
The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Results and Discussion
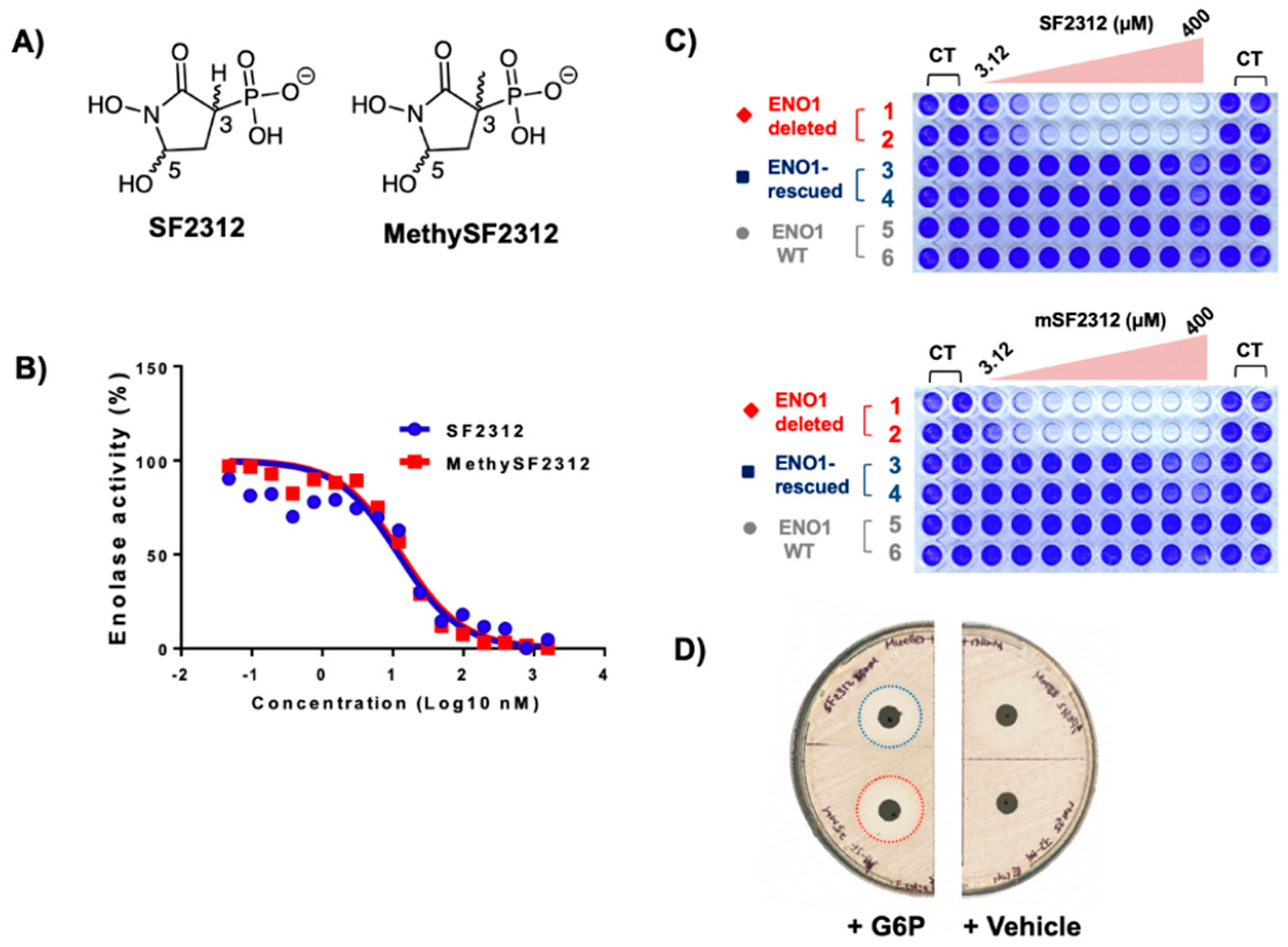
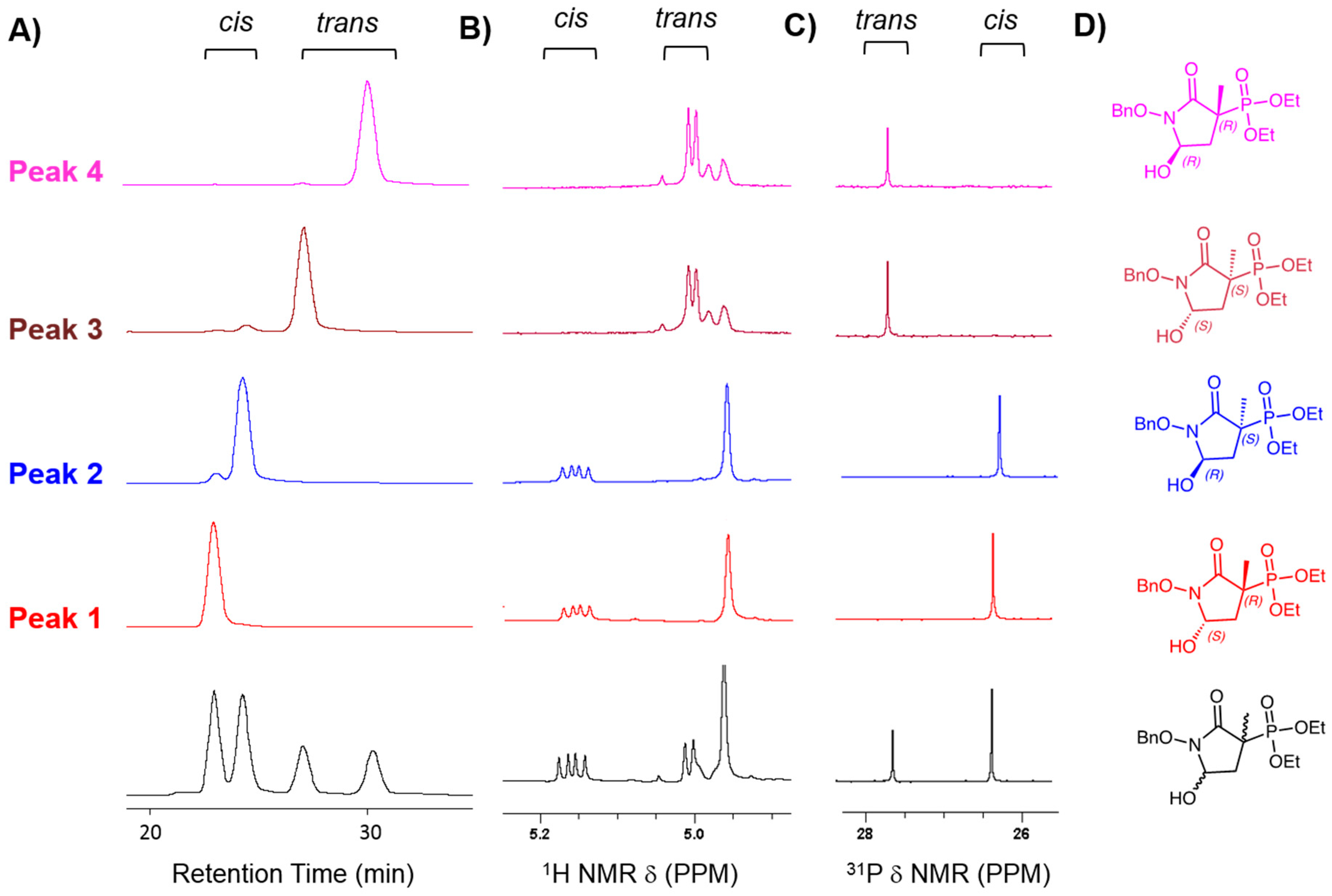
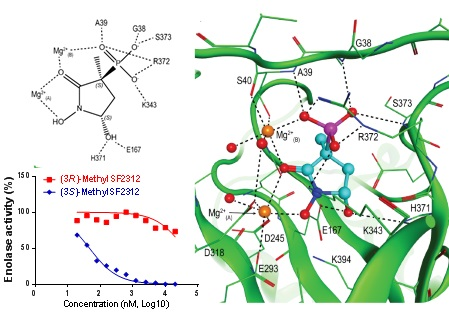
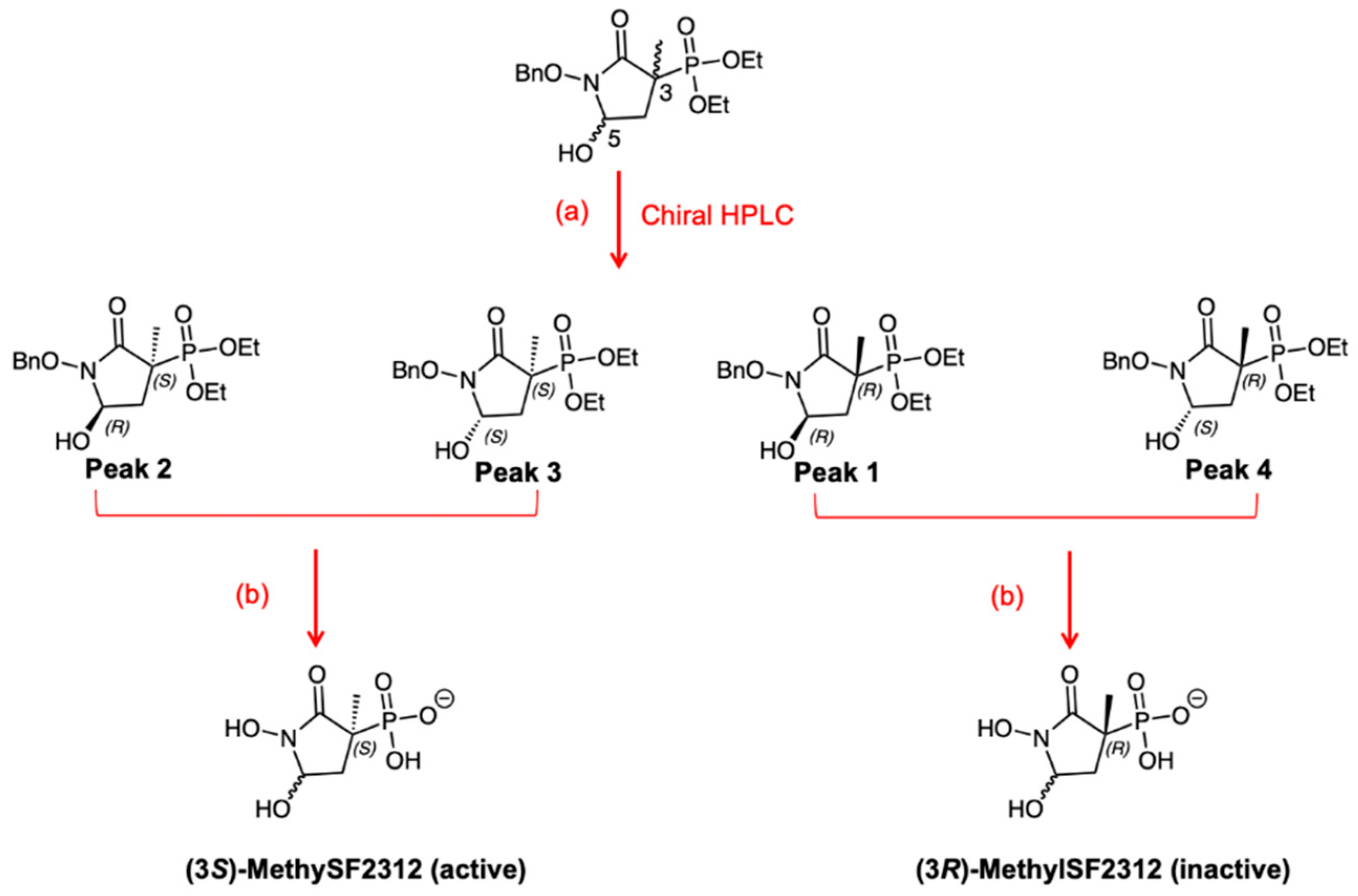
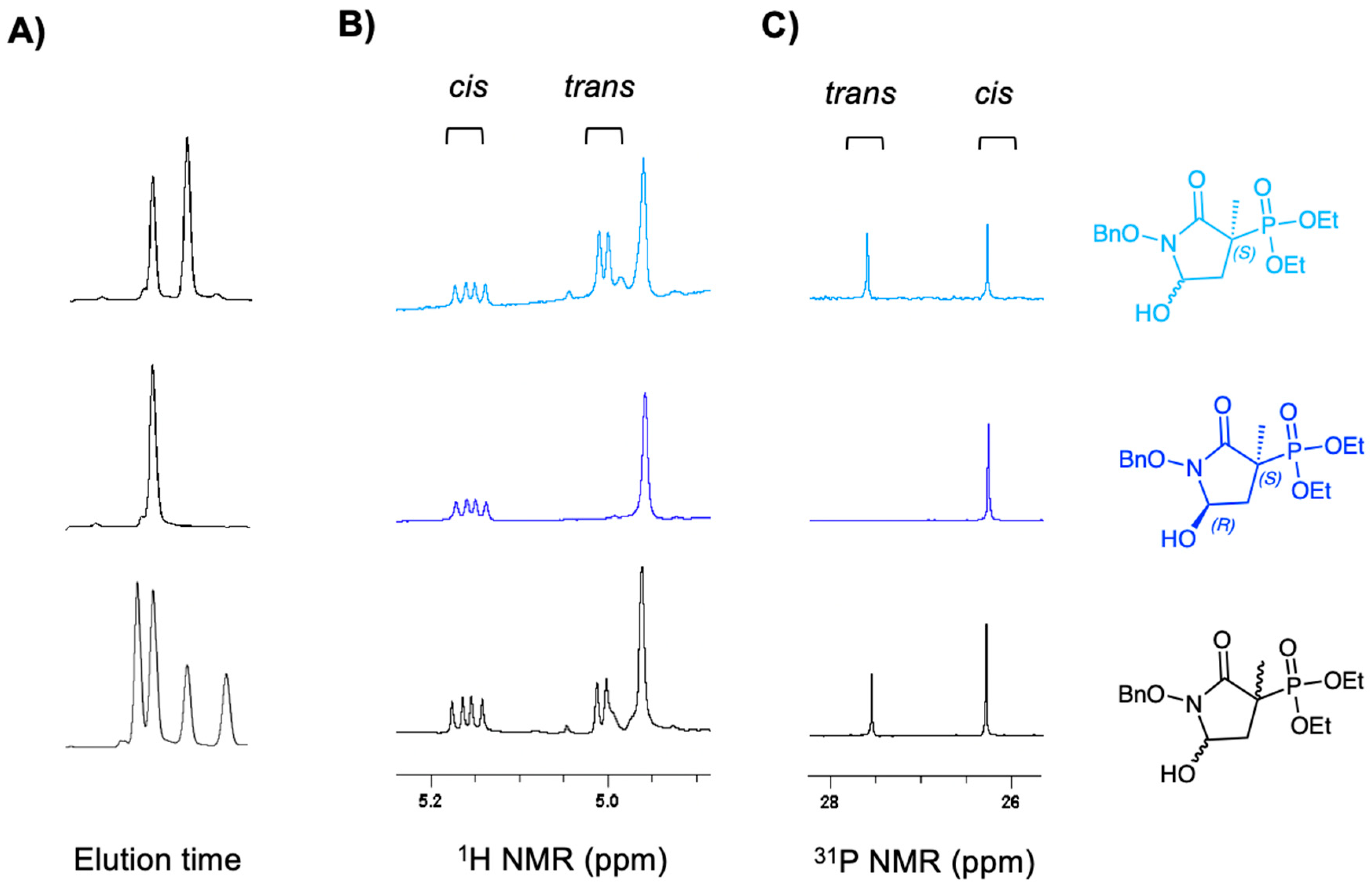
2.1. MethylSF2312 Is a Potent Enolase Inhibitor with a Non-Epimerizable (3S,5S) Stereocenter
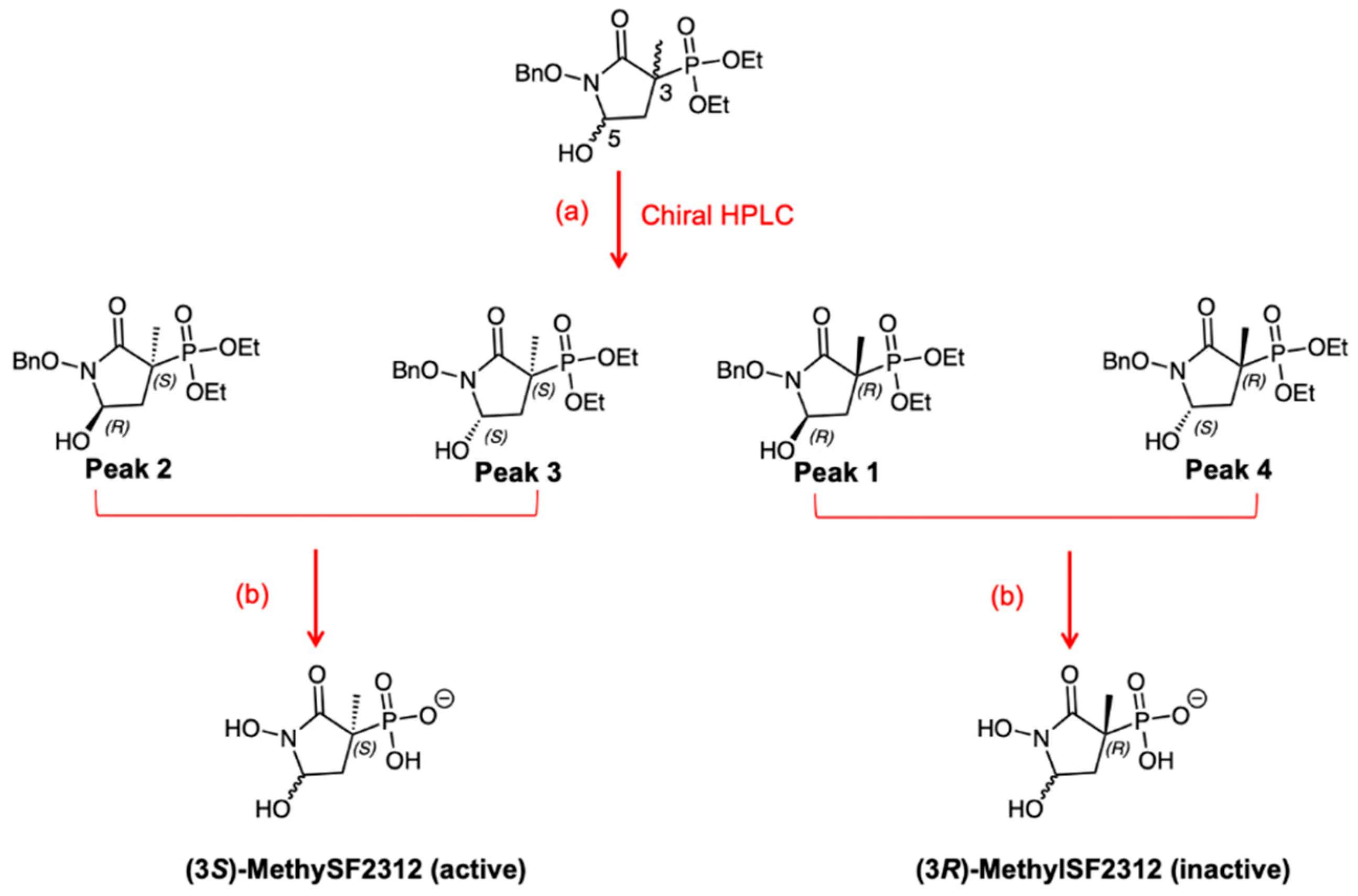
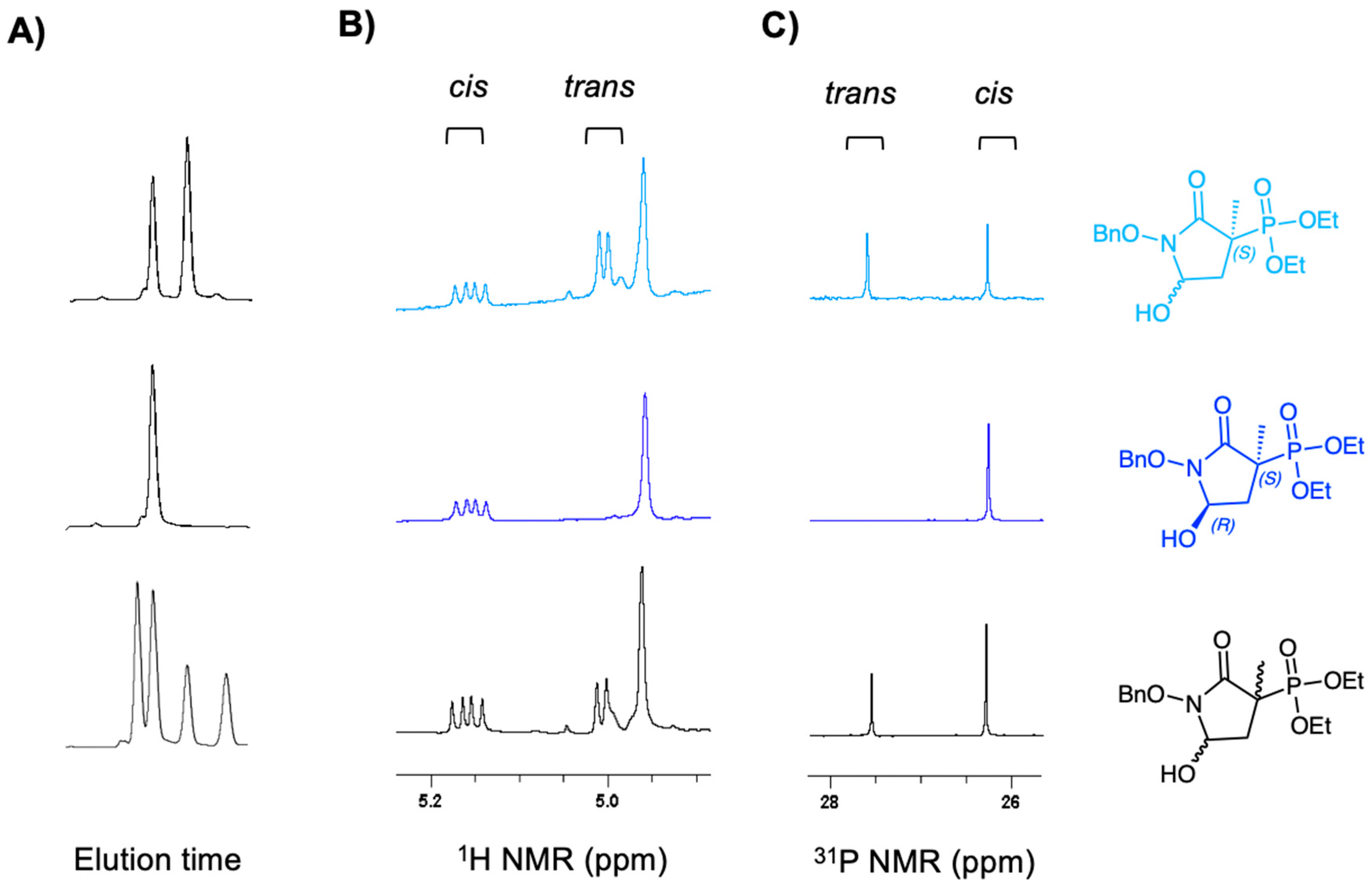
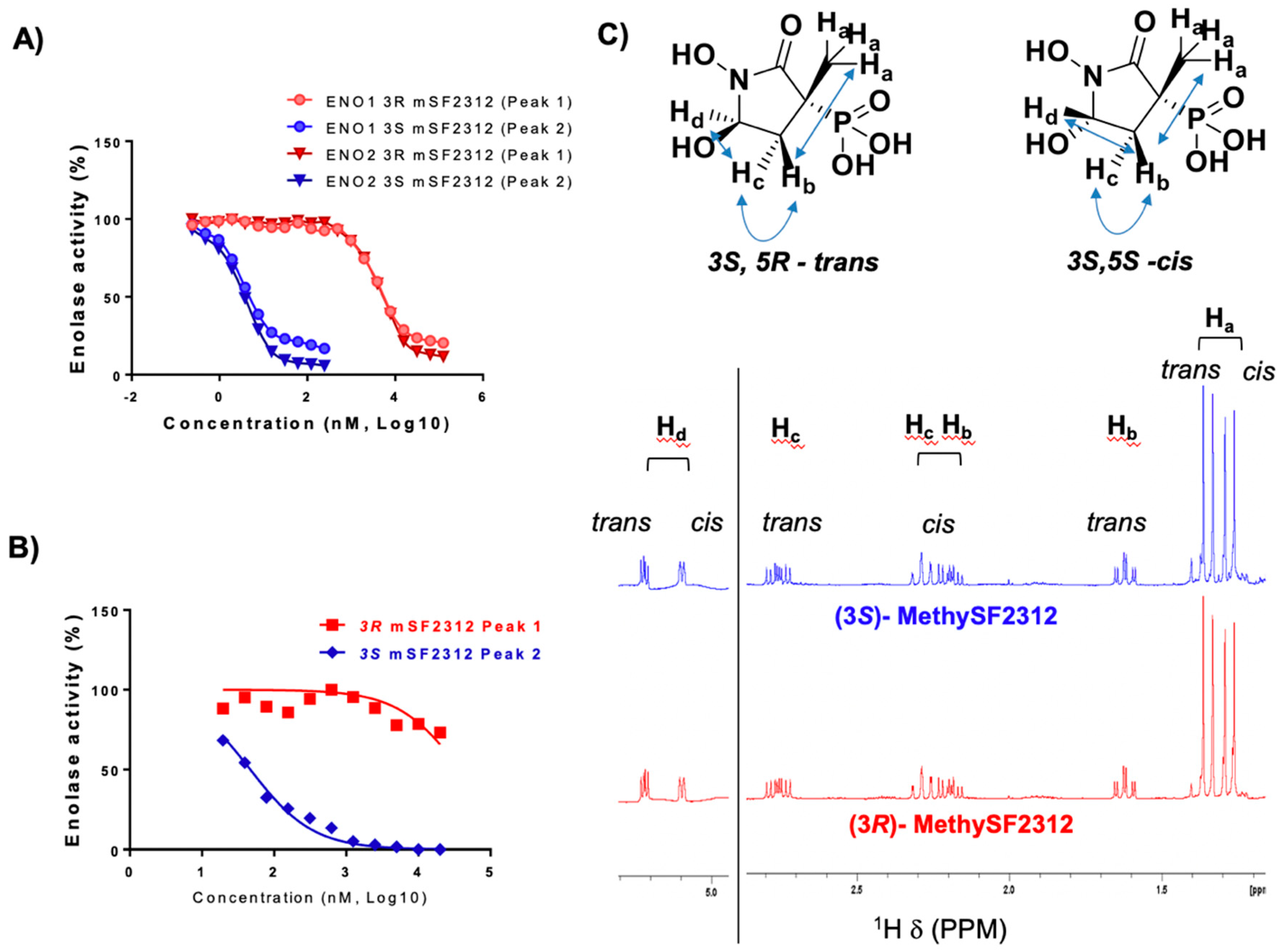
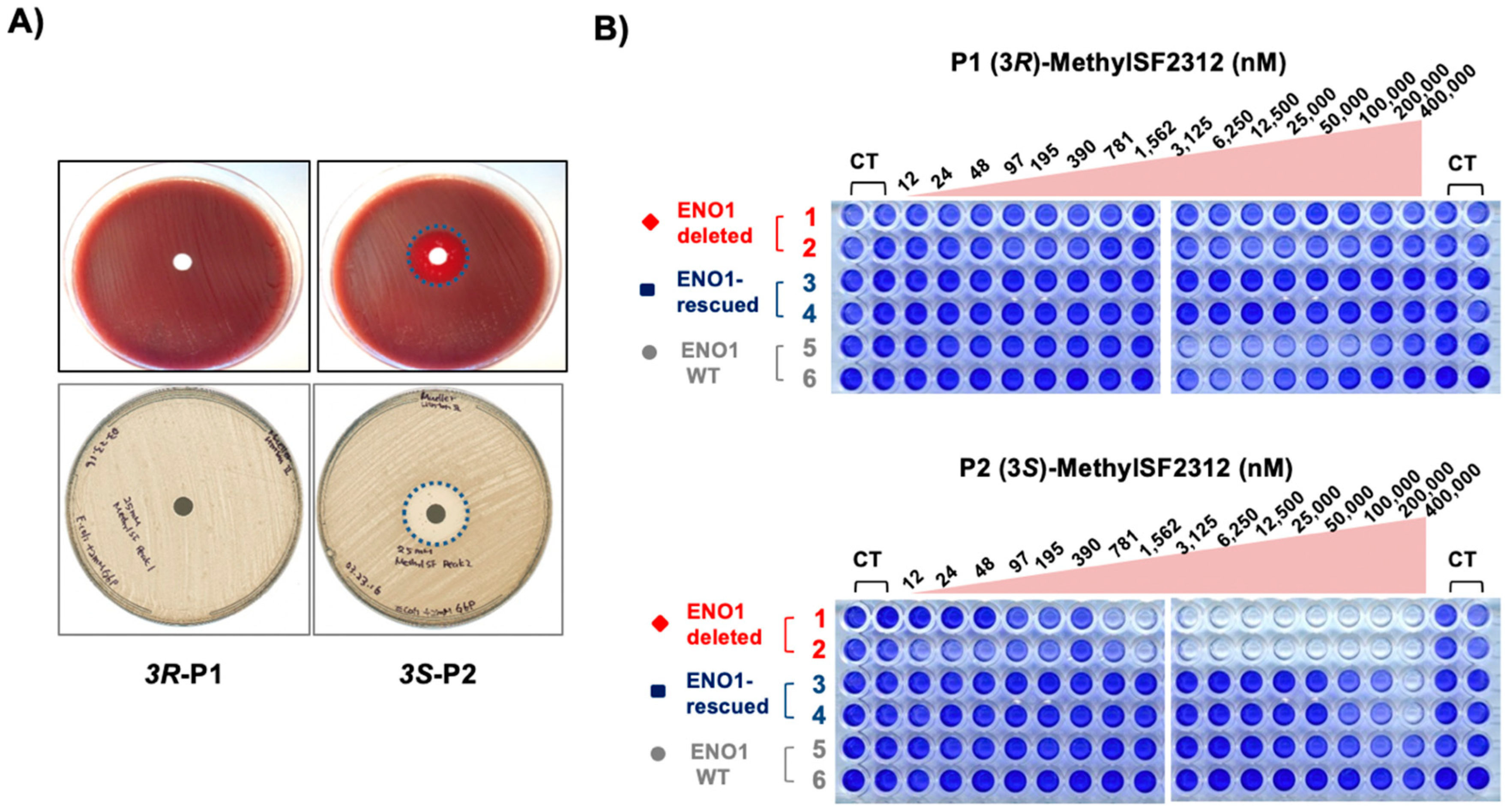
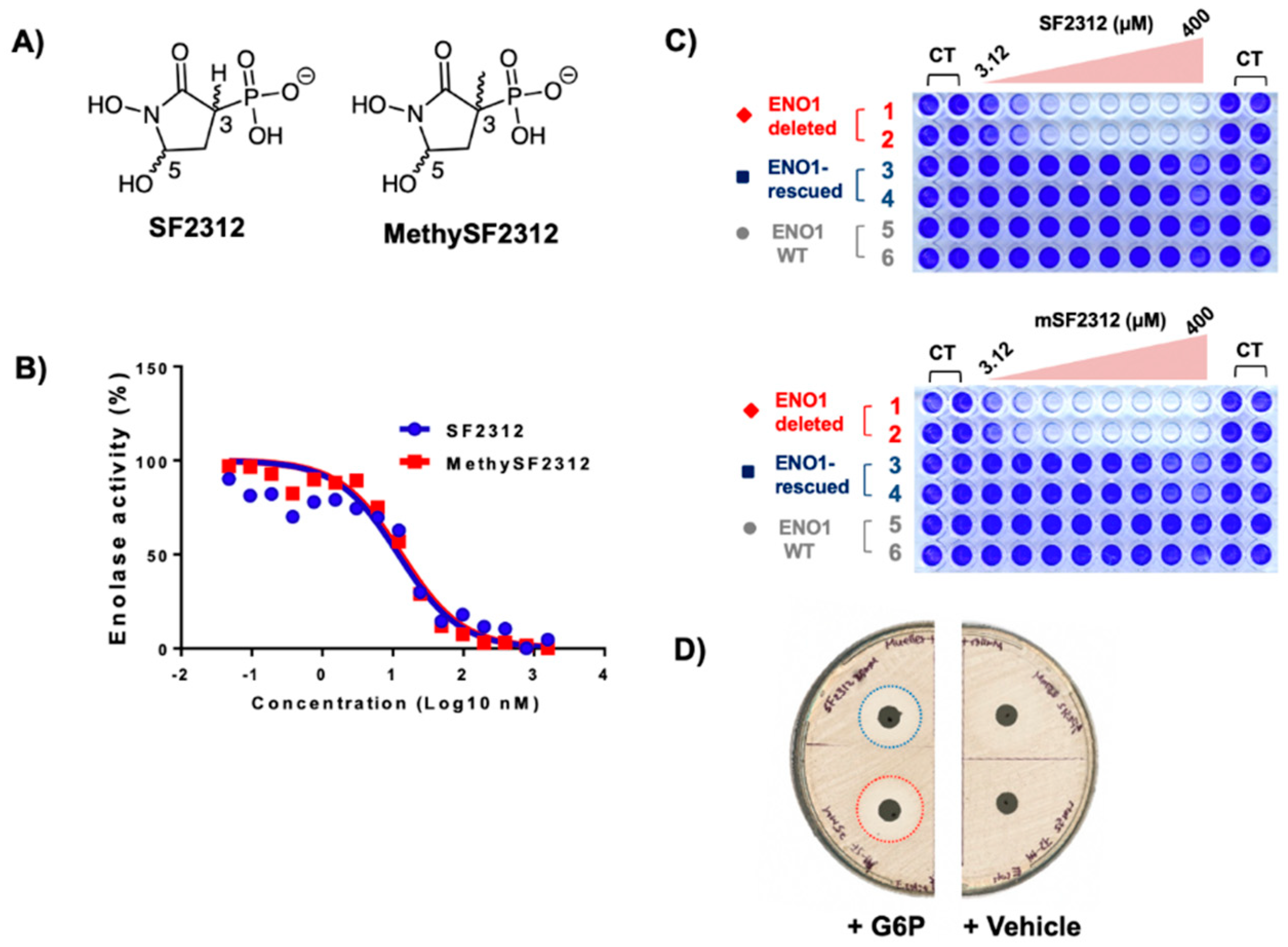
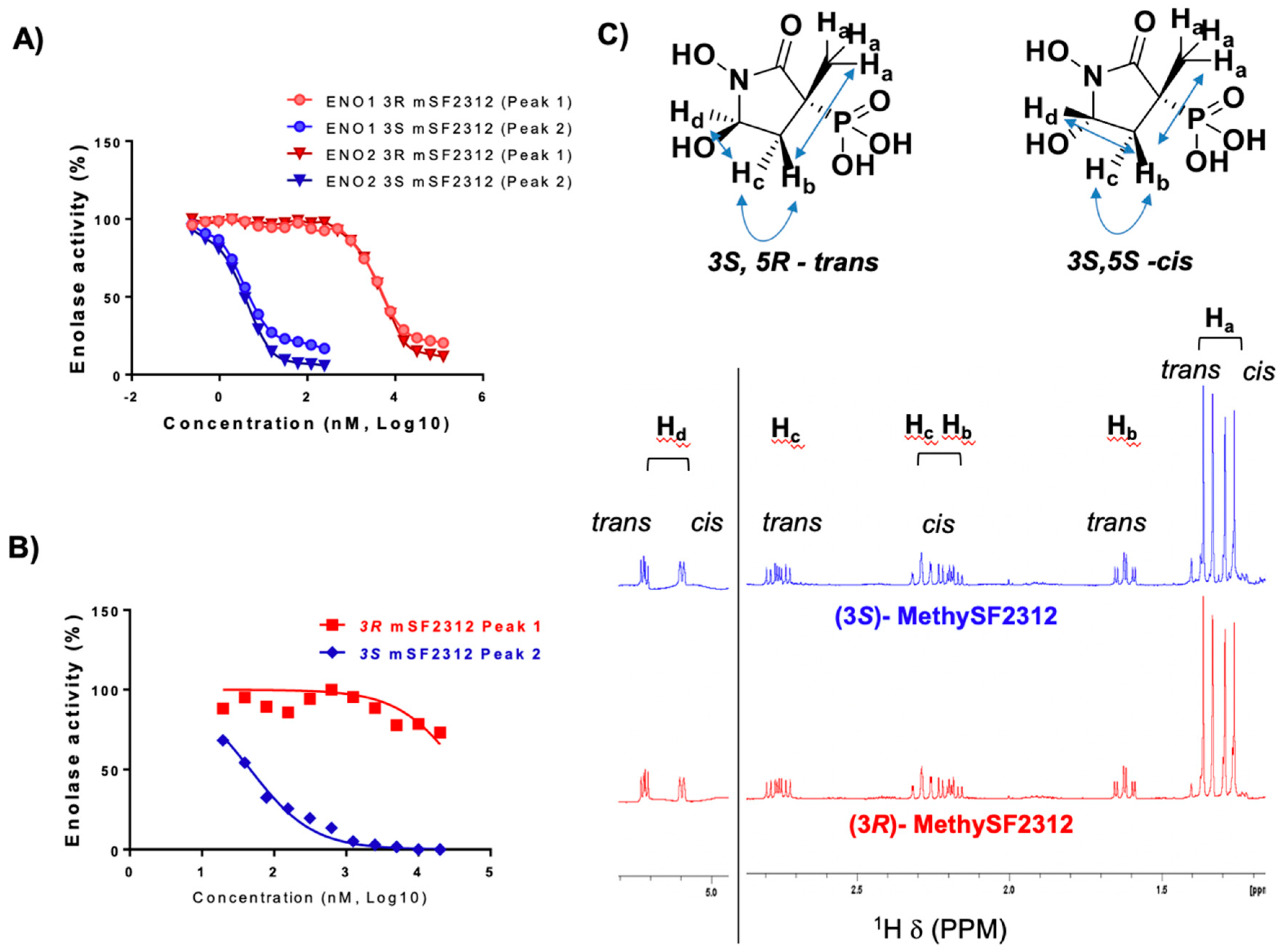
2.2. Chiral Fractions of MethylSF2312 Show Dramatic Differences in Enolase Inhibitory Activity
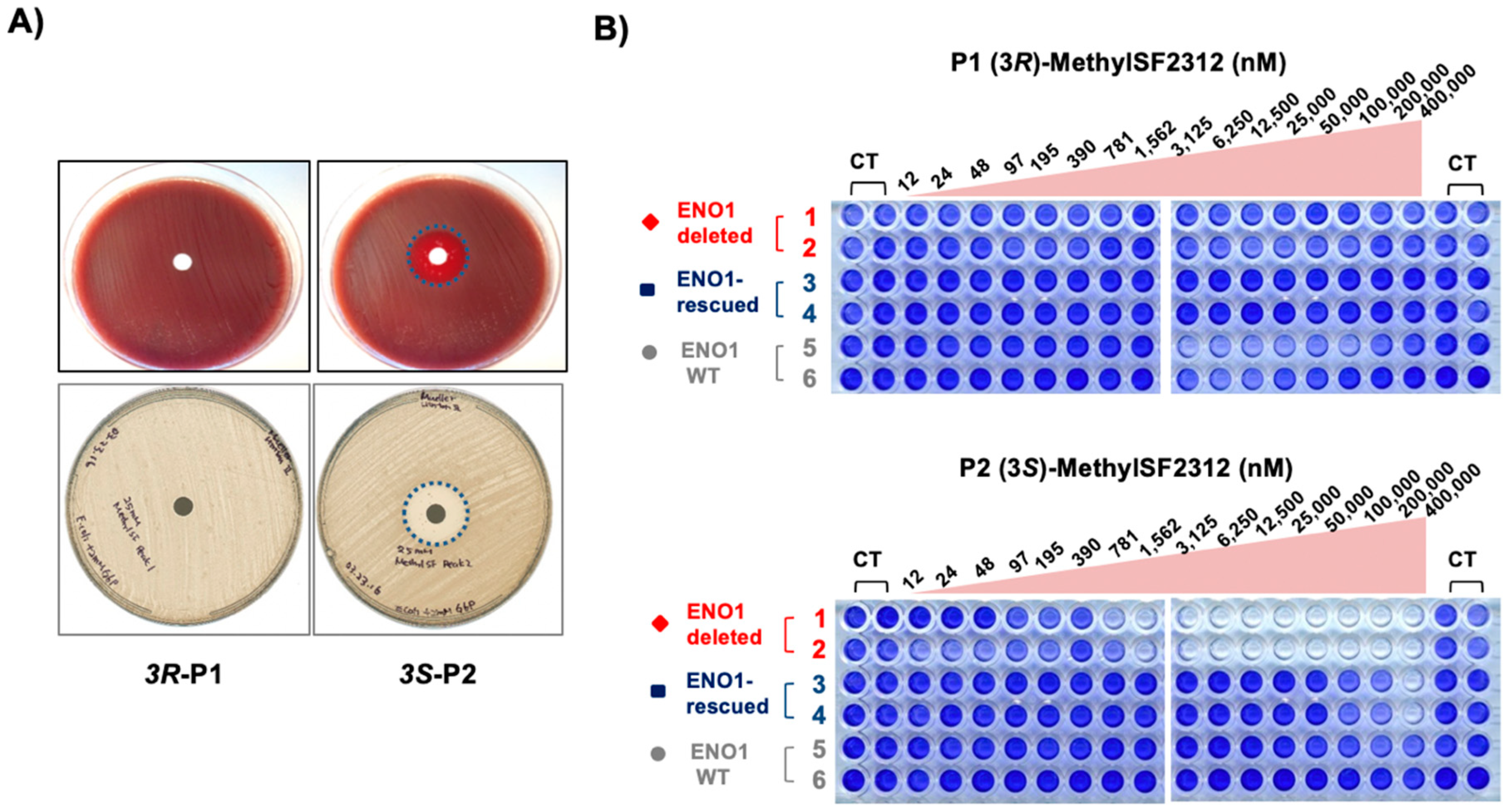
2.3. The Biological Activity of Each Chiral MethylSF2312 Fraction Strongly Correlates with Enolase Inhibitory Activity
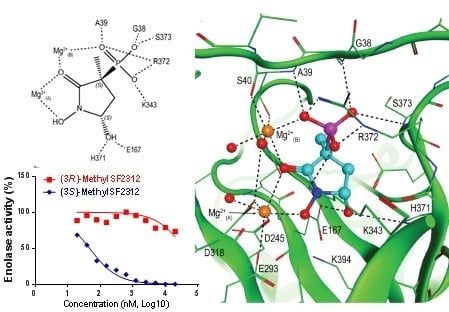
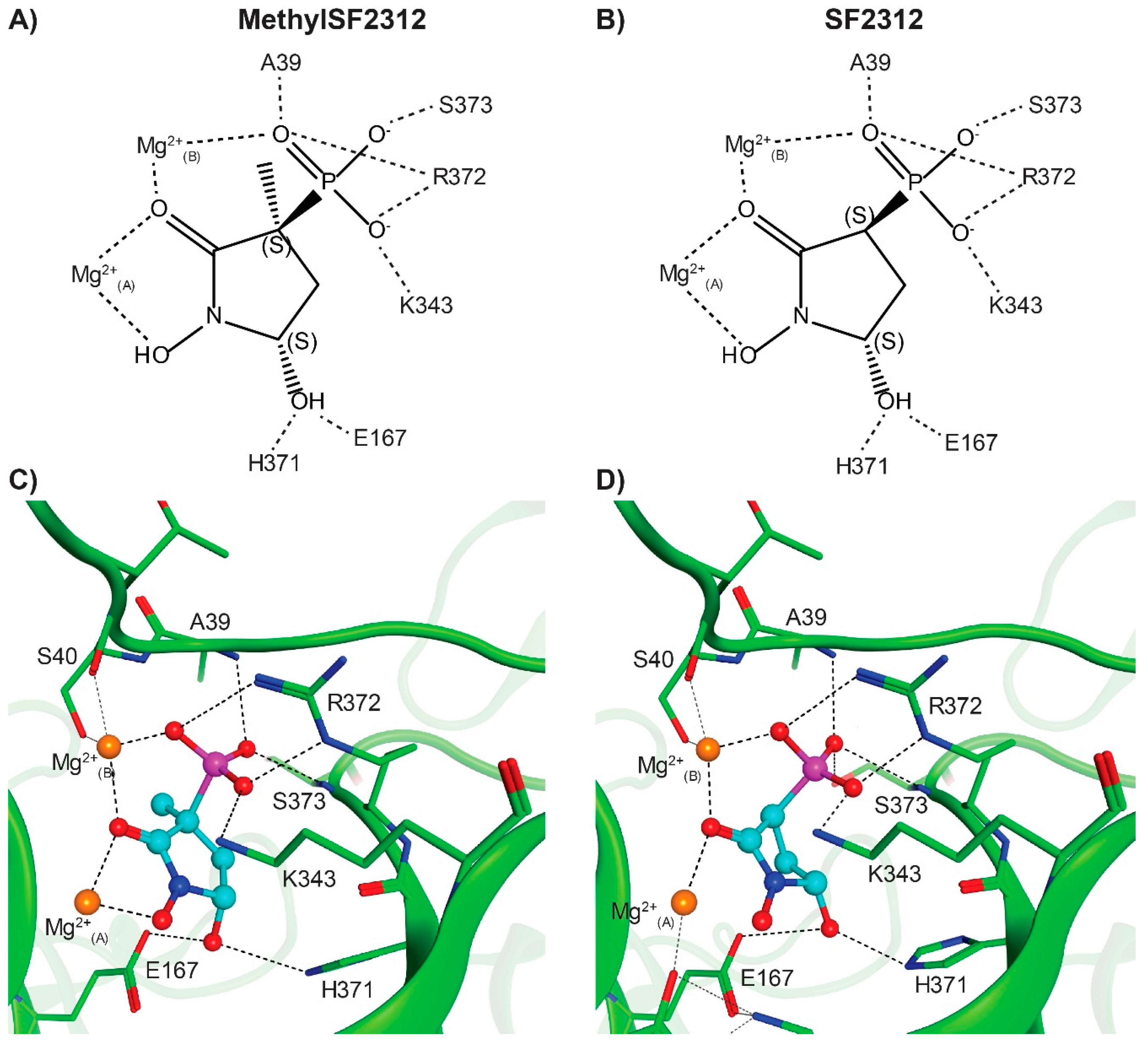
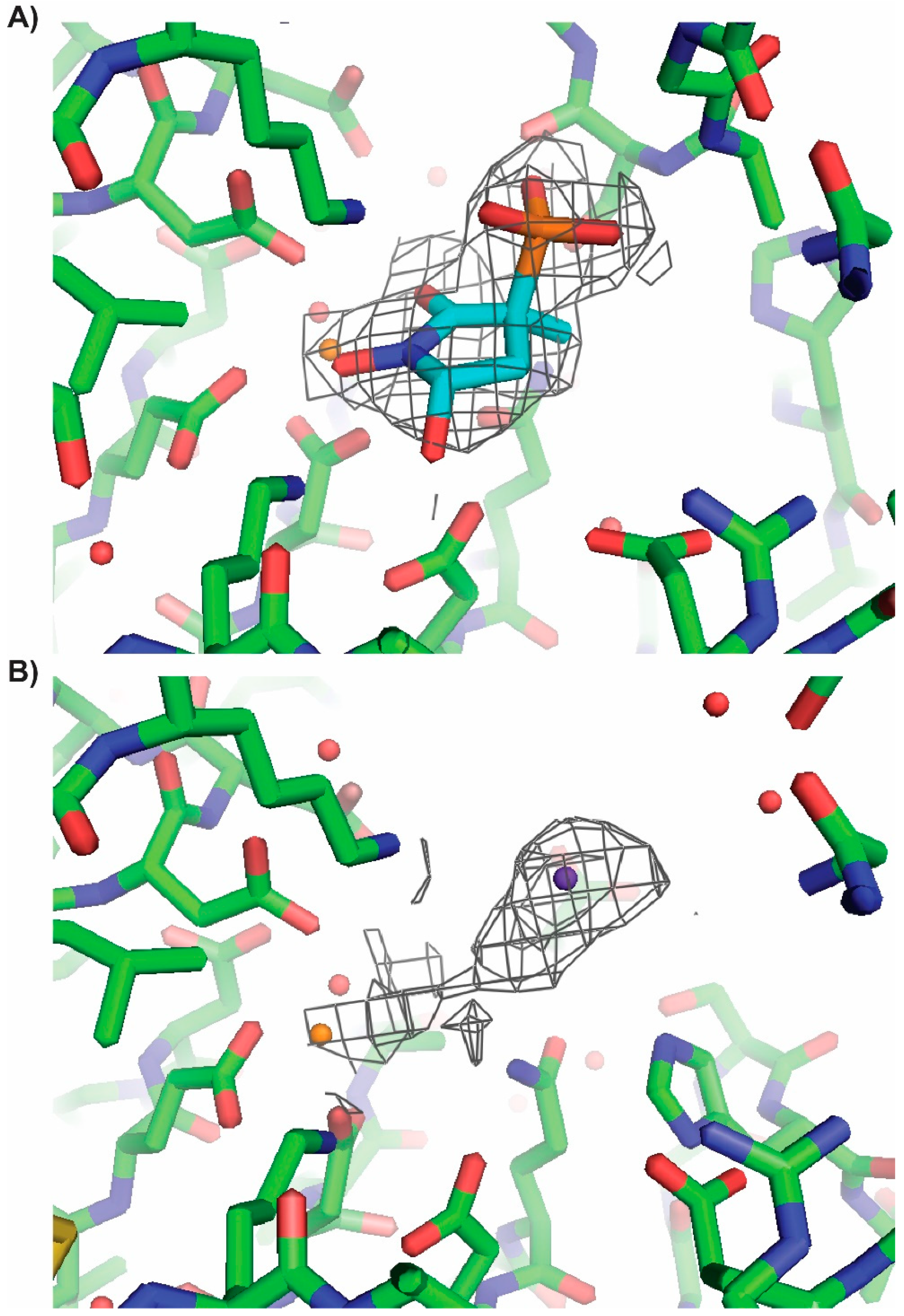
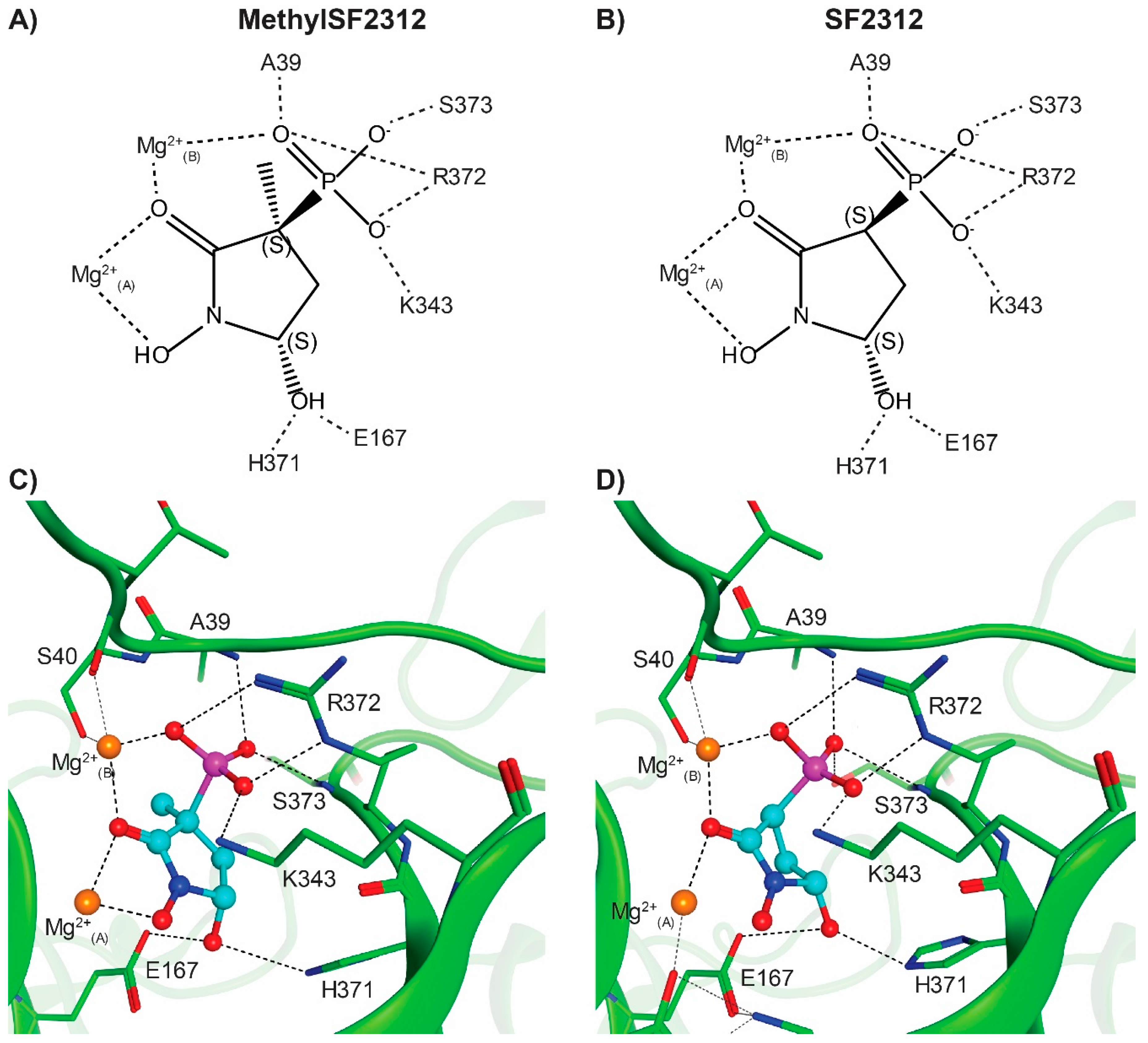
2.4. X-ray Structures of ENO2 are Only Occupied by the S-enantiomer of MethylSF2312
3. Materials and Methods
3.1. Chemistry
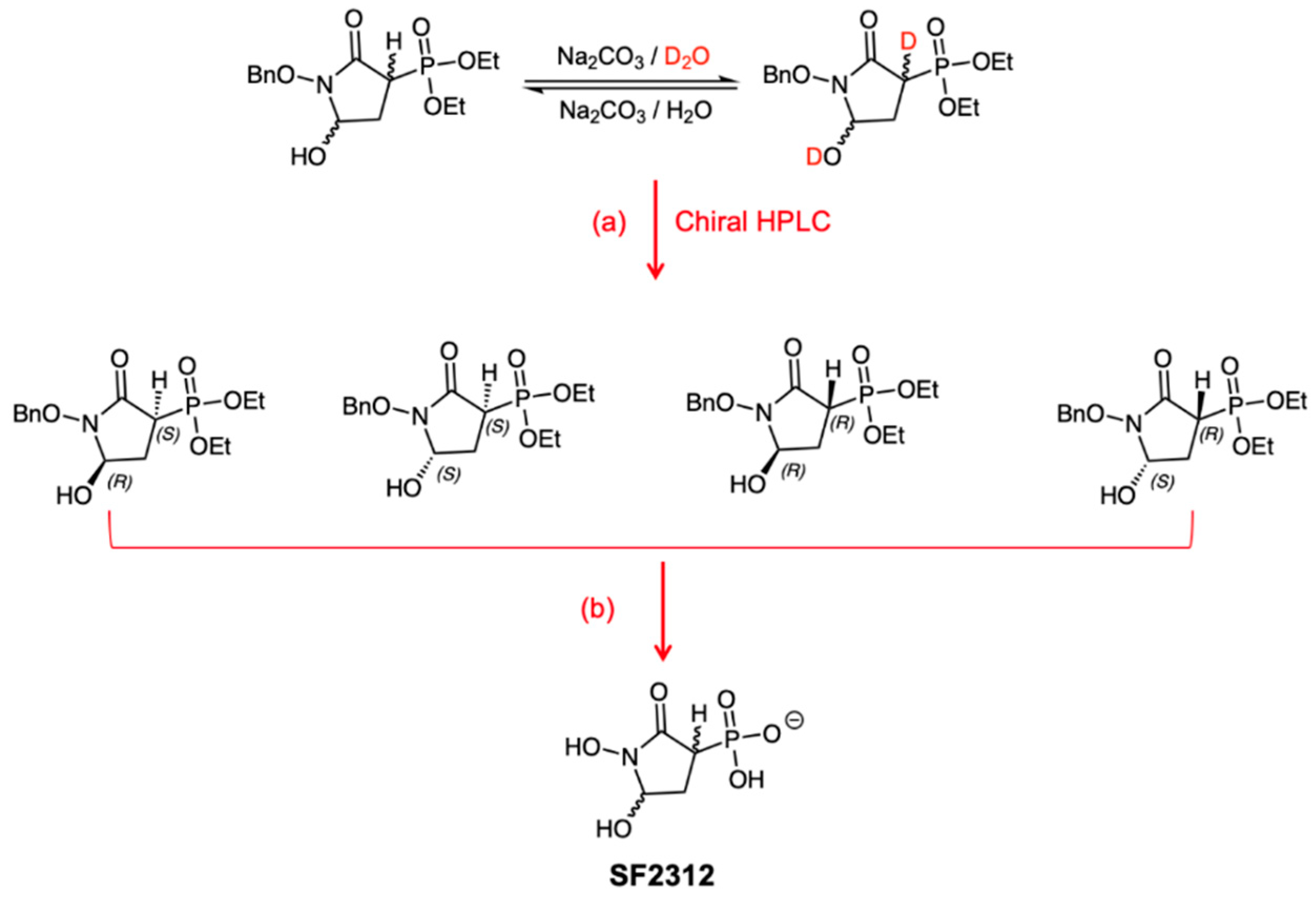
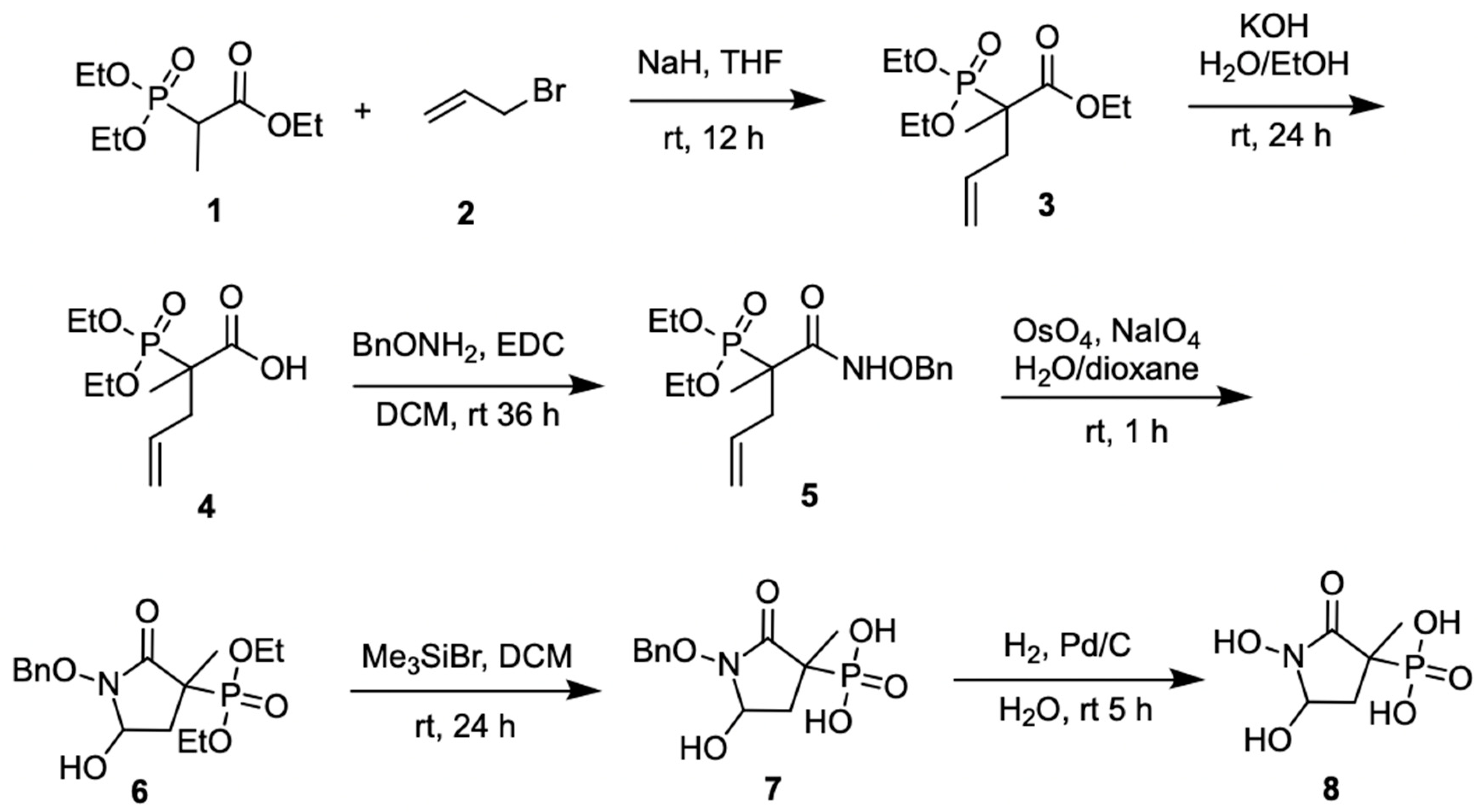
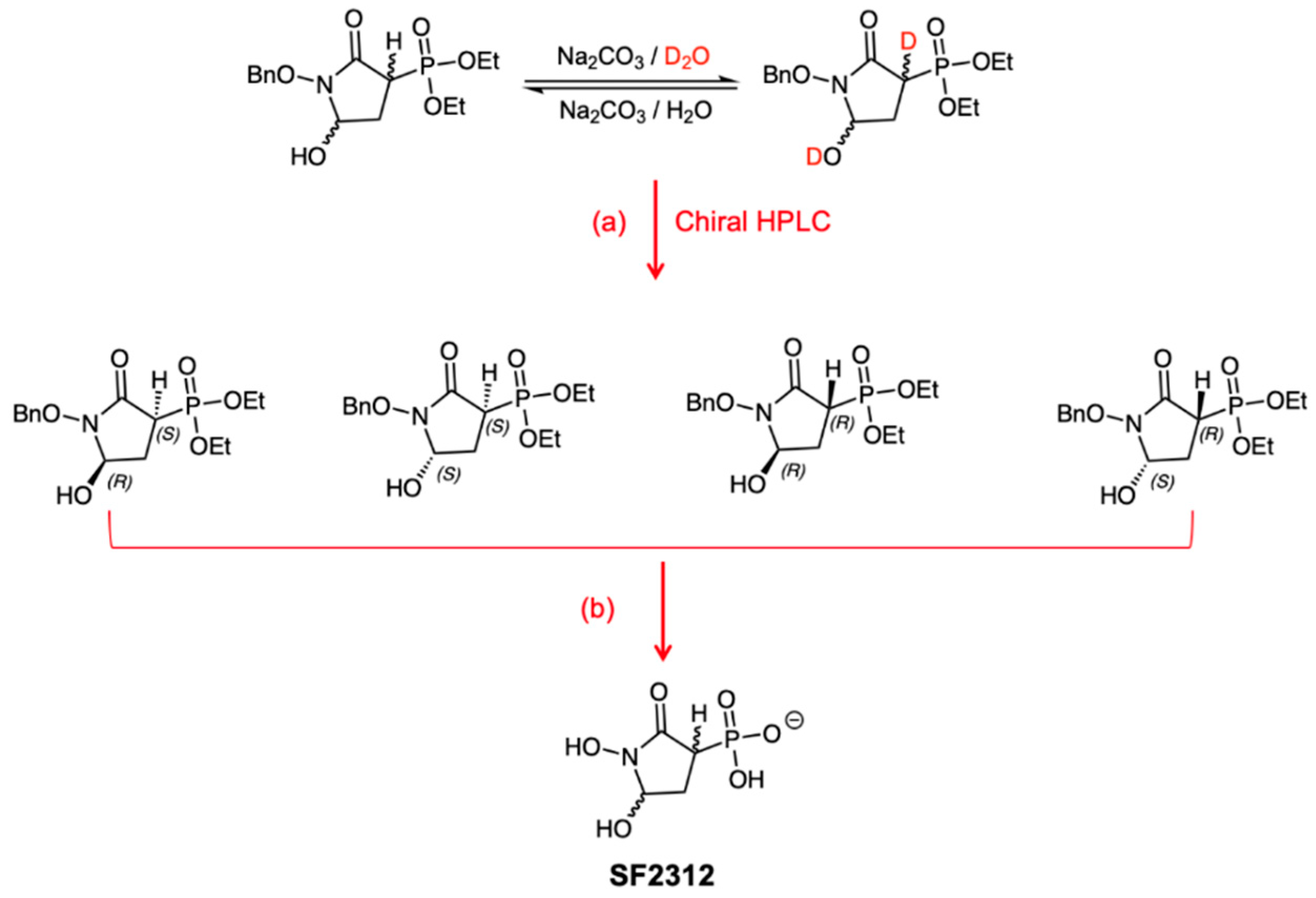
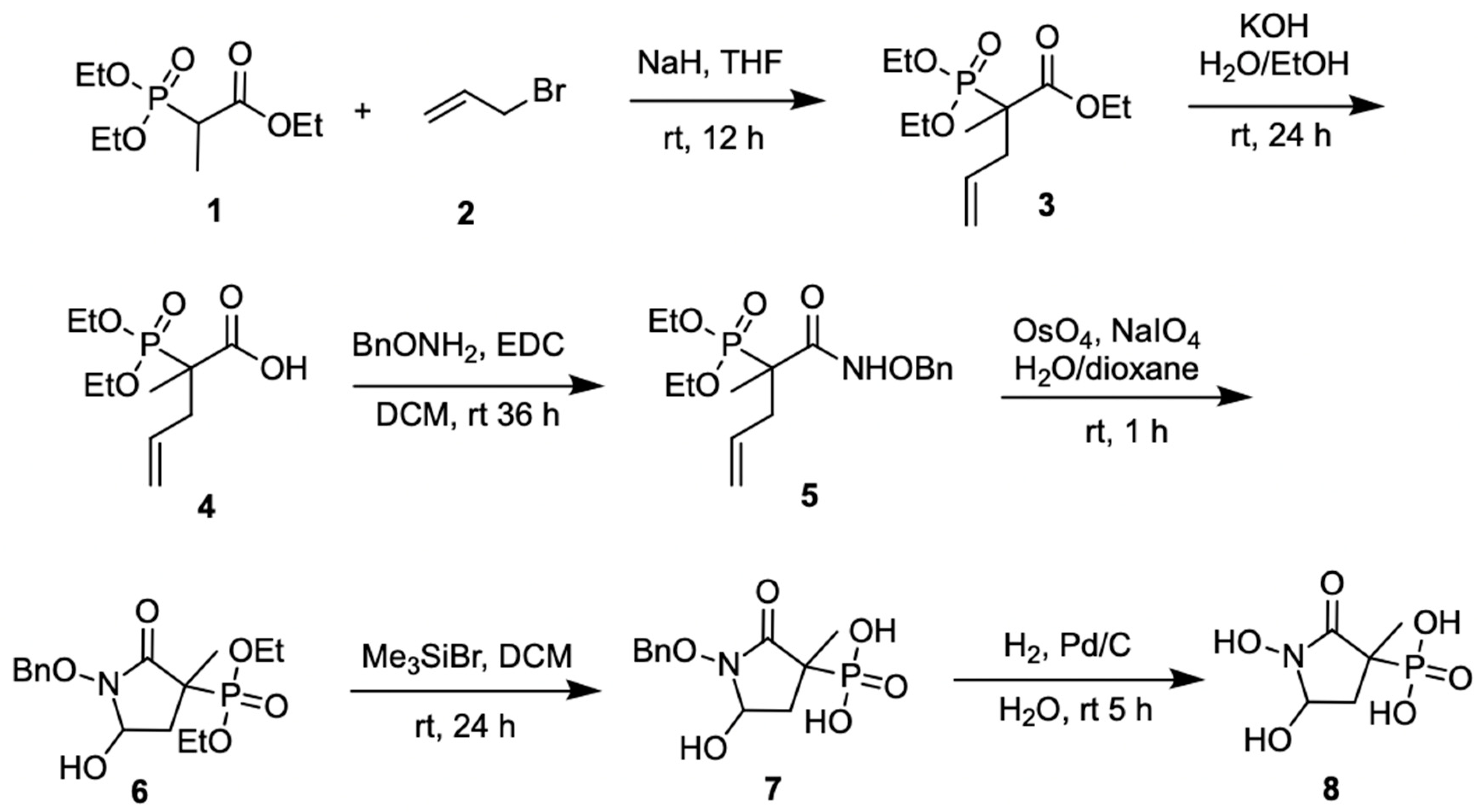
3.1.1. Synthesis of SF2312
3.1.2. Synthesis of MethylSF2312
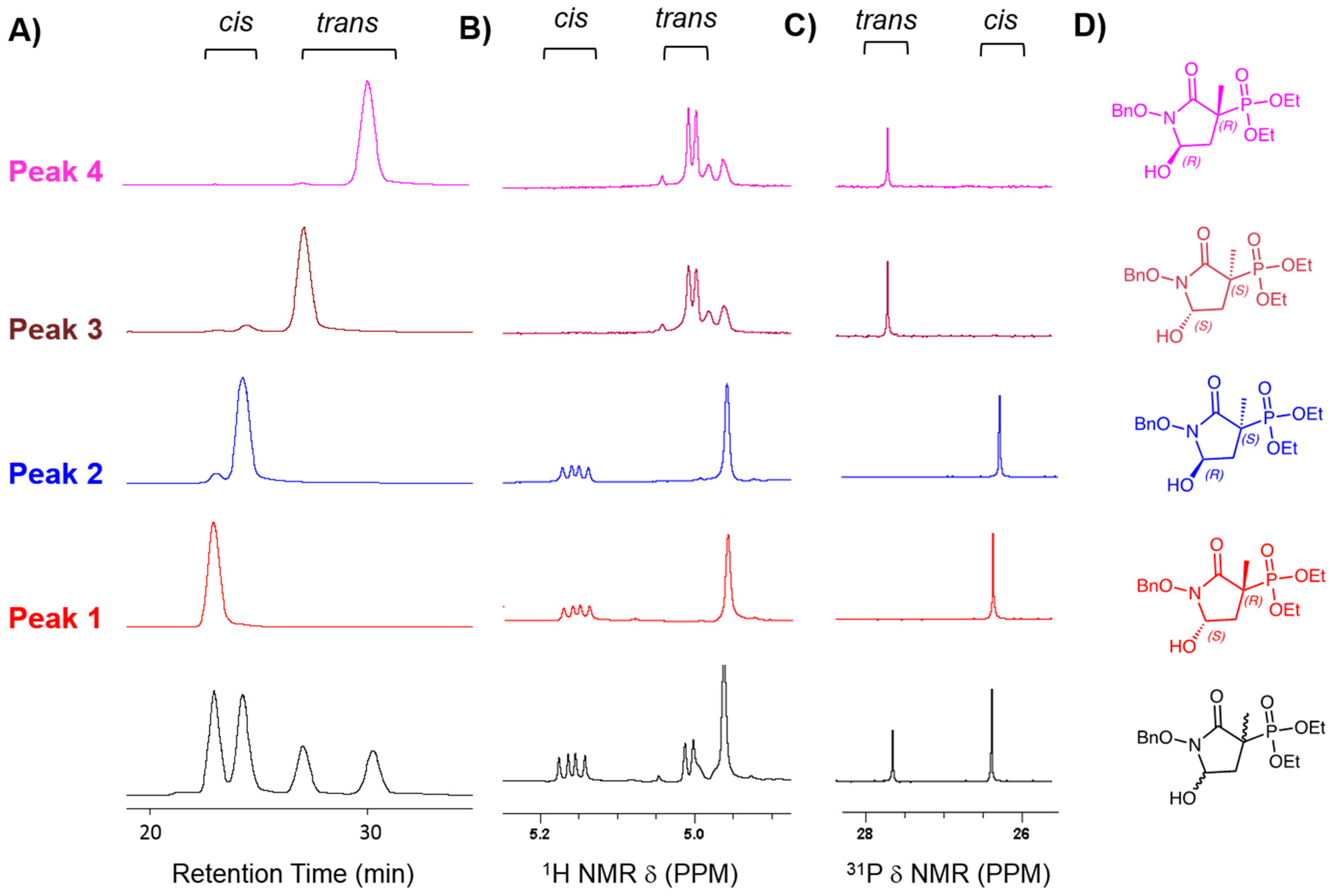
3.1.3. Chiral Chromatography and Synthesis of Chiral mSF2312
3.2. Biology
3.2.1. Enolase Enzymatic Activity
3.2.2. Cell Culture
3.2.3. Proliferation Assays
3.2.4. Antibiotic Activity Determination using the Disc Diffusion Method
3.3. Structure Determination of ENO2 and Its Inhibitors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fothergill-Gilmore, L.A.; Michels, P.A. Evolution of glycolysis. Prog. Biophys. Mol. Biol. 1993, 59, 105–235. [Google Scholar] [CrossRef]
- Deutscher, D.; Meilijson, I.; Kupiec, M.; Ruppin, E. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat. Genet. 2006, 38, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Satani, N.; Hammoudi, N.; Ackroyd, J.J.; Khadka, S.; Yan, V.C.; Georgiou, D.K.; Sun, Y.; Zielinski, R.; Tran, T.; et al. Eradication of ENO1-deleted Glioblastoma through Collateral Lethality. BioRxiv 2018, 331538. [Google Scholar] [Green Version]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Aquilanti, E.A.; Depinho, R.A. Collateral Lethality: A new therapeutic strategy in oncology. Trends Cancer 2015, 1, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Adekola, K.U.A.; Martinez, M.; Rosen, S.T.; Shanmugam, M. Targeting Glycolysis and Compensatory Mitochondrial Metabolism in Multiple Myeloma with FDA Approved Ritonavir and Metformin Via Synthetic Lethality. Blood 2012, 120, 4016. [Google Scholar]
- Van Niekerk, D.D.; Penkler, G.P.; du Toit, F.; Snoep, J.L. Targeting glycolysis in the malaria parasite Plasmodium falciparum. FEBS J. 2016, 283, 634–646. [Google Scholar] [CrossRef]
- Guggisberg, A.M.; Frasse, P.M.; Jezewski, A.J.; Kafai, N.M.; Gandhi, A.Y.; Erlinger, S.J.; John, A.R.O. Suppression of Drug Resistance Reveals a Genetic Mechanism of Metabolic Plasticity in Malaria Parasites. MBio 2018, 9, e01193-18. [Google Scholar] [CrossRef] [Green Version]
- Gavalda, S.; Braga, R.; Dax, C.; Vigroux, A.; Blonski, C. N-Sulfonyl hydroxamate derivatives as inhibitors of class II fructose-1,6-diphosphate aldolase. Bioorg. Med. Chem. Lett. 2005, 15, 5375–5377. [Google Scholar] [CrossRef]
- Navarro, M.V.D.A.S.; Dias, S.M.G.; Mello, L.V.; Giotto, M.T.D.S.; Gavalda, S.; Blonski, C.; Garratt, R.C.; Rigden, D.J. Structural flexibility in Trypanosoma brucei enolase revealed by X-ray crystallography and molecular dynamics. FEBS J. 2007, 274, 5077–5089. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, V.E.; Weiss, P.M.; Cleland, W.W. Reaction intermediate analogues for enolase. Biochemistry 1984, 23, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.; Pisaneschi, F.; Link, T.M.; Lee, G.R.; Sun, D.; et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Methods 2016, 12, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Hanaya, T.; Itoh, C. An Efficient Synthesis of Antibiotic SF-2312 (3-Dihydroxyphosphoryl-1, 5-dihydroxy-2-pyrrolidone). Heterocycles 2010, 82, 1675. [Google Scholar] [CrossRef]
- Zaidan, M.R.S.; Rain, A.N.; Badrul, A.R.; Adlin, A.; Norazah, A.; Zakiah, I. In vitro screening of five local medicinal plants for antibacterial activity using disc diffusion method. Trop. Biomed. 2005, 22, 165–170. [Google Scholar]
- Detter, G.; Knothe, H.; Schönenbach, B.; Plage, G. Comparative study of fosfomycin activity in Mueller–Hinton media and in tissues. J. Antimicrob. Chemother. 1983, 11, 517–524. [Google Scholar] [CrossRef]
- Duncan, C.G.; Killela, P.J.; Payne, C.A.; Lampson, B.; Chen, W.C.; Liu, J.; Solomon, D.; Waldman, T.; Towers, A.J.; Gregory, S.G.; et al. Integrated genomic analyses identify ERRFI1 and TACC3 as glioblastoma-targeted genes. Oncotarget 2010, 1, 265–277. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W.; Battye, T.G.G. IMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Boil. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Boil. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement withphenix.refine. Acta Crystallogr. Sect. D Boil. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.; Merritt, E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar] [CrossRef]
- Watanabe, H.; Yoshida, J.; Tanaka, E.; Ito, M.; Miyadoh, S.; Shomura, T. Studies on a new phosphonic acid antibiotic, SF-2312. Sci. Rep. Meiji Seika Kaisha 1986, 25, 12–17. [Google Scholar]
- Metcalf, W.W.; Van Der Donk, W.A. Biosynthesis of Phosphonic and Phosphinic Acid Natural Products. Annu. Rev. Biochem. 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compound MethylSF2312, as isomeric mixture, is available in milligram quantities and SF2312 is commercially available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enolase 2: MethylSF2312 (PDB 5EU9) | Enolase 2:S-MethylSF2312 (PDB 5TIJ) | Enolase 2: R-MethylSF2312 (PDB 5TD9) | |
|---|---|---|---|
| Data Collection | |||
| Wavelength (Å) | 1.116 | 1.116 | 1.116 |
| Space group | P1 21 1 | P21 21 21 | P2 21 21 |
| Cell dimensions | |||
| a, b, c (Å) | 119.3, 110.3, 136.9 | 68.25, 108.38, 117.48 | 68.32, 112.87, 119.92 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| N°. of unique reflections | 217,213 | 26,354 | 40,898 |
| Resolution (Å) | 89.93–2.05 (2.08–2.05) | 59.01–2.63 (2.76–2.63) | 50.00–2.31 (2.35–2.31) |
| Rmerge (all I+ and I−) | 0.200 (0.994) | 0.110 (0.290) | 0.110 (0.391) |
| I/σI | 6.4 (2.0) | 11.7 (5.2) | 32.2 (5.3) |
| Completeness (%) | 97.6 (91.7) | 99.6 (98.2) | 99.8 (96.4) |
| Redundancy | 6.7 (6.4) | 6.5 (6.1) | 8.0 (7.3) |
| Refinement | |||
| Resolution (Å) | 89.93–2.05 | 59.01–2.63 | 43.51–2.32 |
| σF | 1.34 | 1.38 | 1.34 |
| N°. of reflections | 217,011 | 26,306 | 40,812 |
| Rwork/Rfree | 0.154/0.192 | 0.206/0.267 | 0.200/0.245 |
| Wilson B | 21.4 | 30.0 | 33.4 |
| N°. of atoms | |||
| Protein | 26,693 | 6627 | 6644 |
| Ligands | 164 | 26 | - |
| Ions | 16 | 2 | 4 |
| Water | 2238 | 110 | 86 |
| Average B-factors (Å2) | |||
| Protein | 20.8 | 37.6 | 54.4 |
| Ligands | 23.9 | 33.6 | - |
| Ions | 14.7 | 34.2 | 68.0 |
| Water | 28.7 | 28.2 | 43.1 |
| r.m.s.d. | |||
| Bond lengths (Å) | 0.012 | 0.005 | 0.006 |
| Bond angles (°) | 1.039 | 0.451 | 0.636 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisaneschi, F.; Lin, Y.-H.; Leonard, P.G.; Satani, N.; Yan, V.C.; Hammoudi, N.; Raghavan, S.; Link, T.M.; K. Georgiou, D.; Czako, B.; et al. The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues. Molecules 2019, 24, 2510. https://doi.org/10.3390/molecules24132510
Pisaneschi F, Lin Y-H, Leonard PG, Satani N, Yan VC, Hammoudi N, Raghavan S, Link TM, K. Georgiou D, Czako B, et al. The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues. Molecules. 2019; 24(13):2510. https://doi.org/10.3390/molecules24132510
Chicago/Turabian StylePisaneschi, Federica, Yu-Hsi Lin, Paul G. Leonard, Nikunj Satani, Victoria C. Yan, Naima Hammoudi, Sudhir Raghavan, Todd M. Link, Dimitra K. Georgiou, Barbara Czako, and et al. 2019. "The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues" Molecules 24, no. 13: 2510. https://doi.org/10.3390/molecules24132510
APA StylePisaneschi, F., Lin, Y.-H., Leonard, P. G., Satani, N., Yan, V. C., Hammoudi, N., Raghavan, S., Link, T. M., K. Georgiou, D., Czako, B., & Muller, F. L. (2019). The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues. Molecules, 24(13), 2510. https://doi.org/10.3390/molecules24132510