
- Guidetti, P.; Schwarcz, R. Determination of alpha-aminoadipic acid in brain, peripheral tissues, and body fluids using GC/MS with negative chemical ionization. Brain Res. Mol. Brain Res. 2003, 118, 132–139. [Google Scholar] [CrossRef]
- Swartz, K.J.; During, M.J.; Freese, A.; Beal, M.F. Cerebral synthesis and release of kynurenic acid: An endogenous antagonist of excitatory amino acid receptors. J. Neurosci. 1990, 10, 2965–2973. [Google Scholar] [CrossRef]
- Owe-Young, R.; Webster, N.L.; Mukhtar, M.; Pomerantz, R.J.; Smythe, G.; Walker, D.; Armati, P.J.; Crowe, S.M.; Brew, B.J. Kynurenine pathway metabolism in human blood-brain-barrier cells: Implications for immune tolerance and neurotoxicity. J. Neurochem. 2008, 105, 1346–1357. [Google Scholar] [CrossRef]
- Gál, E.M.; Young, R.B.; Sherman, A.D. Tryptophan loading: Consequent effects on the synthesis of kynurenine and 5-hydroxyindoles in rat brain. J. Neurochem. 1978, 31, 237–244. [Google Scholar] [CrossRef]
- Gál, E.M.; Sherman, A.D. Synthesis and metabolism of L-kynurenine in rat brain. J. Neurochem. 1978, 30, 607–613. [Google Scholar] [CrossRef]
- Maurer, S.V.; Williams, C.L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef]
- Skowrońska, K.; Obara-Michlewska, M.; Zielińska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef]
- Letellier, M.; Park, Y.K.; Chater, T.E.; Chipman, P.H.; Gautam, S.G.; Oshima-Takago, T.; Goda, Y. Astrocytes regulate heterogeneity of presynaptic strengths in hippocampal networks. Proc. Natl. Acad. Sci. USA 2016, 113, E2685–E2694. [Google Scholar] [CrossRef]
- Mei, Y.-Y.; Wu, D.C.; Zhou, N. Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 2018, 9, 544. [Google Scholar] [CrossRef]
- Wu, H.-Q.; Pereira, E.F.R.; Bruno, J.P.; Pellicciari, R.; Albuquerque, E.X.; Schwarcz, R. The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J. Mol. Neurosci. 2010, 40, 204–210. [Google Scholar] [CrossRef]
- Compton, M.T.; Furman, A.C.; Kaslow, N.J. Lower negative symptom scores among cannabis-dependent patients with schizophrenia-spectrum disorders: Preliminary evidence from an African American first-episode sample. Schizophr. Res. 2004, 71, 61–64. [Google Scholar] [CrossRef]
- Dubertret, C.; Bidard, I.; Adès, J.; Gorwood, P. Lifetime positive symptoms in patients with schizophrenia and cannabis abuse are partially explained by co-morbid addiction. Schizophr. Res. 2006, 86, 284–290. [Google Scholar] [CrossRef]
- Grace, A.A. Disruption of cortical-limbic interaction as a substrate for comorbidity. Neurotox. Res. 2006, 10, 93–101. [Google Scholar] [CrossRef]
- Quiroz, C.; Orrú, M.; Rea, W.; Ciudad-Roberts, A.; Yepes, G.; Britt, J.P.; Ferré, S. Local Control of Extracellular Dopamine Levels in the Medial Nucleus Accumbens by a Glutamatergic Projection from the Infralimbic Cortex. J. Neurosci. 2016, 36, 851–859. [Google Scholar] [CrossRef]
- Kaiser, S.; Wonnacott, S. α-Bungarotoxin-Sensitive Nicotinic Receptors Indirectly Modulate [3H]Dopamine Release in Rat Striatal Slices via Glutamate Release. Mol. Pharmacol. 2000, 58, 312–318. [Google Scholar] [CrossRef]
- Rassoulpour, A.; Wu, H.-Q.; Ferre, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef]
- Justinova, Z.; Mascia, P.; Wu, H.-Q.; Secci, M.E.; Redhi, G.H.; Panlilio, L.V.; Scherma, M.; Barnes, C.; Parashos, A.; Zara, T.; et al. Reducing cannabinoid abuse and preventing relapse by enhancing endogenous brain levels of kynurenic acid. Nat. Neurosci. 2013, 16, 1652–1661. [Google Scholar] [CrossRef]
- Benros, M.E.; Nielsen, P.R.; Nordentoft, M.; Eaton, W.W.; Dalton, S.O.; Mortensen, P.B. Autoimmune Diseases and Severe Infections as Risk Factors for Schizophrenia: A 30-Year Population-Based Register Study. Am. J. Psychiatry 2011, 168, 1303–1310. [Google Scholar] [CrossRef]
- Miller, B.J.; Graham, K.L.; Bodenheimer, C.M.; Culpepper, N.H.; Waller, J.L.; Buckley, P.F. A Prevalence Study of Urinary Tract Infections in Acute Relapse of Schizophrenia. J. Clin. Psychiatry 2013, 74, 271–277. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Zimbron, J.; Lewis, G.; Jones, P.B. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2013, 43, 239–257. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Zimbron, J.; Dalman, C.; Lewis, G.; Jones, P.B. Childhood infection and adult schizophrenia: A meta-analysis of population-based studies. Schizophr. Res. 2012, 139, 161–168. [Google Scholar] [CrossRef]
- Benros, M.E.; Eaton, W.W.; Mortensen, P.B. The Epidemiologic Evidence Linking Autoimmune Diseases and Psychosis. Biol. Psychiatry 2014, 75, 300–306. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Niño-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immun. 2014, 20, 401–411. [Google Scholar] [CrossRef]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: A role for IFN-γ? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef]
- Babcock, T.A.; Carlin, J.M. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor α in interferon-treated epithelial cells. Cytokine 2000, 12, 588–594. [Google Scholar] [CrossRef]
- Asp, L.; Johansson, A.-S.; Mann, A.; Owe-Larsson, B.; Urbanska, E.M.; Kocki, T.; Kegel, M.; Engberg, G.; Lundkvist, G.B.; Karlsson, H. Effects of pro-inflammatory cytokines on expression of kynurenine pathway enzymes in human dermal fibroblasts. J. Inflamm. (Lond.) 2011, 8, 25. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef]
- Weis, F.; Beiras-Fernandez, A.; Hauer, D.; Hornuss, C.; Sodian, R.; Kreth, S.; Briegel, J.; Schelling, G. Effect of anaesthesia and cardiopulmonary bypass on blood endocannabinoid concentrations during cardiac surgery. Br. J. Anaesth. 2010, 105, 139–144. [Google Scholar] [CrossRef]
- Knight, J.M.; Szabo, A.; Zhao, S.; Lyness, J.M.; Sahler, O.J.Z.; Liesveld, J.L.; Sander, T.; Rizzo, J.D.; Hillard, C.J.; Moynihan, J.A. Circulating endocannabinoids during hematopoietic stem cell transplantation: A pilot study. Neurobiol. Stress 2015, 2, 44–50. [Google Scholar] [CrossRef][Green Version]
- Suárez-Pinilla, P.; López-Gil, J.; Crespo-Facorro, B. Immune system: A possible nexus between cannabinoids and psychosis. Brain. Behav. Immun. 2014, 40, 269–282. [Google Scholar] [CrossRef]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef]
- Backhaus, C.; Rahman, H.; Scheffler, S.; Laatsch, H.; Hardeland, R. NO scavenging by 3-hydroxyanthranilic acid and 3-hydroxykynurenine: N-nitrosation leads via oxadiazoles to o-quinone diazides. Nitric Oxide 2008, 19, 237–244. [Google Scholar] [CrossRef]
- Hardeland, R.; Zsizsik, B.K.; Poeggeler, B.; Fuhrberg, B.; Holst, S.; Coto-Montes, A. Indole-3-Pyruvic and -Propionic Acids, Kynurenic Acid, and Related Metabolites as Luminophores and Free-Radical Scavengers; Springer: Boston, MA, USA, 1999; pp. 389–395. [Google Scholar]
- Ribeiro, R.; Wen, J.; Li, S.; Zhang, Y. Involvement of ERK1/2, cPLA2 and NF-κB in microglia suppression by cannabinoid receptor agonists and antagonists. Prostaglandins Other Lipid Mediat. 2013, 100–101, 1–14. [Google Scholar] [CrossRef]
- Han, K.H.; Lim, S.; Ryu, J.; Lee, C.-W.; Kim, Y.; Kang, J.-H.; Kang, S.-S.; Ahn, Y.K.; Park, C.-S.; Kim, J.J. CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc. Res. 2009, 84, 378–386. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Forrest, C.M.; Gould, S.R.; Darlington, L.G.; Stone, T.W. Levels of Purine, Kynurenine and Lipid Peroxidation Products in Patients with Inflammatory Bowel Disease. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2003; Volume 527, pp. 395–400. [Google Scholar]
- Dolecka, J.; Urbanik-Sypniewska, T.; SkrzydŁo-Radomañska, B.; Parada-Turska, J. Effect of kynurenic acid on the viability of probiotics in vitro. Pharmacol. Rep. 2011, 63, 548–551. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Taschler, U.; Storr, M.; Schicho, R. The gastrointestinal tract—A central organ of cannabinoid signaling in health and disease. Neurogastroenterol. Motil. 2016, 28, 1765–1780. [Google Scholar] [CrossRef]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tonai-Kachi, H.; Shinjo, K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006, 580, 5003–5008. [Google Scholar] [CrossRef]
- Imielinski, M.; Baldassano, R.N.; Griffiths, A.; Russell, R.K.; Annese, V.; Dubinsky, M.; Kugathasan, S.; Bradfield, J.P.; Walters, T.D.; Sleiman, P.; et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009, 41, 1335–1340. [Google Scholar] [CrossRef]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef]
- Marques, T.R.; Ashok, A.H.; Pillinger, T.; Veronese, M.; Turkheimer, F.E.; Dazzan, P.; Sommer, I.E.C.; Howes, O.D. Neuroinflammation in schizophrenia: Meta-analysis of in vivo microglial imaging studies. Psychol. Med. 2019, 49, 2186–2196. [Google Scholar] [CrossRef]
- Wildenauer, D.B.; Körschenhausen, D.; Hoechtlen, W.; Ackenheil, M.; Kehl, M.; Lottspeich, F. Analysis of cerebrospinal fluid from patients with psychiatric and neurological disorders by two-dimensional electrophoresis: Identification of disease-associated polypeptides as fibrin fragments. Electrophoresis 1991, 12, 487–492. [Google Scholar] [CrossRef]
- Körschenhausen, D.A.; Hampel, H.J.; Ackenheil, M.; Penning, R.; Müller, N. Fibrin degradation products in post mortem brain tissue of schizophrenics: A possible marker for underlying inflammatory processes. Schizophr. Res. 1996, 19, 103–109. [Google Scholar] [CrossRef]
- Aricioglu, F.; Ozkartal, C.S.; Unal, G.; Dursun, S.; Cetin, M.; Müller, N. Neuroinflammation in Schizophrenia: A Critical Review and The Future. Klin. Psikofarmakol. Bülteni-Bull. Clin. Psychopharmacol. 2016, 26, 429–437. [Google Scholar] [CrossRef]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory Cytokine Alterations in Schizophrenia: A Systematic Quantitative Review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Steiner, J.; Bogerts, B. Glial cells in schizophrenia: Pathophysiological significance and possible consequences for therapy. Expert Rev. Neurother. 2009, 9, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Mawrin, C.; Ziegeler, A.; Bielau, H.; Ullrich, O.; Bernstein, H.-G.; Bogerts, B. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol. 2006, 112, 305–316. [Google Scholar] [CrossRef] [PubMed]
- De Picker, L.J.; Morrens, M.; Chance, S.A.; Boche, D. Microglia and Brain Plasticity in Acute Psychosis and Schizophrenia Illness Course: A Meta-Review. Front. Psychiatry 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Kato, T.A.; Mizoguchi, Y.; Horikawa, H.; Seki, Y.; Kasai, M.; Yamauchi, Y.; Yamada, S.; Kanba, S. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 42, 115–121. [Google Scholar] [CrossRef]
- Monji, A.; Kato, T.; Kanba, S. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin. Neurosci. 2009, 63, 257–265. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R.S. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef]
- Steiner, J.; Bogerts, B.; Sarnyai, Z.; Walter, M.; Gos, T.; Bernstein, H.-G.; Myint, A.-M. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: Potential role of glial NMDA receptor modulators and impaired blood–brain barrier integrity. World J. Biol. Psychiatry 2012, 13, 482–492. [Google Scholar] [CrossRef]
- Frank, M.G.; Baratta, M.V.; Sprunger, D.B.; Watkins, L.R.; Maier, S.F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain. Behav. Immun. 2007, 21, 47–59. [Google Scholar] [CrossRef]
- Perry, V.H. Stress primes microglia to the presence of systemic inflammation: Implications for environmental influences on the brain. Brain. Behav. Immun. 2007, 21, 45–46. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Choudhury, S.; Musaelyan, K.; Myint, A.M.; Thuret, S.; Price, J.; Pariante, C.M. Interleukin-1β: A new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 2012, 37, 939–949. [Google Scholar] [CrossRef]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.; Boronow, J.; Stallings, C.; Origoni, A.; Yolken, R. Toxoplasma gondii in individuals with schizophrenia: Association with clinical and demographic factors and with mortality. Schizophr. Bull. 2007, 33, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B.; Nørgaard-Pedersen, B.; Waltoft, B.L.; Sørensen, T.L.; Hougaard, D.; Yolken, R.H. Early infections of Toxoplasma gondii and the later development of schizophrenia. Schizophr. Bull. 2007, 33, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, Z.; Yazar, S.; Gecici, O.; Namli, M.N. Anti-Toxoplasma gondii antibodies in patients with schizophrenia--preliminary findings in a Turkish sample. Schizophr. Bull. 2007, 33, 789–791. [Google Scholar] [CrossRef]
- Schwarcz, R.; Hunter, C.A. Toxoplasma gondii and schizophrenia: Linkage through astrocyte-derived kynurenic acid? Schizophr. Bull. 2007, 33, 652–653. [Google Scholar] [CrossRef]
- Notarangelo, F.M.; Wilson, E.H.; Horning, K.J.; Thomas, M.A.R.; Harris, T.H.; Fang, Q.; Hunter, C.A.; Schwarcz, R. Evaluation of kynurenine pathway metabolism in Toxoplasma gondii-infected mice: Implications for schizophrenia. Schizophr. Res. 2014, 152, 261–267. [Google Scholar] [CrossRef]
- Fujigaki, S.; Saito, K.; Takemura, M.; Maekawa, N.; Yamada, Y.; Wada, H.; Seishima, M. L-tryptophan-L-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: Cross-regulation between inducible nitric oxide synthase and indoleamine-2,3-dioxygenase. Infect. Immun. 2002, 70, 779–786. [Google Scholar] [CrossRef]
- Silva, N.M.; Rodrigues, C.V.; Santoro, M.M.; Reis, L.F.L.; Alvarez-Leite, J.I.; Gazzinelli, R.T. Expression of indoleamine 2,3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: Induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect. Immun. 2002, 70, 859–868. [Google Scholar] [CrossRef]
- Parrott, J.M.; Redus, L.; O’Connor, J.C. Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J. Neuroinflamm. 2016, 13, 124. [Google Scholar] [CrossRef]
- De Campos-Carli, S.M.; Araújo, M.S.; de Oliveira Silveira, A.C.; de Rezende, V.B.; Rocha, N.P.; Ferretjans, R.; Ribeiro-Santos, R.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; Berk, M.; et al. Cannabinoid receptors on peripheral leukocytes from patients with schizophrenia: Evidence for defective immunomodulatory mechanisms. J. Psychiatr. Res. 2017, 87, 44–52. [Google Scholar] [CrossRef]
- Schaefer, C.; Enning, F.; Mueller, J.K.; Bumb, J.M.; Rohleder, C.; Odorfer, T.M.; Klosterkötter, J.; Hellmich, M.; Koethe, D.; Schmahl, C.; et al. Fatty acid ethanolamide levels are altered in borderline personality and complex posttraumatic stress disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, S.; Green-Johnson, J.M.; Murray, L.; Nance, D.M.; Dyck, D.; Anisman, H.; Greenberg, A.H. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994, 643, 40–49. [Google Scholar] [CrossRef]
- Busse, S.; Busse, M.; Schiltz, K.; Bielau, H.; Gos, T.; Brisch, R.; Mawrin, C.; Schmitt, A.; Jordan, W.; Müller, U.J.; et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: Further evidence for disease course-related immune alterations? Brain. Behav. Immun. 2012, 26, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Hickie, I.B.; Banati, R.; Stewart, C.H.; Lloyd, A.R. Are common childhood or adolescent infections risk factors for schizophrenia and other psychotic disorders? Med. J. Aust. 2009, 190, S17–S21. [Google Scholar] [CrossRef]
- Leweke, F.M.; Koethe, D. Cannabis and psychiatric disorders: It is not only addiction. Addict. Biol. 2008, 13, 264–275. [Google Scholar] [CrossRef]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.J.T.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 2009, 284, 19189–19195. [Google Scholar] [CrossRef]
- Gasperi, V.; Evangelista, D.; Chiurchiù, V.; Florenzano, F.; Savini, I.; Oddi, S.; Avigliano, L.; Catani, M.V.; Maccarrone, M. 2-Arachidonoylglycerol modulates human endothelial cell/leukocyte interactions by controlling selectin expression through CB1 and CB2 receptors. Int. J. Biochem. Cell Biol. 2014, 51, 79–88. [Google Scholar] [CrossRef]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef]
- Kianian, M.; Al-Banna, N.A.; Kelly, M.E.M.; Lehmann, C. Inhibition of endocannabinoid degradation in experimental endotoxemia reduces leukocyte adhesion and improves capillary perfusion in the gut. J. Basic Clin. Physiol. Pharmacol. 2013, 24, 27–33. [Google Scholar] [CrossRef]
- Lunn, C.A.; Fine, J.S.; Rojas-Triana, A.; Jackson, J.V.; Fan, X.; Kung, T.T.; Gonsiorek, W.; Schwarz, M.A.; Lavey, B.; Kozlowski, J.A.; et al. A Novel Cannabinoid Peripheral Cannabinoid Receptor-Selective Inverse Agonist Blocks Leukocyte Recruitment in Vivo. J. Pharmacol. Exp. Ther. 2006, 316, 780–788. [Google Scholar] [CrossRef]
- Montecucco, F.; Burger, F.; Mach, F.; Steffens, S. CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. AJP Hear. Circ. Physiol. 2007, 294, H1145–H1155. [Google Scholar] [CrossRef] [PubMed]
- Murikinati, S.; Jüttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010, 24, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Serritella, A.V.; Sedlak, T.W. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Fraguas, D.; Díaz-Caneja, C.M.; Rodríguez-Quiroga, A.; Arango, C. Oxidative Stress and Inflammation in Early Onset First Episode Psychosis: A Systematic Review and Meta-Analysis. Int. J. Neuropsychopharmacol. 2017, 20, 435–444. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 2002, 70, 299–307. [Google Scholar] [CrossRef]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The Endogenous Tryptophan Metabolite and NAD+ Precursor Quinolinic Acid Confers Resistance of Gliomas to Oxidative Stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef]
- Goda, K.; Kishimoto, R.; Shimizu, S.; Hamane, Y.; Ueda, M. Quinolinic acid and active oxygens. Possible contribution of active Oxygens during cell death in the brain. Adv. Exp. Med. Biol. 1996, 398, 247–254. [Google Scholar]
- Rodríguez-Martínez, E.; Camacho, A.; Maldonado, P.D.; Pedraza-Chaverrí, J.; Santamaría, D.; Galván-Arzate, S.; Santamaría, A. Effect of quinolinic acid on endogenous antioxidants in rat corpus striatum. Brain Res. 2000, 858, 436–439. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Iizuka, H.; Yokota, A.; Suzuki, T.; Ohno, C.; Kono, Y.; Nishikiori, M.; Seki, A.; Ichiba, H.; Watanabe, Y.; et al. Quantitative analyses of schizophrenia-associated metabolites in serum: Serum D-lactate levels are negatively correlated with gamma-glutamylcysteine in medicated schizophrenia patients. PLoS ONE 2014, 9, e101652. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 Cannabinoid Receptor Agonist Ameliorates Alzheimer-Like Phenotype in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Mnich, K.; Finn, D.P.; Dowd, E.; Gorman, A.M. Inhibition by Anandamide of 6-Hydroxydopamine-Induced Cell Death in PC12 Cells. Int. J. Cell Biol. 2010, 2010, 818497. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Patel, V.; Kashiwaya, Y.; Liaudet, L.; Evgenov, O.V.; Mackie, K.; Haskó, G.; Pacher, P. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 2010, 85, 773–784. [Google Scholar] [CrossRef]
- Severance, E.G.; Prandovszky, E.; Castiglione, J.; Yolken, R.H. Gastroenterology issues in schizophrenia: Why the gut matters. Curr. Psychiatry Rep. 2015, 17, 27. [Google Scholar] [CrossRef]
- Daneman, R.; Rescigno, M. The gut immune barrier and the blood-brain barrier: Are they so different? Immunity 2009, 31, 722–735. [Google Scholar] [CrossRef]
- Gupta, S.; Masand, P.S.; Kaplan, D.; Bhandary, A.; Hendricks, S. The relationship between schizophrenia and irritable bowel syndrome (IBS). Schizophr. Res. 1997, 23, 265–268. [Google Scholar] [CrossRef]
- Fadgyas-Stanculete, M.; Buga, A.-M.; Popa-Wagner, A.; Dumitrascu, D.L. The relationship between irritable bowel syndrome and psychiatric disorders: From molecular changes to clinical manifestations. J. Mol. Psychiatry 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Petra, A.I.; Panagiotidou, S.; Hatziagelaki, E.; Stewart, J.M.; Conti, P.; Theoharides, T.C. Gut-Microbiota-Brain Axis and Its Effect on Neuropsychiatric Disorders With Suspected Immune Dysregulation. Clin. Ther. 2015, 37, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Han, Y.; Du, J.; Liu, R.; Jin, K.; Yi, W. Microbiota-gut-brain axis and the central nervous system. Oncotarget 2017, 8, 53829–53838. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Salbaum, J.M.; Berthoud, H.-R. Harnessing Gut Microbes for Mental Health: Getting From Here to There. Biol. Psychiatry 2018, 83, 214–223. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, J.; Li, Z.; Huang, Y.; Yuan, Y.; Wang, J.; Zhang, M.; Hu, S.; Liang, Y. Analysis of gut microbiota diversity and auxiliary diagnosis as a biomarker in patients with schizophrenia: A cross-sectional study. Schizophr. Res. 2018, 197, 470–477. [Google Scholar] [CrossRef]
- Yolken, R.H.; Severance, E.G.; Sabunciyan, S.; Gressitt, K.L.; Chen, O.; Stallings, C.; Origoni, A.; Katsafanas, E.; Schweinfurth, L.A.B.; Savage, C.L.G.; et al. Metagenomic Sequencing Indicates That the Oropharyngeal Phageome of Individuals With Schizophrenia Differs From That of Controls. Schizophr. Bull. 2015, 41, 1153–1161. [Google Scholar] [CrossRef]
- Schwarz, E.; Maukonen, J.; Hyytiäinen, T.; Kieseppä, T.; Orešič, M.; Sabunciyan, S.; Mantere, O.; Saarela, M.; Yolken, R.; Suvisaari, J. Analysis of microbiota in first episode psychosis identifies preliminary associations with symptom severity and treatment response. Schizophr. Res. 2018, 192, 398–403. [Google Scholar] [CrossRef]
- Severance, E.G.; Gressitt, K.L.; Stallings, C.R.; Origoni, A.E.; Khushalani, S.; Leweke, F.M.; Dickerson, F.B.; Yolken, R.H. Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr. Res. 2013, 148, 130–137. [Google Scholar] [CrossRef]
- Castro-Nallar, E.; Bendall, M.L.; Pérez-Losada, M.; Sabuncyan, S.; Severance, E.G.; Dickerson, F.B.; Schroeder, J.R.; Yolken, R.H.; Crandall, K.A. Composition, taxonomy and functional diversity of the oropharynx microbiome in individuals with schizophrenia and controls. PeerJ 2015, 3, e1140. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, P.; Wang, Y.; Liu, Y.; Li, X.; Kumar, B.U.; Hei, G.; Lv, L.; Huang, X.-F.; Fan, X.; et al. Changes in metabolism and microbiota after 24-week risperidone treatment in drug naïve, normal weight patients with first episode schizophrenia. Schizophr. Res. 2018, 201, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.P.; Turska, M.; Kocki, T.; Turski, W.A.; Paluszkiewicz, P. Kynurenic Acid Content in Selected Culinary Herbs and Spices. J. Chem. 2015, 2015, 617571. [Google Scholar] [CrossRef]
- Turski, M.P.; Turska, M.; Zgrajka, W.; Kuc, D.; Turski, W.A. Presence of kynurenic acid in food and honeybee products. Amino Acids 2009, 36, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kuc, D.; Rahnama, M.; Tomaszewski, T.; Rzeski, W.; Wejksza, K.; Urbanik-Sypniewska, T.; Parada-Turska, J.; Wielosz, M.; Turski, W.A. Kynurenic acid in human saliva—Does it influence oral microflora? Pharmacol. Rep. 2006, 58, 393–398. [Google Scholar] [PubMed]
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006, 20, 568–570. [Google Scholar] [CrossRef]
- Müller, N. Immunological aspects of the treatment of depression and schizophrenia. Dialogues Clin. Neurosci. 2017, 19, 55–63. [Google Scholar]
- McGuire, P.; Robson, P.; Cubala, W.J.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef]
- Pedrazzi, J.F.C.; Issy, A.C.; Gomes, F.V.; Guimarães, F.S.; Del-Bel, E.A. Cannabidiol effects in the prepulse inhibition disruption induced by amphetamine. Psychopharmacology 2015, 232, 3057–3065. [Google Scholar] [CrossRef]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef]
- Beltramo, M.; de Fonseca, F.R.; Navarro, M.; Calignano, A.; Gorriti, M.A.; Grammatikopoulos, G.; Sadile, A.G.; Giuffrida, A.; Piomelli, D. Reversal of dopamine D(2) receptor responses by an anandamide transport inhibitor. J. Neurosci. 2000, 20, 3401–3407. [Google Scholar] [CrossRef]
- Seillier, A.; Advani, T.; Cassano, T.; Hensler, J.G.; Giuffrida, A. Inhibition of fatty-acid amide hydrolase and CB1 receptor antagonism differentially affect behavioural responses in normal and PCP-treated rats. Int. J. Neuropsychopharmacol. 2010, 13, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, D.D.; Giuffrida, A.; Lodge, D.J. Adolescent Synthetic Cannabinoid Exposure Produces Enduring Changes in Dopamine Neuron Activity in a Rodent Model of Schizophrenia Susceptibility. Int. J. Neuropsychopharmacol. 2018, 21, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, D.D.; Chen, L.; Lodge, D.J. Increasing Endocannabinoid Levels in the Ventral Pallidum Restore Aberrant Dopamine Neuron Activity in the Subchronic PCP Rodent Model of Schizophrenia. Int. J. Neuropsychopharmacol. 2014, 18, pyu035. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, J.; Ma, L.; Wu, M.; Zhang, L.; Zhang, X.; Zhai, Q.; Jiang, T.; Wang, Q.; Xiong, L. Anandamide protects HT22 cells exposed to hydrogen peroxide by inhibiting CB1 receptor-mediated type 2 NADPH oxidase. Oxid. Med. Cell. Longev. 2014, 2014, 893516. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, G.; Karajgi, B.; McCarthy, R. Synthetic Δ-9-Tetrahydrocannabinol (Dronabinol) Can Improve the Symptoms of Schizophrenia. J. Clin. Psychopharmacol. 2009, 29, 255–258. [Google Scholar] [CrossRef]
- Tzavara, E.T.; Degroot, A.; Wade, M.R.; Davis, R.J.; Nomikos, G.G. CB1 receptor knockout mice are hyporesponsive to the behavior-stimulating actions of d-amphetamine: Role of mGlu5 receptors. Eur. Neuropsychopharmacol. 2009, 19, 196–204. [Google Scholar] [CrossRef]
- Tzavara, E.T.; Davis, R.J.; Perry, K.W.; Li, X.; Salhoff, C.; Bymaster, F.P.; Witkin, J.M.; Nomikos, G.G. The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: Implications for therapeutic actions. Br. J. Pharmacol. 2003, 138, 544–553. [Google Scholar] [CrossRef]
- Boggs, D.L.; Kelly, D.L.; McMahon, R.P.; Gold, J.M.; Gorelick, D.A.; Linthicum, J.; Conley, R.R.; Liu, F.; Waltz, J.; Huestis, M.A.; et al. Rimonabant for neurocognition in schizophrenia: A 16-week double blind randomized placebo controlled trial. Schizophr. Res. 2012, 134, 207–210. [Google Scholar] [CrossRef]
- Crismon, L.; Argo, T.R.; Buckley, P.F. Schizophrenia. In Pharmacotherapy: A Pathophysiologic Approach; McGraw-Hill: New York, NY, USA, 2014; pp. 1019–1046. [Google Scholar]
- Tandon, R. Antipsychotics in the Treatment of Schizophrenia. J. Clin. Psychiatry 2011, 72, 4–8. [Google Scholar] [CrossRef]
- Seeman, P. Dopamine receptor sequences. Therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacology 1992, 7, 261–284. [Google Scholar]
- Miyamoto, S.; Miyake, N.; Jarskog, L.F.; Fleischhacker, W.W.; Lieberman, J.A. Pharmacological treatment of schizophrenia: A critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 2012, 17, 1206–1227. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Duncan, G.E.; Marx, C.E.; Lieberman, J.A. Treatments for schizophrenia: A critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 2005, 10, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Fleischhacker, W.W. Emerging drugs for schizophrenia. Expert Opin. Emerg. Drugs 2011, 16, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.F.; Miyamoto, S.; Lieberman, J.A. Schizophrenia: New Pathological Insights and Therapies. Annu. Rev. Med. 2007, 58, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.K.; Bishop, J.R.; Palumbo, D.; Sweeney, J.A. Effect of second-generation antipsychotics on cognition: Current issues and future challenges. Expert Rev. Neurother. 2010, 10, 43–57. [Google Scholar] [CrossRef]
- Kuroki, T.; Nagao, N.; Nakahara, T. Neuropharmacology of second-generation antipsychotic drugs: A validity of the serotonin–dopamine hypothesis. Prog. Brain Res. 2008, 172, 199–212. [Google Scholar]
- Meltzer, H.Y.; Matsubara, S.; Lee, J.C. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 1989, 251, 238–246. [Google Scholar]
- MacKenzie, N.E.; Kowalchuk, C.; Agarwal, S.M.; Costa-Dookhan, K.A.; Caravaggio, F.; Gerretsen, P.; Chintoh, A.; Remington, G.J.; Taylor, V.H.; Müeller, D.J.; et al. Antipsychotics, Metabolic Adverse Effects, and Cognitive Function in Schizophrenia. Front. Psychiatry 2018, 9, 622. [Google Scholar] [CrossRef]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar]
- Lieberman, J.A. Dopamine Partial Agonists. CNS Drugs 2004, 18, 251–267. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.-X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Horacek, J.; Bubenikova-Valesova, V.; Kopecek, M.; Palenicek, T.; Dockery, C.; Mohr, P.; Höschl, C. Mechanism of Action of Atypical Antipsychotic Drugs and the Neurobiology of Schizophrenia. CNS Drugs 2006, 20, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Amato, D.; Kruyer, A.; Samaha, A.-N.; Heinz, A. Hypofunctional Dopamine Uptake and Antipsychotic Treatment-Resistant Schizophrenia. Front. Psychiatry 2019, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Coyle, J.T. The Emerging Role of Glutamate in the Pathophysiology and Treatment of Schizophrenia. Am. J. Psychiatry 2001, 158, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Tsai, G.; Lin, P.-Y. Strategies to Enhance N-Methyl-D-Aspartate Receptor-Mediated Neurotransmission in Schizophrenia, a Critical Review and Meta-Analysis. Curr. Pharm. Des. 2010, 16, 522–537. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Silipo, G.; Lichtenstein, M. Efficacy of High-Dose Glycine in the Treatment of Enduring Negative Symptoms of Schizophrenia. Arch. Gen. Psychiatry 1999, 56, 29–36. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zylberman, I.; Zukin, S.R.; Heresco-Levy, U.; Lindenmayer, J.P. Amelioration of negative symptoms in schizophrenia by glycine. Am. J. Psychiatry 1994, 151, 1234–1236. [Google Scholar]
- Diaz, P.; Bhaskara, S.; Dursun, S.M.; Deakin, B. Double-blind, placebo-controlled, crossover trial of clozapine plus glycine in refractory schizophrenia negative results. J. Clin. Psychopharmacol. 2005, 25, 277–278. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Javitt, D.C.; Marder, S.R.; Schooler, N.R.; Gold, J.M.; McMahon, R.P.; Heresco-Levy, U.; Carpenter, W.T. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): The Efficacy of Glutamatergic Agents for Negative Symptoms and Cognitive Impairments. Am. J. Psychiatry 2007, 164, 1593–1602. [Google Scholar] [CrossRef]
- Cain, C.K.; McCue, M.; Bello, I.; Creedon, T.; Tang, D.; Laska, E.; Goff, D.C. d-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr. Res. 2014, 153, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C. D-cycloserine in Schizophrenia: New Strategies for Improving Clinical Outcomes by Enhancing Plasticity. Curr. Neuropharmacol. 2017, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, A.; Pakseresht, S.; Haghdoost, M.R.; Hekmatkhah, N.; Torkashvand, M.; Ghorbanzadeh, B. Memantine Enhances the Effect of Olanzapine in Patients With Schizophrenia: A Randomized, Placebo-Controlled Study. Acta Med. Iran. 2016, 54, 696–703. [Google Scholar] [PubMed]
- De Lucena, D.; Fernandes, B.S.; Berk, M.; Dodd, S.; Medeiros, D.W.; Pedrini, M.; Kunz, M.; Gomes, F.A.; Giglio, L.F.; Lobato, M.I.; et al. Improvement of Negative and Positive Symptoms in Treatment-Refractory Schizophrenia. J. Clin. Psychiatry 2009, 70, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Marenco, S.; Egan, M.F.; Goldberg, T.E.; Knable, M.B.; McClure, R.K.; Winterer, G.; Weinberger, D.R. Preliminary experience with an ampakine (CX516) as a single agent for the treatment of schizophrenia: A case series. Schizophr. Res. 2002, 57, 221–226. [Google Scholar] [CrossRef]
- Tsai, G.; Lane, H.-Y.; Yang, P.; Chong, M.-Y.; Lange, N. Glycine transporter I inhibitor, N-Methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 2004, 55, 452–456. [Google Scholar] [CrossRef]
- Lane, H.-Y.; Huang, C.-L.; Wu, P.-L.; Liu, Y.-C.; Chang, Y.-C.; Lin, P.-Y.; Chen, P.-W.; Tsai, G. Glycine Transporter I Inhibitor, N-methylglycine (Sarcosine), Added to Clozapine for the Treatment of Schizophrenia. Biol. Psychiatry 2006, 60, 645–649. [Google Scholar] [CrossRef]
- Lane, H.-Y.; Lin, C.-H.; Huang, Y.-J.; Liao, C.-H.; Chang, Y.-C.; Tsai, G.E. A randomized, double-blind, placebo-controlled comparison study of sarcosine ( N-methylglycine) and d-serine add-on treatment for schizophrenia. Int. J. Neuropsychopharmacol. 2010, 13, 451–460. [Google Scholar] [CrossRef]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef]
- Lewis, D.A.; Cho, R.Y.; Carter, C.S.; Eklund, K.; Forster, S.; Kelly, M.A.; Montrose, D. Subunit-Selective Modulation of GABA Type A Receptor Neurotransmission and Cognition in Schizophrenia. Am. J. Psychiatry 2008, 165, 1585–1593. [Google Scholar] [CrossRef]
- Heyes, M.P.; Chen, C.Y.; Major, E.O.; Saito, K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem. J. 1997, 326 Pt 2, 351–356. [Google Scholar] [CrossRef]
- Stone, T.W.; Darlington, L.G. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br. J. Pharmacol 2013, 169, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Elmer, G.I.; Schwarcz, R. Inhibition of Kynurenine Aminotransferase II Attenuates Hippocampus-dependent Memory Deficit in Adult Rats Treated Prenatally with Kynurenine. Hippocampus 2019, 29, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Wu, H.-Q.; Marinozzi, M.; Pellicciari, R.; Guidetti, P.; Schwarcz, R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience 2009, 159, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Rizzo, R.C.; Costantino, G.; Marinozzi, M.; Amori, L.; Guidetti, P.; Wu, H.-Q.; Schwarcz, R. Modulators of the Kynurenine Pathway of Tryptophan Metabolism: Synthesis and Preliminary Biological Evaluation of (S)-4-(Ethylsulfonyl)benzoylalanine, a Potent and Selective Kynurenine Aminotransferase II (KAT II) Inhibitor. ChemMedChem 2006, 1, 528–531. [Google Scholar] [CrossRef]
- Konradsson-Geuken, Å.; Wu, H.Q.; Gash, C.R.; Alexander, K.S.; Campbell, A.; Sozeri, Y.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience 2010, 169, 1848–1859. [Google Scholar] [CrossRef]
- Zmarowski, A.; Wu, H.-Q.; Brooks, J.M.; Potter, M.C.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur. J. Neurosci. 2009, 29, 529–538. [Google Scholar] [CrossRef]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Claffey, M.M.; Evdokimov, A.; Evrard, E.; Fonseca, K.R.; Gan, X.; Ghosh, S.; et al. Discovery of Brain-Penetrant, Irreversible Kynurenine Aminotransferase II Inhibitors for Schizophrenia. ACS Med. Chem. Lett. 2012, 3, 187–192. [Google Scholar] [CrossRef]
- Koshy Cherian, A.; Gritton, H.; Johnson, D.E.; Young, D.; Kozak, R.; Sarter, M. A systemically-available kynurenine aminotransferase II (KAT II) inhibitor restores nicotine-evoked glutamatergic activity in the cortex of rats. Neuropharmacology 2014, 82, 41–48. [Google Scholar] [CrossRef]
- Linderholm, K.R.; Alm, M.T.; Larsson, M.K.; Olsson, S.K.; Goiny, M.; Hajos, M.; Erhardt, S.; Engberg, G. Inhibition of kynurenine aminotransferase II reduces activity of midbrain dopamine neurons. Neuropharmacology 2016, 102, 42–47. [Google Scholar] [CrossRef]
- Henderson, J.L.; Sawant-Basak, A.; Tuttle, J.B.; Dounay, A.B.; McAllister, L.A.; Pandit, J.; Rong, S.; Hou, X.; Bechle, B.M.; Kim, J.-Y.; et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. Med. Chem. Commun. 2013, 4, 125–129. [Google Scholar] [CrossRef]
- Bortz, D.M.; Wu, H.-Q.; Schwarcz, R.; Bruno, J.P. Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology 2017, 121, 69–78. [Google Scholar] [CrossRef]
- Réus, G.Z.; Becker, I.R.T.; Scaini, G.; Petronilho, F.; Oses, J.P.; Kaddurah-Daouk, R.; Ceretta, L.B.; Zugno, A.I.; Dal-Pizzol, F.; Quevedo, J.; et al. The inhibition of the kynurenine pathway prevents behavioral disturbances and oxidative stress in the brain of adult rats subjected to an animal model of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 81, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Morrison, P.D.; Fusar-Poli, P.; Martin-Santos, R.; Borgwardt, S.; Winton-Brown, T.; Nosarti, C.; O’ Carroll, C.M.; Seal, M.; Allen, P.; et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 2010, 35, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanuš, L.; de Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Seeman, P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl. Psychiatry 2016, 6, e920. [Google Scholar] [CrossRef]
- Ibeas Bih, C.; Chen, T.; Nunn, A.V.W.; Bazelot, M.; Dallas, M.; Whalley, B.J. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar] [CrossRef]
- Moreira, F.A.; Guimarães, F.S. Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur. J. Pharmacol. 2005, 512, 199–205. [Google Scholar] [CrossRef]
- Zuardi, A.W.; Morais, S.L.; Guimarães, F.S.; Mechoulam, R. Antipsychotic effect of cannabidiol. J. Clin. Psychiatry 1995, 56, 485–486. [Google Scholar]
- Zuardi, A.W.; Crippa, J.A.S.; Hallak, J.E.C.; Moreira, F.A.; Guimarães, F.S. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz. J. Med. Biol. Res. 2006, 39, 421–429. [Google Scholar] [CrossRef]
- Batalla, A.; Bhattacharyya, S.; Yücel, M.; Fusar-Poli, P.; Crippa, J.A.; Nogué, S.; Torrens, M.; Pujol, J.; Farré, M.; Martin-Santos, R. Structural and functional imaging studies in chronic cannabis users: A systematic review of adolescent and adult findings. PLoS ONE 2013, 8, e55821. [Google Scholar] [CrossRef]
- Hallak, J.E.C.; Machado-de-Sousa, J.P.; Crippa, J.A.S.; Sanches, R.F.; Trzesniak, C.; Chaves, C.; Bernardo, S.A.; Regalo, S.C.; Zuardi, A.W. Performance of schizophrenic patients in the Stroop Color Word Test and electrodermal responsiveness after acute administration of cannabidiol (CBD). Rev. Bras. Psiquiatr. 2010, 32, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Hallak, J.E.C.; Dursun, S.M.; Morais, S.L.; Sanches, R.F.; Musty, R.E.; Crippa, J.A.S. Cannabidiol monotherapy for treatment-resistant schizophrenia. J. Psychopharmacol. 2006, 20, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B. The Potential of Cannabidiol Treatment for Cannabis Users With Recent-Onset Psychosis. Schizophr. Bull. 2018, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Seillier, A.; Giuffrida, A. Distinct neuronal activation patterns are associated with PCP-induced social withdrawal and its reversal by the endocannabinoid-enhancing drug URB597. Neurosci. Res. 2016, 110, 49–58. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Arvanitis, L.; Bauer, D.; Rein, W.; Group, M.-T.S. Placebo-Controlled Evaluation of Four Novel Compounds for the Treatment of Schizophrenia and Schizoaffective Disorder. Am. J. Psychiatry 2004, 161, 975–984. [Google Scholar] [CrossRef]
- Bisogno, T.; Di Marzo, V. The role of the endocannabinoid system in Alzheimer’s disease: Facts and hypotheses. Curr. Pharm. Des. 2008, 14, 2299–3305. [Google Scholar] [CrossRef]
- Dezsi, L.; Tuka, B.; Martos, D.; Vecsei, L. Alzheimer’s disease, astrocytes and kynurenines. Curr. Alzheimer Res. 2015, 12, 462–480. [Google Scholar] [CrossRef]
- Miller, L.K.; Devi, L.A. The Highs and Lows of Cannabinoid Receptor Expression in Disease: Mechanisms and Their Therapeutic Implications. Pharmacol. Rev. 2011, 63, 461–470. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Points and Therapeutics | Kynurenines | ECS |
|---|---|---|
| Glutamatergic, dopaminergic, and GABAergic systems | [7,8] | [9,10] |
| Astrocytes | [11] | [12] |
| Inflammation | [13,14,15,16,17] | [18,19,20,21] |
| Therapeutics | [7,22,23,24,25,26,27] | [28,29,30] |
| Members | References |
|---|---|
| Kynurenines and associated elements | |
| KYNA | [7,8] |
| α7nAChR | [121] |
| ECS | |
| AEA, 2-AG | [122,123,124] |
| CB1R | [10,125,126,127] |
| Members and Features | References |
|---|---|
| Kynurenines and associated elements | |
| KYNA | [137] |
| KAT II | [35] |
| α7nAChR 1 | [138] |
| ECS | |
| AEA, 2-AG | [122,123] |
| DAGL, MAGL | [139,140] |
| CB1R | [141] |
| Common points | |
| Involved in the THC-induced enhanced glutamate release | [142] |
| Co-localized CB1R and α7nAChRs mRNA | [142] |
| Members and Features | References |
|---|---|
| Kynurenines and associated elements | |
| l-KYN, KYNA, 3-HK | [15,35,172] |
| KAT, IDO, KMO | [173,174,175] |
| GPR35 1, AHR 1 | [14,15] |
| ECS | |
| AEA, 2-AG | [176,177,178] |
| CB2R, CB1R | [19,179] |
| Common points | |
| Cytokine regulation, microglial activation | [19,35,175,177,178,179] |
| Oxidative stress | [16,17,20,180,181,182,183,184] |
| KYNA and endocannabinoids communicate with gut microbiome | [14,18,185,186,187] |
| Involvement in IBD | [18,186,188] |
| Common features of GPR35 and CBRs | [49,78,189,190,191] |
| Approaches | References |
|---|---|
| Kynurenine pathway | |
| KAT II inhibition | [22,23,24,25,27] |
| IDO, TDO KMO inhibition | [7,26,263] |
| ECS | |
| FAAH inhibition (including CBD) | [264,265,266,267,268,269,270,271] |
| CB1R activation | [272] |
| CB1R blockade | [273,274,275] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zádor, F.; Nagy-Grócz, G.; Kekesi, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Horvath, G.; Benyhe, S.; Vécsei, L. Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules 2019, 24, 3709. https://doi.org/10.3390/molecules24203709
Zádor F, Nagy-Grócz G, Kekesi G, Dvorácskó S, Szűcs E, Tömböly C, Horvath G, Benyhe S, Vécsei L. Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules. 2019; 24(20):3709. https://doi.org/10.3390/molecules24203709
Chicago/Turabian StyleZádor, Ferenc, Gábor Nagy-Grócz, Gabriella Kekesi, Szabolcs Dvorácskó, Edina Szűcs, Csaba Tömböly, Gyongyi Horvath, Sándor Benyhe, and László Vécsei. 2019. "Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions" Molecules 24, no. 20: 3709. https://doi.org/10.3390/molecules24203709
APA StyleZádor, F., Nagy-Grócz, G., Kekesi, G., Dvorácskó, S., Szűcs, E., Tömböly, C., Horvath, G., Benyhe, S., & Vécsei, L. (2019). Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules, 24(20), 3709. https://doi.org/10.3390/molecules24203709