X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid

 ,
,  , , , , and
, , , , and
Abstract
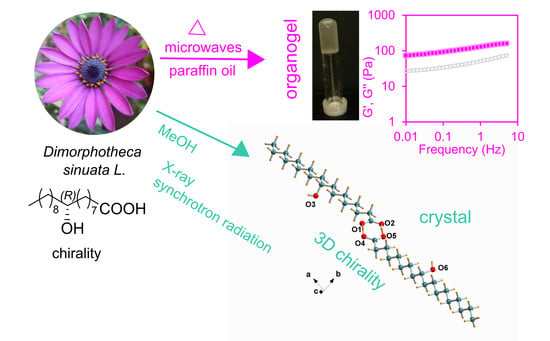
1. Introduction
2. Results
2.1. (R)-9-HSA Organogel
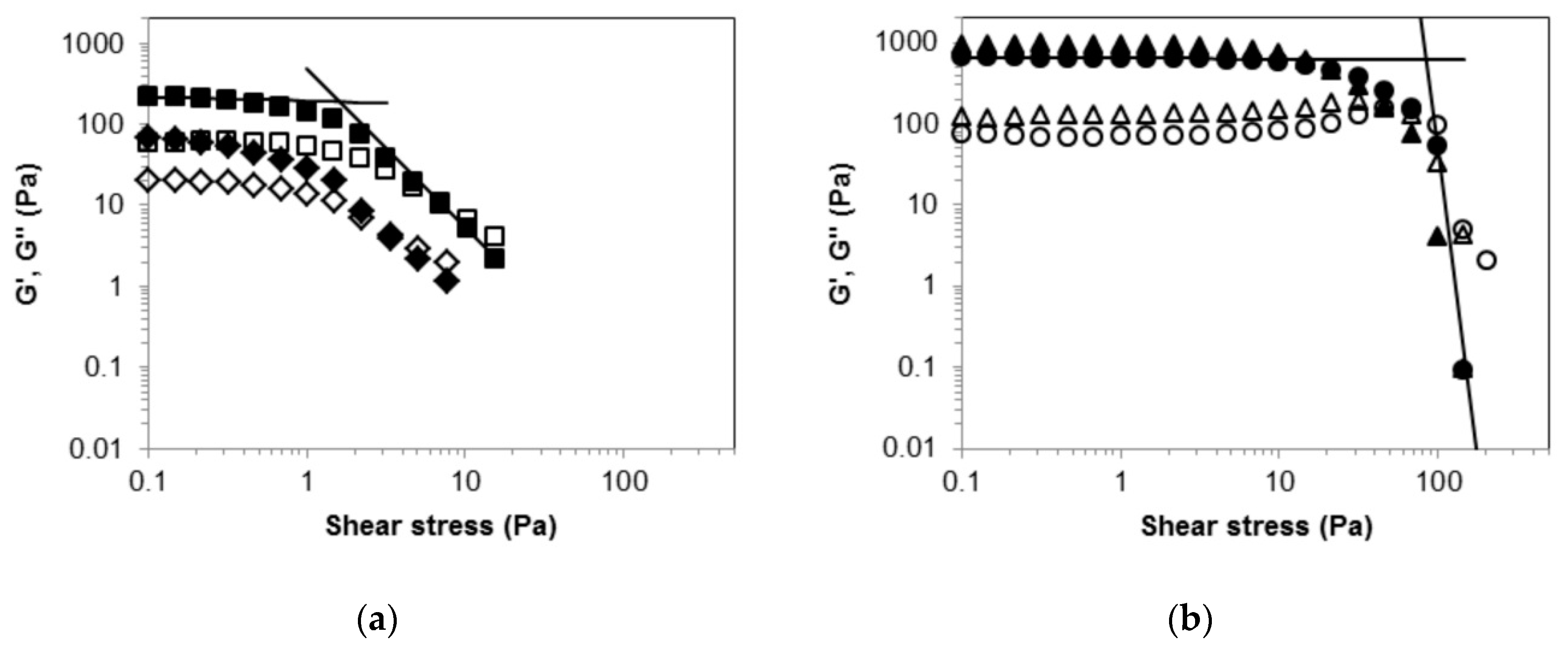
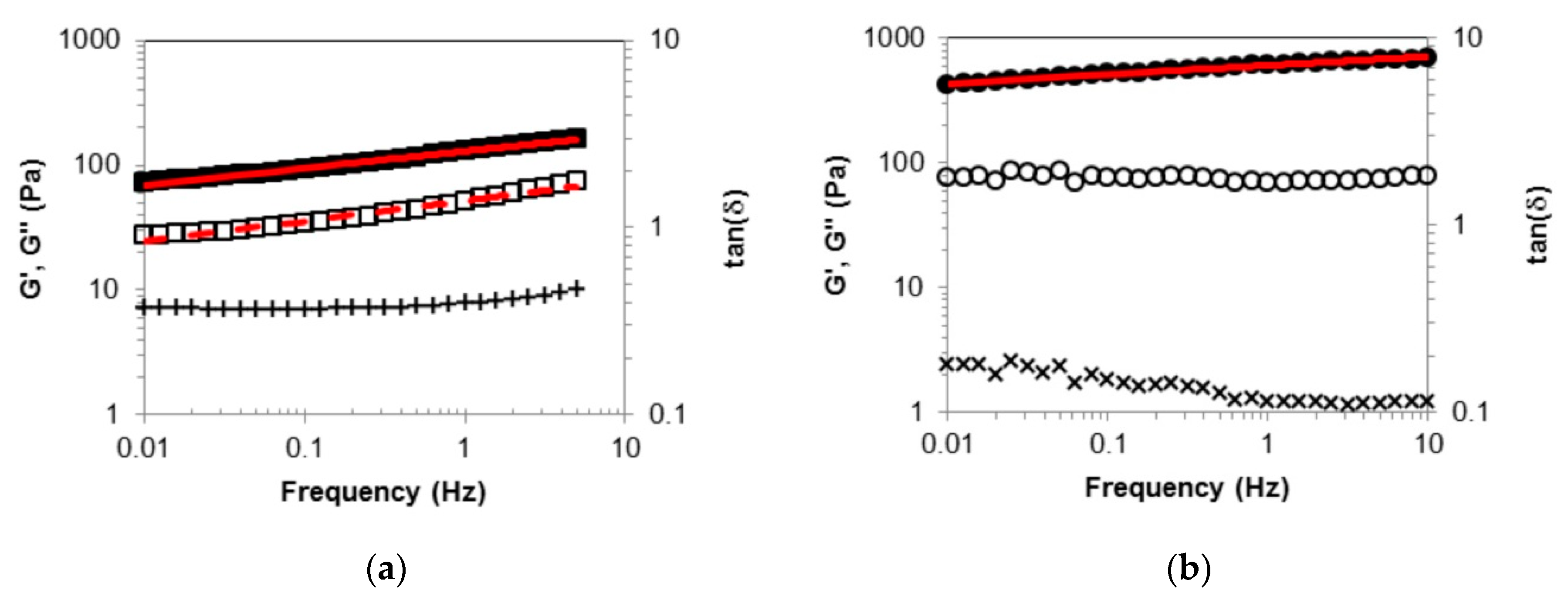
2.2. Rheological Measurements
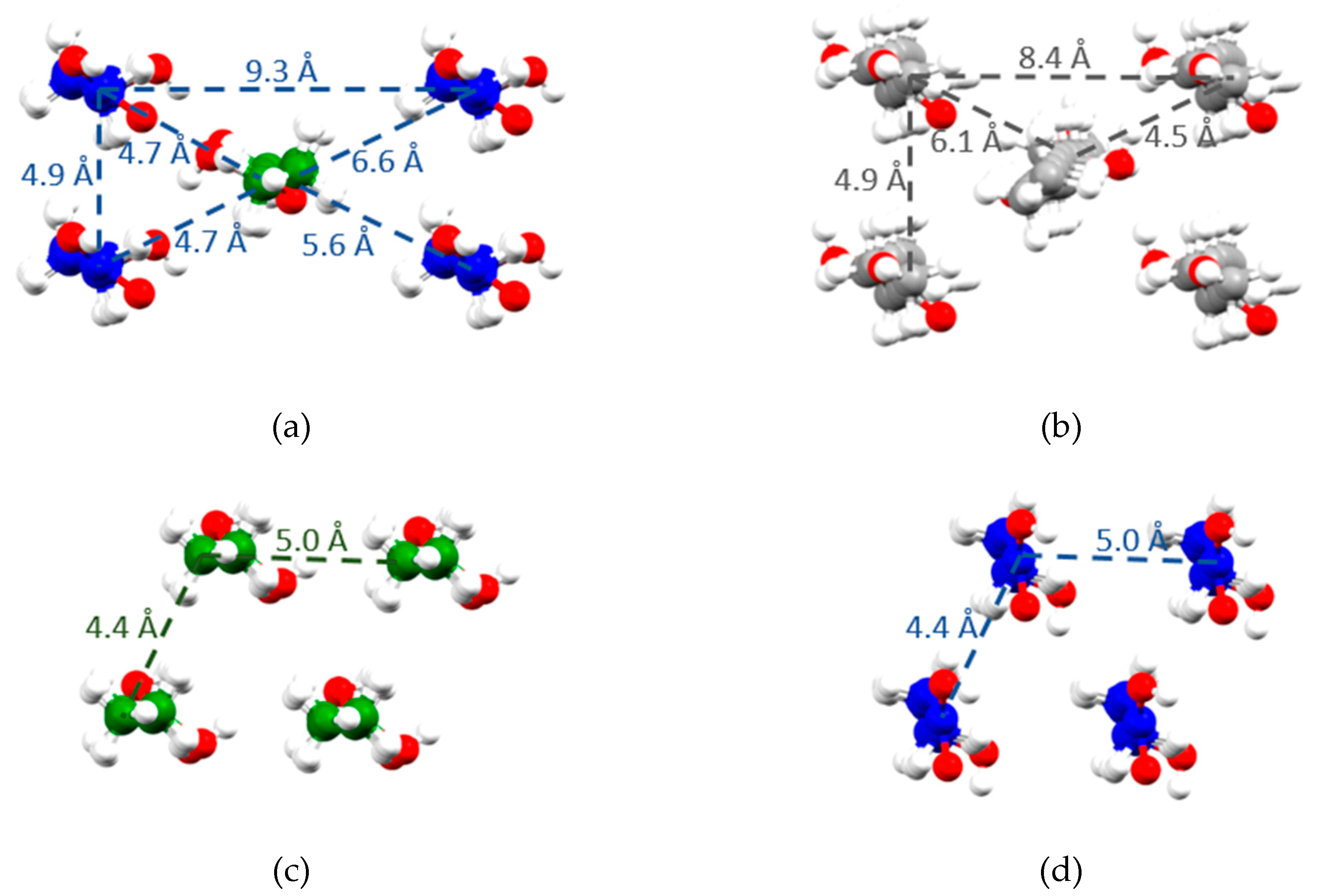
2.3. X-Ray Single Crystal Structure
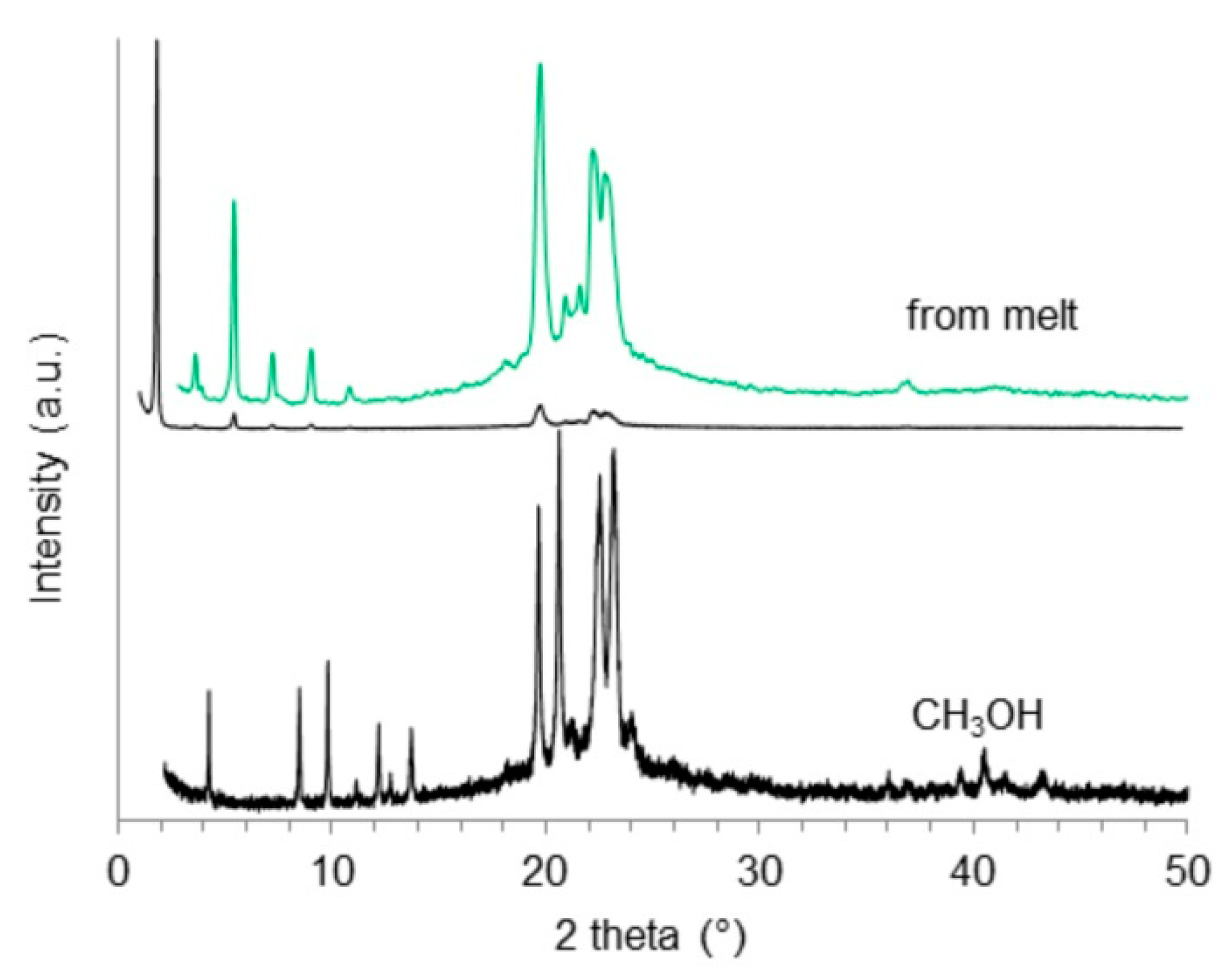
2.4. X-Ray Powder Diffraction
3. Discussion
4. Materials and Methods
4.1. Synthesis of (R)-9-Hydroxystearic Acid
4.2. High-Temperature Treatment of (R)-9-Hydroxystearic Acid
4.3. Gel Preparation
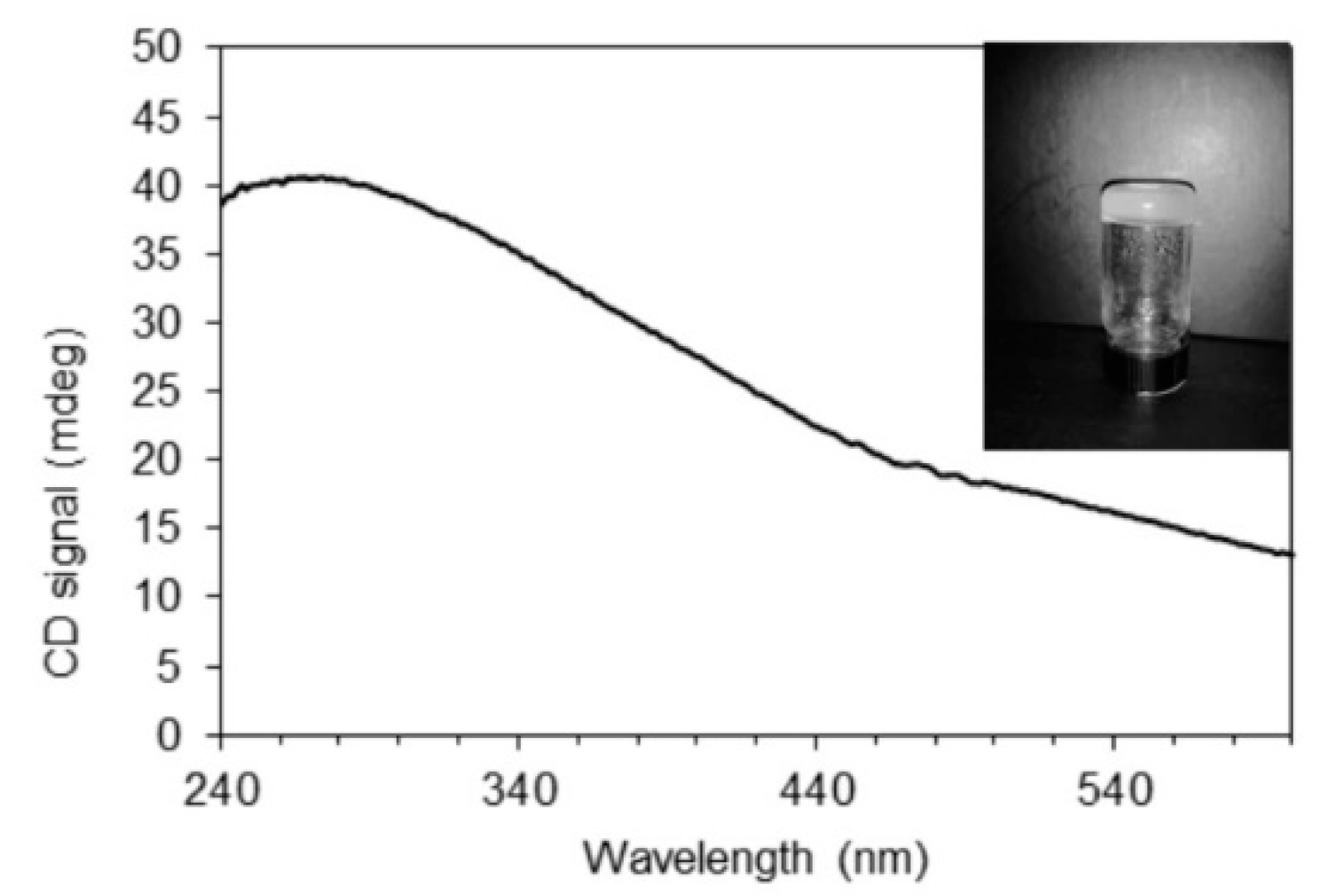
4.4. Circular Dichroism Measurements
4.5. Rheological Measurements
4.6. X-Ray Structural Analysis
4.7. Synchrotron X-Ray Powder Diffraction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mubofu, E.B. Castor oil as a potential renewable resource for the production of functional materials. Sustain. Chem. Process. 2016, 4, 11. [Google Scholar] [CrossRef]
- Ahmad, I.; Jie, M.S.F.L.K. Oleochemicals from Isoricinoleic Acid (Wrightia t inctoria Seed Oil). Ind. Eng. Chem. Res. 2008, 47, 2091–2095. [Google Scholar] [CrossRef]
- Pabiś, S.; Kula, J. Synthesis and Bioactivity of (R)-Ricinoleic Acid Derivatives: A Review. Curr. Med. Chem. 2016, 23, 4037–4056. [Google Scholar] [CrossRef] [PubMed]
- Sutradhar, M.; Fernandes, A.R.; Paradinha, F.; Rijo, P.; Garcia, C.; Roma-Rodrigues, C.; Pombeiro, A.J.L.; Charmier, A.J. A new Cu(II)-O-Carvacrotinate complex: Synthesis, characterization and biological activity. J. Inorg. Biochem. 2019, 190, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Farruggia, G.; Buontempo, F.; Boga, C.; Brusa, G.L.; Santucci, M.A.; Masotti, L. 9-Hydroxystearic acid upregulates p21WAF1 in HT29 cancer cells. Biochem. Biophys. Res. Commun. 2004, 314, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Boga, C.; Bertucci, C.; Fiori, J.; Tasco, G.; Casadio, R.; Masotti, L. Histone deacetylase 1: A target of 9-hydroxystearic acid in the inhibition of cell growth in human colon cancer. J. Lipid Res. 2005, 46, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Molinari, C.; Boga, C.; Dal Piaz, F.; Brusa, G.L.; Santucci, M.A.; Masotti, L. Modulation of apoptotic signalling by 9-hydroxystearic acid in osteosarcoma cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 139–146. [Google Scholar] [CrossRef]
- Albadri, S.; Naso, F.; Thauvin, M.; Gauron, C.; Parolin, C.; Duroure, K.; Vougny, J.; Fiori, J.; Boga, C.; Vriz, S.; et al. Redox Signaling via Lipid Peroxidation Regulates Retinal Progenitor Cell Differentiation. Dev. Cell 2019, 50, 73–89.e6. [Google Scholar] [CrossRef]
- Parolin, C.; Calonghi, N.; Presta, E.; Boga, C.; Caruana, P.; Naldi, M.; Andrisano, V.; Masotti, L.; Sartor, G. Mechanism and stereoselectivity of HDAC I inhibition by (R)-9-hydroxystearic acid in colon cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 1334–1340. [Google Scholar] [CrossRef]
- Badami, R.; Patil, K. Structure and occurrence of unusual fatty acids in minor seed oils. Prog. Lipid Res. 1980, 19, 119–153. [Google Scholar] [CrossRef]
- Ebert, C.; Felluga, F.; Forzato, C.; Foscato, M.; Gardossi, L.; Nitti, P.; Pitacco, G.; Boga, C.; Caruana, P.; Micheletti, G.; et al. Enzymatic kinetic resolution of hydroxystearic acids: A combined experimental and molecular modelling investigation. J. Mol. Catal. B Enzym. 2012, 83, 38–45. [Google Scholar] [CrossRef]
- Boanini, E.; Torricelli, P.; Boga, C.; Micheletti, G.; Cassani, M.C.; Fini, M.; Bigi, A. (9R)-9-Hydroxystearate-Functionalized Hydroxyapatite as Antiproliferative and Cytotoxic Agent toward Osteosarcoma Cells. Langmuir 2016, 32, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Boanini, E.; Cassani, M.; Rubini, K.; Boga, C.; Bigi, A. (9R)-9-Hydroxystearate-Functionalized Anticancer Ceramics Promote Loading of Silver Nanoparticles. Nanomaterials 2018, 8, 390. [Google Scholar] [CrossRef]
- Busi, A.; Aluigi, A.; Guerrini, A.; Boga, C.; Sartor, G.; Calonghi, N.; Sotgiu, G.; Posati, T.; Corticelli, F.; Fiori, J.; et al. Unprecedented Behavior of (9 R)-9-Hydroxystearic Acid-Loaded Keratin Nanoparticles on Cancer Cell Cycle. Mol. Pharm. 2019, 16, 931–942. [Google Scholar] [CrossRef]
- Terech, P.; Weiss, R.G. Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels. Chem. Rev. 1997, 97, 3133–3160. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, S.; Rogers, M.A. Harnessing Hansen solubility parameters to predict organogel formation. J. Mater. Chem. 2012, 22, 12651. [Google Scholar] [CrossRef]
- Weiss, R.G. The Past, Present, and Future of Molecular Gels. What Is the Status of the Field, and Where Is It Going? J. Am. Chem. Soc. 2014, 136, 7519–7530. [Google Scholar] [CrossRef]
- Abraham, S.; Lan, Y.; Lam, R.S.H.; Grahame, D.A.S.; Kim, J.J.H.; Weiss, R.G.; Rogers, M.A. Influence of Positional Isomers on the Macroscale and Nanoscale Architectures of Aggregates of Racemic Hydroxyoctadecanoic Acids in Their Molecular Gel, Dispersion, and Solid States. Langmuir 2012, 28, 4955–4964. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Abraham, S.; Rogers, M.A.; Dey, J.; Weiss, R.G. Comparison of Dipolar, H-Bonding, and Dispersive Interactions on Gelation Efficiency of Positional Isomers of Keto and Hydroxy Substituted Octadecanoic Acids. Langmuir 2013, 29, 6467–6475. [Google Scholar] [CrossRef]
- Terech, P.; Rodriguez, V.; Barnes, J.D.; McKenna, G.B. Organogels and Aerogels of Racemic and Chiral 12-Hydroxyoctadecanoic Acid. Langmuir 1994, 10, 3406–3418. [Google Scholar] [CrossRef]
- Tachibana, T.; Kambara, H. Studies of Helical Aggregates of Molecules. I. Enantiomorphism in the Helical Aggregates of Optically Active 12-Hydroxystearic Acid and Its Lithium Salt. Bull. Chem. Soc. Jpn. 1969, 42, 3422–3424. [Google Scholar] [CrossRef]
- Takeno, H.; Yanagita, M.; Motegi, Y.; Kondo, S. Relationship between helical aggregates and polymorphs in a 12-hydroxystearic acid gel: Their thermal stability and formation kinetics. Colloid Polym. Sci. 2015, 293, 199–207. [Google Scholar] [CrossRef]
- Zhang, M.; Weiss, R.G. Mechano-Responsive, Thermo-Reversible, Luminescent Organogels Derived from a Long-Chained, Naturally Occurring Fatty Acid. Chem. Eur. J. 2016, 22, 8262–8272. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Nagase, H.; Endo, T.; Ueda, H.; Nakagaki, M. Crystal Structure of DL-12-Hydroxystearic Acid. Chem. Lett. 1996, 25, 435–436. [Google Scholar] [CrossRef]
- Kamijo, M.; Nagase, H.; Endo, T.; Ueda, H.; Nakagaki, M. Polymorphic Structure of dl-12Hydroxystearic Acid. Anal. Sci. 1999, 15, 1291–1292. [Google Scholar] [CrossRef][Green Version]
- Sakurai, T.; Masuda, Y.; Sato, H.; Yamagishi, A.; Kawaji, H.; Atake, T.; Hori, K. A Comparative Study on Chiral and Racemic 12-Hydroxyoctadecanoic Acids in the Solutions and Aggregation States: Does the Racemic Form Really Form a Gel? Bull. Chem. Soc. Jpn. 2010, 83, 145–150. [Google Scholar] [CrossRef]
- Lundén, B.-M. The crystal structure of 12- D -hydroxyoctadecanoic acid methyl ester. Acta Crystallogr. B 1976, 32, 3149–3153. [Google Scholar] [CrossRef]
- Lundén, B.-M.; Lófgren, H.; Pascher, I. Accommodation of hydroxyl groups and their hydrogen bond system in a hydrocarbon matrix. Chem. Phys. Lipids 1977, 20, 263–271. [Google Scholar] [CrossRef]
- Rogers, M.A.; Weiss, R.G. Systematic modifications of alkane-based molecular gelators and the consequences to the structures and properties of their gels. New, J. Chem. 2015, 39, 785–799. [Google Scholar] [CrossRef]
- Sato, H.; Sakurai, T.; Yamagishi, A. Comparison of Vibrational Circular Dichroism between the Langmuir–Blodgett Films and Gels of 12-Hydroxyoctadecanoic Acid. Chem. Lett. 2011, 40, 25–27. [Google Scholar] [CrossRef]
- Sato, K. Polymorphism of Lipid Crystals. In Crystallization of Lipids; Sato, K., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2018; pp. 17–60. ISBN 978-1-118-59388-2. [Google Scholar]
- Moreno, E.; Cordobilla, R.; Calvet, T.; Cuevas-Diarte, M.A.; Gbabode, G.; Negrier, P.; Mondieig, D.; Oonk, H.A.J. Polymorphism of even saturated carboxylic acids from n-decanoic to n-eicosanoic acid. New J. Chem. 2007, 31, 947. [Google Scholar] [CrossRef]
- Tachibana, T.; Mori, T.; Hori, K. Chiral Mesophases of 12-Hydroxyoctadecanoic Acid in Jelly and in the Solid State. II. A New Type of Mesomorphic Solid State. Bull. Chem. Soc. Jpn. 1981, 54, 73–80. [Google Scholar] [CrossRef]
- Wu, S.; Gao, J.; Emge, T.J.; Rogers, M.A. Solvent-Induced Polymorphic Nanoscale Transitions for 12-Hydroxyoctadecanoic Acid Molecular Gels. Cryst. Growth Des. 2013, 13, 1360–1366. [Google Scholar] [CrossRef]
- Toro-Vazquez, J.F.; Morales-Rueda, J.; Torres-Martínez, A.; Charó-Alonso, M.A.; Mallia, V.A.; Weiss, R.G. Cooling Rate Effects on the Microstructure, Solid Content, and Rheological Properties of Organogels of Amides Derived from Stearic and (R)-12-Hydroxystearic Acid in Vegetable Oil. Langmuir 2013, 29, 7642–7654. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D. Lateral order in gel, subgel and crystalline phases of lipid membranes: Wide-angle X-ray scattering. Chem. Phys. Lipids 2012, 165, 59–76. [Google Scholar] [CrossRef]
- Watanabe, J.; Nagase, T. Thermotropic polypeptides. 5. Temperature dependence of cholesteric pitches exhibiting a cholesteric sense inversion. Macromolecules 1988, 21, 171–175. [Google Scholar] [CrossRef]
- Tachibana, T.; Kitazawa, S.; Takeno, H. Studies of Helical Aggregates of Molecules. II. The Sense of Twist in the Fibrous Aggregates from the Alkali Metal Soaps of Optically Active 12-Hydroxystearic Acid. Bull. Chem. Soc. Jpn. 1970, 43, 2418–2421. [Google Scholar] [CrossRef]
- Hyun, K.; Wilhelm, M.; Klein, C.O.; Cho, K.S.; Nam, J.G.; Ahn, K.H.; Lee, S.J.; Ewoldt, R.H.; McKinley, G.H. A review of nonlinear oscillatory shear tests: Analysis and application of large amplitude oscillatory shear (LAOS). Prog. Polym. Sci. 2011, 36, 1697–1753. [Google Scholar] [CrossRef]
- Bonn, D.; Denn, M.M.; Berthier, L.; Divoux, T.; Manneville, S. Yield stress materials in soft condensed matter. Rev. Mod. Phys. 2017, 89, 035005. [Google Scholar] [CrossRef]
- Rouyer, F.; Cohen-Addad, S.; Höhler, R. Is the yield stress of aqueous foam a well-defined quantity? Colloids Surf. Physicochem. Eng. Asp. 2005, 263, 111–116. [Google Scholar] [CrossRef]
- Dinkgreve, M.; Paredes, J.; Denn, M.M.; Bonn, D. On different ways of measuring “the” yield stress. J. Non-Newton. Fluid Mech. 2016, 238, 233–241. [Google Scholar] [CrossRef]
- Almdal, K.; Dyre, J.; Hvidt, S.; Kramer, O. Towards a phenomenological definition of the term ‘gel’. Polym. Gels Netw. 1993, 1, 5–17. [Google Scholar] [CrossRef]
- Laupheimer, M.; Preisig, N.; Stubenrauch, C. The molecular organogel n-decane/12-hydroxyoctadecanoic acid: Sol–gel transition, rheology, and microstructure. Colloids Surf. Physicochem. Eng. Asp. 2015, 469, 315–325. [Google Scholar] [CrossRef]
- Terech, P.; Pasquier, D.; Bordas, V.; Rossat, C. Rheological Properties and Structural Correlations in Molecular Organogels. Langmuir 2000, 16, 4485–4494. [Google Scholar] [CrossRef]
- Burkhardt, M.; Kinzel, S.; Gradzielski, M. Macroscopic properties and microstructure of HSA based organogels: Sensitivity to polar additives. J. Colloid Interface Sci. 2009, 331, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Hanabusa, K.; Matsumoto, M.; Kimura, M.; Kakehi, A.; Shirai, H. Low Molecular Weight Gelators for Organic Fluids: Gelation Using a Family of Cyclo(dipeptide)s. J. Colloid Interface Sci. 2000, 224, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Zurcher, D.M.; McNeil, A.J. Tools for Identifying Gelator Scaffolds and Solvents. J. Org. Chem. 2015, 80, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Chen, D.; Maclennan, J.E.; Shao, R.; Yoon, D.K.; Wang, H.; Korblova, E.; Walba, D.M.; Glaser, M.A.; Clark, N.A. Chirality-Preserving Growth of Helical Filaments in the B4 Phase of Bent-Core Liquid Crystals. J. Am. Chem. Soc. 2011, 133, 12656–12663. [Google Scholar] [CrossRef]
- Sollich, P.; Lequeux, F.; Hébraud, P.; Cates, M.E. Rheology of Soft Glassy Materials. Phys. Rev. Lett. 1997, 78, 2020–2023. [Google Scholar] [CrossRef]
- Hansen, C.M. 50 Years with solubility parameters—past and future. Prog. Org. Coat. 2004, 51, 77–84. [Google Scholar] [CrossRef]
- Raynal, M.; Bouteiller, L. Organogel formation rationalized by Hansen solubility parameters. Chem. Commun. 2011, 47, 8271. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; De Simone, A.; Fitzpatrick, A.W.; Baldwin, A.; Meehan, S.; Rajah, L.; Vendruscolo, M.; Welland, M.E.; Dobson, C.M.; Terentjev, E.M. Twisting Transition between Crystalline and Fibrillar Phases of Aggregated Peptides. Phys. Rev. Lett. 2012, 109, 158101. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Rebuffi, L.; Plaisier, J.R.; Abdellatief, M.; Lausi, A.; Scardi, P. MCX: A Synchrotron Radiation Beamline for X-ray Diffraction Line Profile Analysis. Z. Für Anorg. Allg. Chem. 2014, 640, 3100–3106. [Google Scholar] [CrossRef]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the crystallography beamlines at Elettra. Eur. Phys. J. Plus 2015, 130, 43. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound (R)-9-hydroxystearic acid are available from the authors. |










{kind=link}
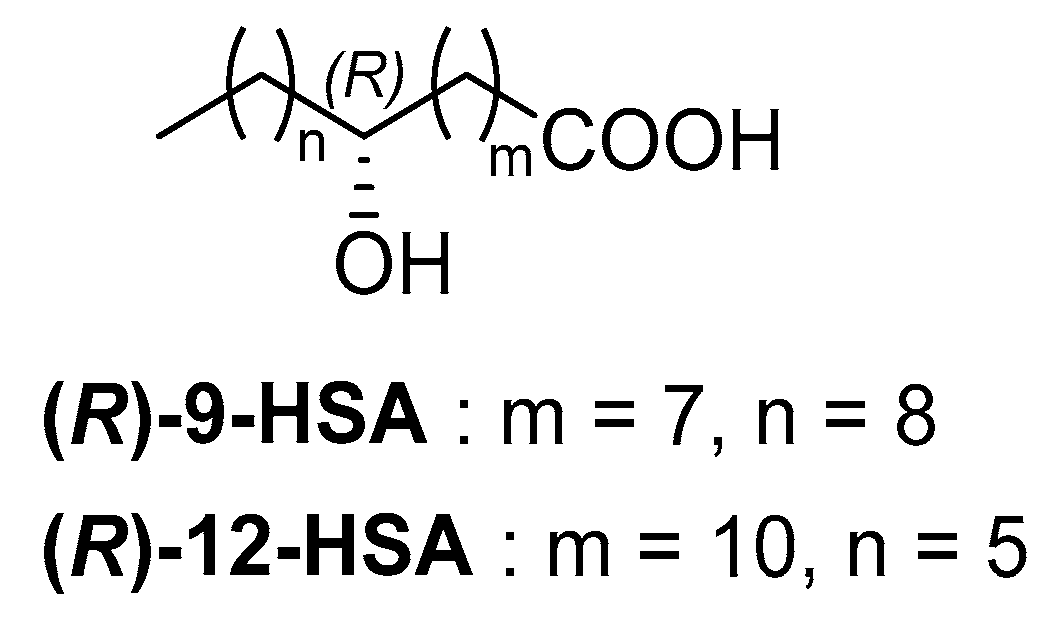{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH3OH—Single Crystal | ||
|---|---|---|
| Molecule 1 | Molecule 2 | |
| O1-C1-C2-C3/O5-C21-C22-C23 | −21.0(3) | 41.3(3) |
| O2-C1-C2-C3/O4-C21-C22-C23 | 160.4(2) | −141.2(2) |
| C1-C2-C3-C4/C21-C22-C23-C24 | −179.12(15) | −174.25(15) |
| Donor-H | D-H | H...A | D...A | D-H...A | Acceptor |
|---|---|---|---|---|---|
| O(2)-H(2) | 0.89(4) Å | 1.76(4) Å | 2.645(3) Å | 171(4)° | O(5) |
| O(4)-H(4) | 1.26(4) Å | 1.43(4) Å | 2.675(3) Å | 169(4)° | O(1) |
| O(3)-H(3) | 0.77(3) Å | 2.06(3) Å | 2.795(2) Å | 163(3)° | O(6) * |
| O(6)-H(6) | 0.85(3) Å | 1.97(3) Å | 2.791(2) Å | 164(3)° | O(3) * |
| CH3OH—Single Crystal | CH3OH–Powder | CH3CN–Powder | CCl4–Powder | Crystallized from Melt–Powder | |
|---|---|---|---|---|---|
| S.G. | P1 | P1 | P1 | P1 | P1 |
| a (Å) | 4.9300(15) | 4.926(2) | 4.982(4) | 4.982(4) | 4.454(1) |
| b | 9.2106(25) | 9.210(2) | 9.217(4) | 9.217(4) | 5.02(3) |
| c | 21.089(4) | 21.084(4) | 21.158(8) | 21.158(8) | 49.280(1) |
| α (°) | 83.62(5) | 83.62(1) | 83.54(1) | 83.54(2) | 89.610(3) |
| β | 92.20(6) | 92.20(9) | 91.93(9) | 91.93(9) | 89.655(5) |
| γ | 82.38(5) | 82.38(19 | 82.86(1) | 82.86(1) | 64.739(3) |
| V (Å3) | 941.9(3) | 941.9(1) | 956.3(1) | 956.3(1) | 996.39(3) |
| d (g/cm3) 1 | 1.059 | 1.059 | 1.044 | 1.044 | 1.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asaro, F.; Boga, C.; Demitri, N.; De Zorzi, R.; Drioli, S.; Gigli, L.; Micheletti, G.; Nitti, P.; Zangrando, E. X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid. Molecules 2019, 24, 2854. https://doi.org/10.3390/molecules24152854
Asaro F, Boga C, Demitri N, De Zorzi R, Drioli S, Gigli L, Micheletti G, Nitti P, Zangrando E. X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid. Molecules. 2019; 24(15):2854. https://doi.org/10.3390/molecules24152854
Chicago/Turabian StyleAsaro, Fioretta, Carla Boga, Nicola Demitri, Rita De Zorzi, Sara Drioli, Lara Gigli, Gabriele Micheletti, Patrizia Nitti, and Ennio Zangrando. 2019. "X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid" Molecules 24, no. 15: 2854. https://doi.org/10.3390/molecules24152854
APA StyleAsaro, F., Boga, C., Demitri, N., De Zorzi, R., Drioli, S., Gigli, L., Micheletti, G., Nitti, P., & Zangrando, E. (2019). X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid. Molecules, 24(15), 2854. https://doi.org/10.3390/molecules24152854