Abstract
Intracellular synthesis, folding, trafficking and degradation of proteins are controlled and integrated by proteostasis. The frequency of protein misfolding disorders in the human population, e.g., in Alzheimer’s disease (AD), is increasing due to the aging population. AD treatment options are limited to symptomatic interventions that at best slow-down disease progression. The key biochemical change in AD is the excessive accumulation of per-se non-toxic and soluble amyloid peptides (Aβ(1-37/44), in the intracellular and extracellular space, that alters proteostasis and triggers Aβ modification (e.g., by reactive oxygen species (ROS)) into toxic intermediate, misfolded soluble Aβ peptides, Aβ dimers and Aβ oligomers. The toxic intermediate Aβ products aggregate into progressively less toxic and less soluble protofibrils, fibrils and senile plaques. This review focuses on peptides that inhibit toxic Aβ oligomerization, Aβ aggregation into fibrils, or stabilize Aβ peptides in non-toxic oligomers, and discusses their potential for AD treatment.
1. The Social and Economic Relevance of Alzheimer’s Disease
The increase in life expectancy is accompanied with an increased number of patients suffering from Alzheimer’s disease (AD). Patients with AD experience a progressive memory loss combined with a decline in cognitive function and ultimately a premature death 3–9 years after diagnosis [1]. AD is the most common cause of dementia, with a prevalence that increases with age to about 50% in people aged 85 or older. In practical terms, the majority of the population will have AD provided he or she has a sufficiently long life. The worldwide number of patients with AD is projected to increase to more than 140 million in 2050 [2].The total estimated worldwide cost of dementia in 2015 was US $818 billion, will reach US $1 trillion in 2018 and US $2 trillion by 2030. The true number of patients and the associated cost of dementia are likely to be considerably higher, since a huge majority of people with dementia have not received a diagnosis, have not been registered, and are thus unable to access care and treatment [3].
2. Etiology of AD
The characteristic changes in the brain tissue of AD patients are: (a) hyperphosphorylated tau protein rich intra-neuronal neurofibrillary tangles (NFT); (b) extracellular senile plaques of aggregated amyloid β (Aβ) peptides; and (c) brain tissue atrophy, starting in the entorhinal region and the temporal lobe, and progressing to the limbic system and the neocortex [4,5,6,7,8,9]. Most patients with AD have a sporadic, late onset form, where the major risk factors are aging, type 2 diabetes (T2D) and apolipoprotein E ε4 (APOE-ε4) [10,11,12,13,14,15]. A minority of AD patients have an early onset, genetic, familial form of AD due to autosomal dominant mutations in amyloid β precursor protein (AβPP), presenilin-1 (PS1), and presenilin-2 (PS2) [16]. The initiating step that leads to the disease in most AD patients is not known. The number and extent of amyloid plaques have only a week correlation with the degree of cognitive decline; they can also be found in individuals with mild cognitive impairment and in people with no cognitive symptoms [17]. There are at least three possible, and non-excluding, explanations for the lack of correlation between Aβ plaques and cognitive impairment in patients with AD: (a) person-to-person differences in the ability of inflammatory cells to effectively remove senile plaques from the brain [18]; (b) the high neurotoxic properties of Aβ42 oligomers, which precede the less neurotoxic senile plaques, are the main cause of cognitive impairment; and (c) person-to-person variations in brain plasticity, the ability to restore brain function after injury that operates over long [19] and short [20] timescales.
2.1. Amyloid Precursor Protein (APP)
The human amyloid precursor protein (APP) is a single-pass transmembrane protein in nerve cells. The physiological significance of APP is not fully understood, although it has a fast turnover rate in the mature, normal CNS [21,22]. APP is first processed in the endoplasmic reticulum and Golgi apparatus, and then transported either to synaptic terminals where it is inserted into the synaptic cell membrane [23] or into endosomes. The cell-membrane bound APP is processed by three competing pathways: (a) two amyloidogenic pathways; and (b) a non-amyloidogenic pathway (Figure 1). In the amyloidogenic pathways, APP is: (i) either sequentially cleaved by β- and γ-secretase leading to direct extracellular production of amyloid β-peptides (Aβps); or (ii) re-internalized into endosomes containing the β- and γ-secretases, leading to intracellular production of Aβps, that are either exported to the extracellular space (via vesicular transport) or degraded in lysosomes [21]. The APP re-internalization pathway substantially contributes to production of Aβps since: (a) β-secretase is located predominantly in endosomes [24]; and (b) 80% of Aβp release into the extracellular space is blocked by inhibiting endocytosis [23]. The amyloidogenic pathway first proteolyses APP with β-secretase into a soluble APPβ peptide and a 99-amino acid membrane bound C-terminal fragment (99-CTF)—and then proteolyses the 99-CTF with γ-secretase into a APP intracellular domain fragment (APP-ICD) and Aβps with 37–44 amino acid residues [21]. Low concentrations of Aβps are present in the central nervous system of non-demented individuals, therefore, Aβps could present a negative feedback loop in the regulation of synaptic plasticity and neuronal survival [25]. For example, the Aβ(1-42) monomer’s neuroprotective role is mediated by activation of the phosphatidylinositol-3-kinase pathway, and the stimulation of receptors of the insulin superfamily [26]. APP is cleaved by α- and then γ-secretase in the non-amyloidogenic pathway; γ-secretase activity is present on the cell surface and in endosomes [27,28] while α-secretase proteolysis of APP occurs mainly on the cell surface, and to a small extent in the Golgi apparatus [21]. APPα, the soluble product of α-secretase action on APP, has neuroprotective and memory-enhancing effects [29,30]. In physiological conditions, APP is preferentially processed by the non-amyloidogenic pathway and increased Aβp production correlates with decreased sAPPα levels [31,32,33,34]. Amyloid processing of APP is concentrated in lipid rafts, and non-amyloid processing in the non-raft regions of the cell membrane [35,36,37,38,39]. Proteolytic enzymes of both APP processing pathways are modulated by external factors. For example, some metalloproteases have an α secretase activity, contributing to the production of soluble sAPPα [40]. The amyloidogenic pathway activity is attenuated by cholesterol lowering drugs, metal chelators, steroid hormones or non-steroidal anti-inflammatory drugs [41]. Decreased cellular cholesterol levels disrupt the lipid raft’s structure and function, leading to a concomitant decrease in Aβp and an increase in sAPPα formation [42,43].
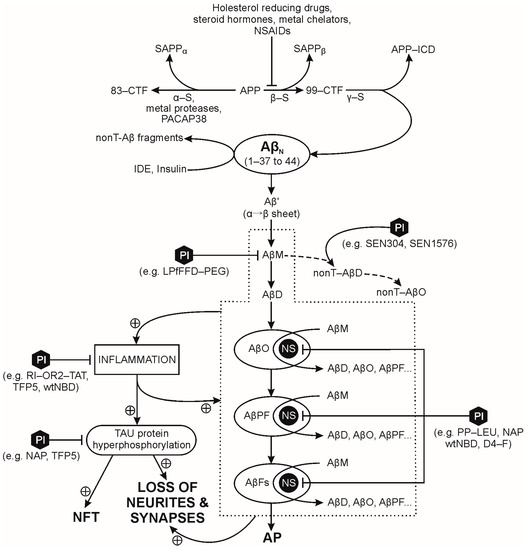
Figure 1.
Pathways of amyloid precursor protein proteolysis, amyloid plaque formation and targets of selected peptide inhibitors. Abbreviations: 83-CTF (83-amino acid membrane bound C-terminal fragment); 99-CTF (99-amino acid membrane bound C-terminal fragment); AP (amyloid plaque); APP (amyloid precursor protein); APP-ICD (APP intracellular domain); Aβ′ (misfolded Aβ peptide with first α-sheet, then β-sheet structure); Aβn (native Aβ peptide with α-helix structure); AβD (amyloid β dimer); AβFs (amyloid β fibrils); AβM (amyloid β monomer); AβO (amyloid β oligomer); AβPF (amyloid β protofibril); IDE (insulin degrading enzyme); NFT (neurofibrillary tangles); nonT-AβD (non-toxic amyloid β dimer); nonT-AβO (non-toxic amyloid β oligomer); NS (nucleation site); NSAIDs (nonsteroidal anti-inflammatory drugs); PI (peptide inhibitor with examples in parentheses); SAPPα (soluble peptide APPα); SAPPβ (soluble peptide APPβ); ⊥ attenuates/inhibits, α-S (α-secretase); β-S (β-secretase); γ-S (γ-secretase); ⊕ promotes/accelerates.
2.2. Pathological Processing of Soluble Amyloid β-Peptides (Aβps)
Aβ peptides are useful for neurons [26,44]. The key biochemical change in AD is the excessive accumulation of per-se non-toxic and soluble amyloid peptides (Aβ(1-37/44), in the intracellular and extracellular spaces, that alters proteostasis and triggers Aβ modification (e.g., by reactive oxygen species—ROS) into toxic intermediate Aβp products, misfolded soluble Aβps, Aβ dimers and Aβ oligomers. For example, intracellular accumulation of Aβ(1-42) per-se promotes increased ROS production and p53 mediated apoptosis [45,46,47,48]. The toxic intermediate Aβp products further aggregate into progressively less toxic and less soluble protofibrils, fibrils and finally extracellular brain senile plaques composed of proteinaceous deposit with β sheet structure. The most common Aβ is Aβ(1-40); Aβ(1-42) is the most susceptible to toxic conformational changes leading to nerve death and amyloid plaque formation. Aβ(1-42) monomers with intermediate conformations and Aβ(1-42) oligomers are the most neurotoxic and amyloid plaques the least [49,50]. Modified Aβ(1-42) peptides enter the cells via endocytosis and lead to lysosomal fusion dysfunction. The overall effect of lysosomal fusion dysfunction is an enhanced transport of vesicles by the exosomal pathway with increased shedding of modified Aβps into the extracellular space and a reduced Aβp digestion rate by macroautophagy [51]. Aβ(1-42) oligomers interfere with synaptic transmission by: (a) promoting neuronal death by attenuating NMDAR desensitization thus increasing the probability of intracellular Ca2+ overload [52,53]; (b) decreasing the density of AMPA synaptic receptors [54]; (c) uncoupling metabotropic glutamate receptors’ (mGluR5) dependent activation of PKC [55]; and (d) reducing glutamate reuptake thus promoting an increased NMDAR and mGluR5 mediated entry of Ca2+ [56]. AD studies on patients and animal models associated Aβ oligomers with synaptic dysfunction, cognitive decline, inhibition of hippocampal long-term potentiation (LTP) component in memory, and learning and memory impairment [57,58,59,60,61,62,63,64,65,66]. Aβ oligomers were better correlated with dementia and synaptic loss then Aβps in amyloid plaques [57,58].
2.3. Factors Promoting and/or Sustaining Pathological Processing of Amyloid β-Peptides (Aβps)
2.3.1. Aβp Oxidation
Aβp oxidation promotes toxic misfolded Aβ monomers, oligomers and intermediate products [67]. For example, oxidation of Aβ(1-42) at the methionine residue 35, Aβ1-42-MET35-OX, promoted by H2O2 or copper ions, accelerates the production of toxic Aβ(1-42) products directly and indirectly by increasing oxidative stress, protein oxidation and lipid peroxidation [67,68]. In a cell model, ROS stabilize Aβ oligomers, by dityrosine cross-links in Aβ(1-42), and promote internalization of toxic Aβps into lysosomes [68]. Dityrosine crosslinked Aβ oligomers self-assemble to form amyloid fibrils; their presence was detected within plaques in brain samples of patients with AD [68].
2.3.2. Mitochondrial (MITO) Dysfunction
Aβps and Aβ oligomers accumulate in MITO samples from transgenic mice overexpressing mutant AβPP and in post-mortem brains and from AD patients [69,70,71,72]. In human and animal studies, increased Aβp levels either preceded or followed MITO dysfunction implying a positive feedback loop. MITO dysfunction was due to: (a) oxidative modifications of key MITO enzymes (e.g., pyruvate dehydrogenase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase and cytochrome c oxidase) [70,72,73,74,75,76]; (b) reduced antioxidant defenses [77]; and (c) increased production of ROS [78]. Human and animal studies are in agreement that Aβp binds with MITO Aβ-binding alcohol dehydrogenase (ABAD) precipitating increased ROS generation, MITO dysfunction and cell death [79]. MITO dysfunction can also stimulate the amyloidogenic APP pathway; in a transgenic AD mouse model, knockout of manganese superoxide dismutase, a major MITO antioxidant enzyme, increased Aβp levels and amyloid plaque formation in the brain [77].
2.3.3. Oxidative Stress
In AD, oxidative stress supports a self-sustained process of increased production of soluble Aβ oligomers from Aβs with a concomitant progressive failure of macroautophagy (reduced clearance of Aβs) and mitochondrial function (increased production of ROS). Oxidative stress upregulates β-secretase and γ-secretase expression thus promoting Aβ production [80,81,82,83,84,85]. Studies on human autopsy brain samples from patients with AD and on animal models of AD imply that oxidative damage occurs before Aβ plaque formation [86,87,88]. For example, an increase in reactive nitrogen species coincided with the onset of Aβ deposition in a transgenic AD mouse model [89].
2.3.4. Advanced Glycation End Products (AGEs)
Patients with AD had more AGEs in brain samples than age-matched controls [90]; AGEs were co-localized with NFT and amyloid plaques [91], implying they accelerate aggregation of soluble Aβps and tau into amyloid plaques and NFTs respectively [90,92]. In a cell model, AGEs promoted oxidative stress and inflammation by activation of kappa-light-chain-enhancer of activated B cells (NF-κB) and increased cytokine IL6 gene expression with a concomitant increased release of Aβps [93].
2.3.5. Apolipoprotein E (ApoE) Polymorphism and Cholesterol Levels
ApoE, the principal cholesterol carrier in the brain, is synthetized in astrocytes and transports cholesterol to neurons [94,95]. Persons with two APO-ε4 alleles have the single largest known genetic risk factor for late-onset sporadic AD [10,96,97,98] since APOE-ε4 does not promote the extra- and intra-cellular proteolysis of Aβps as efficiently as the APOE-ε2 or -ε3 isoforms [99]. This is consistent with the finding that MITO dysfunction in AD patients with ApoE-ε4 allele correlates better with cognitive dysfunction, than in AD patients carrying the ApoE-ε3 allele [100]. High serum total cholesterol is an independent risk factor for late onset AD in persons with any combination of ApoE alleles [13]. Supporting experimental evidence is provided by: (a) reduced Aβp production in cultured cells when cholesteryl-ester levels were reduced by inhibiting Acyl-CoA cholesterol acyltransferase (ACAT) [101]; (b) reduced amyloid and tau deposition, by enhancing autophagy with ACAT1 inhibition, in cell culture and whole animal mouse models of AD [102,103,104]; and (c) cholesterol depletion decreased Aβp production in rat hippocampal neurons [105].
2.3.6. Inflammation, Vascular Pathology and Cellular Immunity
Human and animal model studies imply that inflammation promotes Aβ plaque deposition and tau hyperphosphorylation [22,106,107,108,109,110,111,112,113,114,115,116]. For example, post-mortem human AD brains have an increased activation of inflammatory and immune pathways with upregulated levels of pro-inflammatory cytokines, chemokines and complement proteins [108] consistent with microarray studies of brain samples from humans with AD or from animal models of AD that have an increased expression of genes involved in inflammation [109,111]. The intensity of inflammation varies over the course of AD; cytokine activity is the highest in the early clinical course of AD. The reduction of cytokine activity, in the late clinical course of AD, is concomitant with the increased brain levels of Aβ plaque, hyperphosphorylated tau, and AGEs [117]. Animal studies of AD transgenic mice imply that the intra-neuronal Aβ(1-42) is concentrated in neurites and synapses leading to their destruction and leaving a surviving nerve cell with a reduced number of neurites and synapses with adjacent extracellular accumulations of Aβ(1-42) [118]. The magnitude of brain synapse loss in AD correlates well with the degree of cognitive decline [119,120]. This could explain short term memory binding impairment in patients with AD, an early sign of the disease, since the patient’s capacity to form new memories is degraded by loss of synapses [121]. Chronic brain inflammation, as a result of extracellular Aβ(1-42) accumulation following destruction of synapses, neurites and finally neurons, is sustained by an inflammatory response of the surrounding astrocytes and microglia [122,123]. Inflammation increases ROS production that oxidize prostaglandins into F2α-isoprostanes. Both H2O2 and F2α-isoprostanes accelerate aggregation of Aβps into toxic oligomers [49,124]. Extracellular Aβ(1-42): (a) propagates its neurotoxic effects to neighboring nerve cells, an increased extracellular Aβ(1-42) leads to an increased intracellular Aβ(1-42) [18,125,126]; and (b) is further metabolized into less neurotoxic Aβ amyloid plaques by the concomitant inflammatory process [18,127,128,129]. Chronic inflammation, vascular pathology and cellular immune response in the brain are interdependent in AD and tend to re-inforce each other. Aβ(1-42) deposition in brain blood vessel walls accelerates its own brain deposition by promoting vascular damage with subsequent brain hypoperfusion, hypoxia and reduced Aβ(1-42) brain clearance. Thus, the observed vascular defects in AD can be either the cause or consequence of amyloid β accumulation [130]. The role of astroglia and microglia in the evolution of AD is under-investigated, although these cells are vital for normal nerve cell function [130]. Astroglia, a part of the tripartite synapse formed with the pre- and postsynaptic neurons, participates in synapse formation, pruning, maintenance and modulation of synaptic transmission; astroglia cells are also in contact with blood vessels and form, together with neurons, discrete neurovascular clusters [131,132]. Astroglia responds to Aβ(1-42) with an increased, NF-κB mediated, C3 release; C3 binds to neurons and promotes atrophy and elimination of synapses [133]. Astrogliosis, a reaction to brain damage, is present in patients with AD and can be reproduced in animal models of AD. Microglia cells also respond to Aβ(1-42) by promoting inflammation and Aβ(1-42) phagocytosis in the brain [130]. In addition to phagocytosis, microglia also regulate brain nerve cells’ function with synaptic pruning and apoptosis [134]. The role, or different roles over time, of astroglia and microglia on the progression of AD is not well understood; for a recent comprehensive review, see De Strooper and Karran, 2016 [130].
2.3.7. Tau Processing in Alzheimer’s Disease
Tau phosphoprotein promotes tubulin assembly into microtubules and microtubule stability. In the brain, tau is concentrated in neurons and expressed at very low levels in astrocytes and oligodendrocytes [135]. In nerve cells, tau activity is not evenly distributed, but is concentrated at the distal end of axons, presumably to enable optimal stability and plasticity of synapses, the cell-to-cell connections between axon terminals of the upstream nerve and dendrites of the downstream nerve, that propagate the electrochemical stimulation. The breakdown of microtubules is promoted by tau phosphorylation with kinases or caspases; microtubule stability and growth are enhanced by phosphatases, promoting tau dephosphorylation [21,136,137]. Tau hyperphosphorylation leads to the disappearance of microtubules, the breakdown of intracellular traffic, the “dying back” of axons and tau redistribution from an axonal to a somato-dendritic pattern, presumably due to an increased tau synthesis as a response to increased tau hyperphosphorylation [138]. The degree of tau phosphorylation increases with age and is further accelerated by high blood glucose [139,140]. Additional factors that also promote tau hyperphosphorylation are: (a) Aβ(1-42) (via activation of kinases and caspases) [141,142,143]; (b) serine protease inhibitor α1-antichymotrypsin ACT [123]; and (c) oxidative stress [84,144,145,146]. Tau hyperphosphorylation promotes tau self-assembly and NFT formation [147].
2.4. Association of NFTs and Aβ Plaques with the Severity of Cognitive Decline in AD
Post-mortem studies on patients with AD imply a strong association among NFTs, neuronal loss and the severity of cognitive decline [148,149,150]. On the other hand, Aβ plaques seem to appear at the pre-symptomatic stage of AD and their levels stabilize relatively early in the disease process [151].
3. Peptides for Modifying Alzheimer’s Disease
Thus far, the efforts to develop AD therapies, based on “amyloid cascade hypothesis” and NFTs hypotheses, have been disappointing, even when the effective clearing of Aβ deposits in AD brain was demonstrated [152,153,154,155,156,157,158,159,160]. Alternative therapies with antioxidants [161] and anti-inflammatory agents [162,163] were also ineffective. Current drug therapy of AD is symptomatic, to support normal brain synaptic transmission with NMDAR antagonists (to attenuate excessive glutamate release and related nerve cell death rate) and with cholinesterase inhibitors, to conserve acetylcholine synaptic concentration [8,164].
The key issues for development of an efficient AD peptide therapy include: (a) selecting a target for peptide therapy; (b) understanding the relationships among protein sequence, structure, solubility and aggregation [165,166]; (c) integration of chemical kinetics into the drug discovery process to characterize and quantify the inhibition of protein aggregation [167]; (d) development of either Aβ peptide-sequence based or non-Aβ peptide sequence based peptide inhibitors of Aβ aggregation [168]; (e) selection of a drug delivery route [169]; and (f) development of a personalized therapy for AD patients [8,164,168].
3.1. Selecting a Target for Peptide Therapy
3.1.1. Ameliorating AD Cognitive Impairment with Insulin
Brain regions with the highest concentration of insulin receptors are the olfactory bulb, hippocampus and hypothalamus [170,171]. The hippocampus, involved in long-term memory formation, spatial cognition, and conflict processing [172,173,174], has insulin receptors preferentially localized to nerve synapses [175,176]. Research on animal models implies that insulin receptor signaling contributes to long-term memory consolidation and improved spatial learning [171,177,178,179]. Insulin treatment improved memory and cognitive function in patients with AD and in patients with mild cognitive impairment [180,181,182,183,184]. The neuroprotective effects of insulin are mediated by several pathways. (a) Insulin binds to brain receptors and their activation: (i) increases transcription of anti-amyloidogenic proteins insulin-degrading enzyme and α-secretase; and (ii) decreases transcription of pro-amyloidogenic proteins AβPP, β-secretase and glycogen synthase kinase 3 α (Gsk3α) [185,186]. (b) Insulin attenuates Aβ oligomers’ binding to neurons thus protecting synapses against the toxic effects of Aβ oligomers [175]. (c) Insulin increases clearance of brain Aβ(1-42) into the CSF thus reducing Aβ(1-42)’s intracellular accumulation [187]. Insulin improved memory and cognition in patients with AD after a single dose and also after up-to four-months treatment [188,189,190,191,192]. The positive effects of insulin therapy diminish with the progression of AD when increased Aβp levels promote brain insulin resistance [193].
3.1.2. Inhibition of Aβ Peptide Synthesis and Therapies with Tau Inhibitors, Aβ Peptide Chelators or Antibodies
Inhibitors were designed and tested that block APP expression or prevent proteolytic cleavage of APP into Aβps. Inhibition of γ-secretase, an enzyme involved in many physiological reactions, also lead to impaired lymphocyte differentiation and altered intestinal development [194,195]. β-secretase inhibitors are large molecules that are prevented by the blood–brain barrier (BBB) to reach their target [196,197]. Thus far, all inhibitors of β- or γ-secretase failed during clinical trials [198,199,200,201,202,203,204,205,206]. Chelation therapies, to disrupt interactions between Aβps and metal, were also not successful [207,208,209,210]. The only tau inhibitor molecule under clinical trials is phenothiazine methylene blue [211,212,213,214]. Antibody therapies for AD failed due to the antibodies’ inability to cross the BBB [215,216,217,218].
3.1.3. Inhibition of Aβ Peptide Aggregation and Amyloid Formation
In principle, small molecules that inhibit Aβ aggregation [219,220,221,222,223,224] should not disrupt the normal biochemical processes in the body. However, their design is challenging for several reasons: (a) in contrast to protein-protein interactions that occur over large surface areas [225,226], protein-small molecule interactions occur over 3–5 times smaller contact surfaces [227,228]; and (b) regions of protein-protein interactions are relatively featureless and variable [229,230,231]. The discovery of key regions in the Aβ peptide sequence (i.e., N-terminus, hydrophobic core, hinge/turn region and C-terminus), responsible for Aβ plaque formation, was essential for the rational development of peptide inhibitors of amyloidogenesis [232,233,234,235,236,237,238].
Aβ peptides undergo several conformational changes: from an α-helix (i.e., random coil) native structure ⇒ to an intermediate α-sheet ⇒ to a β-sheet (i.e., highly ordered, with exposed hydrophobic amino acid residues that promote Aβp aggregation) structure present in amyloid fibrils, the building blocks of Aβ plaques. The intermediate α-sheet configuration has the propensity to aggregate into colloidal spheres that spontaneously form linear chains, the first step in amyloid fibril formation [239]. The conversion from the intermediate α-sheet to the β-sheet conformation has been inferred from protein crystal structure analysis [240,241,242] and studies with inhibitors of Aβ peptide aggregation [243]. Aβp intermediates and oligomers, that chronologically precede amyloid fibril formation, have heterogeneous conformations [244,245,246] that also change with age [247]. The Aβ plaques also contain a diverse population of truncated or modified Aβ peptides [248]. The Aβ peptide’s α-helix native structure can be stabilized or its β-sheet structure destabilized which has important implications for development of AD modifying therapies [196,249,250,251].
1. Aβp Sequence Derived Peptides
This group of peptides is based on the central hydrophobic core (CHC) sequence Aβ(16-20/22) and includes peptides with natural or modified amino-acids. The natural amino-acid peptides inhibit Aβ aggregation, but have the disadvantages of self-association into fibrils and low proteolytic stability. These disadvantages were ameliorated by various modifications including N-methylation [252], d-amino acid incorporation, retro-inverso peptides, replacement of amide by ester bonds, and cyclization [168].
• Natural Aβp sequence derived peptides
An example of a natural Aβ sequence derived peptide is the oligopeptide Ac-Leu-Pro-Phe-Phe-Asp-NH2 (iAb5p), where a proline and an aspartic acid amide substitute the valine and the alanine, respectively. This peptide crosses the BBB; in vivo, it inhibits formation of amyloid fibrils and promotes disassembly of formed amyloid plaques. The efficacy of this synthetic peptide is contributed to its proline residues that attenuate the β-sheet confirmation propensity of the natural Aβ peptides [249].
• Modified Aβp sequence derived peptides
Fluorinated Aβ sequence derived peptides are produced by fluorination of hydrophobic amino acids valine or phenylalanine. Fluorinated amino acids bind with hydrophobic residues of Aβps, thus preventing contact between Aβ peptide molecules and inhibiting their aggregation [253,254].
d-peptides are more resistant to proteolytic enzymes than their l-isomers. d-peptides inhibited Aβ aggregation in an animal model [255].
Retro-Inverso modified Aβ sequence derived peptides are derived from peptides by substituting the l-amino acids for their d-counterparts and reversing the sequence to mimic the original peptide since they retain the same spatial positioning of the side chains and 3D structure [244,256,257]. These peptides combine the advantage of the original peptide (i.e., inhibition of aggregation) with an increased resistance to enzymatic proteolysis, a reduced self-aggregation and an improved ability to cross the BBB in an animal model [258,259,260,261,262].
Cyclic peptides were developed from KLVFF derivatives. They are strong and specific inhibitors of amyloid formation. Compared to their noncyclic equivalents, they are metabolized more slowly due to their resistance to enzymatic degradation [244,263,264].
N-methylated peptides (e.g., SEN304 and SEN1576) inhibit Aβp mediated toxicity in an animal model. They do not inhibit Aβp aggregation, they divert Aβp aggregation into non-toxic forms and remove toxic oligomers [265,266].
α sheet peptides have alternating d-amino acids and l-amino acids; they reduce Aβ aggregation and toxicity but lack the ability to cross the BBB [267].
2. Peptides not derived from Aβp sequence
Peptides not derived from Aβp sequence and peptides derived from Aβ sequence share the qualities of hydrophobicity and the ability to incorporate into the β-sheets structure, thus inhibiting Aβp aggregation.
• Peptides not derived from Aβp sequence containing natural amino acids
Examples are NAP [268], carnosine [269,270] and hexapeptides [271]. All of them inhibit Aβ peptide aggregation. NAP also attenuated mild cognitive impairment in phase II clinical trials [272,273].
• Peptides not derived from Aβp sequence containing modified amino acids
D-4F improved cognitive function when administered orally to mice and also inhibited Aβp deposition [274].
Cyclic peptide PP-Leu is a tridecapeptide analogue of theta-defensins. This peptide combines the ability to inhibit Aβ oligomer and amyloid fibril formation, by sequestration of amyloidogenic Aβ peptides into colloid-like assemblies, with a high resistance to endoproteinase K [275].
3.2. Relationship between Protein Sequence, Structure, Solubility and Aggregation
Protein aggregation is regulated by the same physicochemical laws as normal protein folding and shares many common features with other thermodynamically stable protein conformations [276]. Protein solubility is an essential feature for successful development of AD peptide therapy; proteins in aggregates lose their therapeutic effect, can become toxic or increase the probability for an immune response in the treated patient [277,278].
Protein aggregation prediction methods are based on the analysis of physicochemical factors that determine protein thermodynamics and kinetics of unfolded polypeptide aggregation [279,280], the discovery of key regions in the Aβ peptide sequence responsible for Aβ plaque formation [281,282,283,284,285,286,287,288] and advances in modelling and simulation of aggregation-prone regions, recently reviewed by Trainor et al. [166]. The discovery of aggregation-prone regions in Aβ peptides is essential for the rational development of peptides that reduce the Aβ peptides’ tendency to form ordered intermolecular assemblies [289,290,291]. The regions of Aβ(1-42) and Aβ(1-40), with a high aggregation propensity, are the central region (residues 18–22) and the C-terminal region (residues 32–42) [236,292,293]. In solution, Aβ peptides are largely unstructured with no stable folded structure [165].
Aggregation prediction algorithms were classified by Trainor et al. into amino acid composition-based algorithms, sliding window/pattern-based algorithms and tertiary/quaternary structure-based algorithms [166].
3.2.1. Amino Acid Composition-Based Algorithms
Amino acid composition-based algorithms use statistical analyses and machine learning algorithms to predict/generalize aggregation-prone regions based on amino acid sequences of proteins with known aggregation propensities. These algorithms use amino acid and/or dipeptide frequencies to determine peptide solubility in a binary manner (either soluble or insoluble) and disregard the position of each amino acid in the studied peptide sequence. Therefore, they cannot pinpoint specific amino acid changes with the greatest aggregation impact [3,166,279,294,295,296,297,298,299]. To improve solubility predictions of amino acid composition-based algorithms, parameters are included that reflect the order of the amino acids in the studied protein sequence, for example Shannon entropy and parameters derived from Chaos Game Representations of amino acid sequences [296,300]. To summarize, these methods are more appropriate for binary, proteome wide, solubility surveys and less for optimizing protein solubility of AD modifying peptides [166].
3.2.2. Sliding Window/Pattern-Based Algorithms
The sliding window methods can analyze amino acid sequences for aggregation-prone regions that can be as short as five amino acid residues. They are based on the theoretically and experimentally verified premise that 5–7 residue sequence segments can exert a disproportionate influence on protein solubility. Protein solubility is estimated by considering the contributions of charge, hydrophobicity/hydrophilicity ratio, secondary structure propensity, statistical analysis of residue pairings between adjacent β-strands in known structures and patterns of residue distribution in amyloidogenic hexapeptides [165,282,301,302,303,304,305,306,307,308,309,310,311,312,313,314,315,316,317]. CamSOL incorporates the sliding window method to develop soluble antibodies targeting Aβ peptides [313]. Other examples of tools using the sliding window method to study Aβ peptides aggregation are AmyloidMutants [310], STITCHER [318], AGGRESCAN [301,316] and Ziggregator [165]. The sliding window based methods can recognize amino acid sequence features relevant to aggregation of Aβ peptides. However, the composition of Aβ peptide aggregates varies, is not homogenous [287]. In addition, most sliding window method based tools do not include information on protein structure that influences exposure of aggregation prone regions; a limited exposure of aggregation prone regions can change the agreement between aggregation propensity predictions and experimentally measured aggregation [166]. None of the sliding window methods explicitly take into account the tertiary/quaternary structure of a native protein although associations between natively folded proteins can indirectly impact aggregation kinetics of partially or fully unfolded proteins [277]; in addition, aggregates may be formed by partially unfolded/structured proteins [319,320,321].
3.2.3. Tertiary/Quaternary Structure-Based Algorithms
Structure based methods [313,322,323,324,325] reliably predict protein aggregation even when a significant number of aggregation-prone regions is inaccessible; for example when transient (α-sheet) or stable (β-sheet) associations between folded Aβ proteins accelerate aggregation [166]. Examples of structure based methods are SAP [326], the upgraded version of CamSol [165] and AGGRESCAN3D [166]. The SAP method can grade cytotoxicity of Aβ(1-42) peptide variants [326].
3.3. Integration of Chemical Kinetics into the Drug Discovery Process to Charaterize and Quantify the Inhibition of Protein Aggregation
3.3.1. Challenges in Understanding Aggregation Inhibition Mechanisms at the Molecular Level
The key step in Aβ plaque formation are conformational changes of Aβ peptides into reactive, metastable oligomers (i.e., the primary nucleation processes) that assemble into protofibrils and later fibrils [327]. Aggregation of Aβ peptides is further accelerated by the secondary nucleation processes triggered by fibril fragmentation and fibril surface catalyzed nucleation [328,329]. The kinetics of fibril formation follow a sigmoidal curve, preceded by a lag phase and terminated by a plateau after Aβ monomer depletion [330,331]. Most inhibitors attenuate Aβ peptide aggregation by forming covalent or noncovalent bonds with one or more products of the aggregation pathway. For example, aromatic compounds sequester protein monomers and/or oligomers in a nonspecific manner [332,333]. A variety of methods is necessary to quantify the binding characteristics of Aβ peptides’ monomers and oligomers with peptide inhibitors [50,333,334,335,336,337,338,339].
3.3.2. Kinetic Analysis of Aggregation Inhibition Mechanisms Is Necessary for Development and Evaluation of Drug-Like Small Molecules
Arosio et al. present a strategy that integrates experimental characterization of Aβ peptide products with chemical kinetics (i.e., reaction kinetics) to facilitate a rational design and evaluation of peptides for AD therapy [167]. The advantage of chemical kinetics is the method’s ability to measure very weak binding events and their effects, an important characteristic of inhibitor–protein interactions. This method evaluates how different experimental conditions influence the speed of reactions that generate Aβ oligomers, protofibrils and fibrils, yielding information about the reactions’ mechanisms and transition states [340,341]. Chemical kinetics can model the effect of: (a) protein structural changes due to binding [342]; (b) covalent reactions between proteins and inhibitors [343]; (c) off-pathways (i.e., non-amyloidogenic pathways) that also produce toxic soluble oligomers [50]; and (d) Aβ aggregation inhibitors that reduce the amount of toxic Aβ oligomers by redirecting the aggregation process to alternative off-pathway products [334,339,343,344,345].
Kinetic analysis of Aβ peptide aggregation inhibition mechanisms evaluates how the inhibitions of primary nucleation, secondary nucleation or elongation individually contribute to Aβ fibril formation. For example: (a) inhibition of monomer formation decreases the rate of primary and secondary nucleation and fibril elongation; (b) inhibition of oligomer production reduces primary and secondary nucleation rates; (c) inhibitor binding to the fibril ends slows down the fibril elongation process; and (d) inhibitor binding to amyloid fibrils decreases only the surface catalyzed secondary nucleation rate [167]. In addition, kinetic analysis can: (a) distinguish between the effects of fibril elongation and secondary nucleation on lag-time phases and growth rates of fibril mass fraction; (b) quantify the effect of primary nucleation inhibition on the lag-time preceding the fibril mass growth phase; (c) demonstrate that the final quantity of fibril mass fraction is reduced only by the inhibition of primary nucleation and fibril elongation processes; and (d) demonstrate that the inhibition of primary nucleation and fibril elongation processes delay formation of the toxic Aβ oligomer intermediates, but only the inhibition of secondary nucleation attenuates the maximum amount of toxic Aβ oligomers [167].
In summary, the chemical kinetics data can be used to build mathematical models of Aβp forming reactions to: (a) better understand and describe chemical processes governing peptide inhibitor-Aβ peptide products interactions; and (b) to design or modify peptide inhibitors of Aβ peptide aggregation [330,331,337,346,347,348].
3.4. Selection of Drug Delivery Route
Efficient brain delivery over the BBB is essential for peptide based AD treatment [169]. Intravenous peptide administration, in an animal model, leads to a brain peptide content of less <1% of the administered dose [349]. Therefore, intraperitoneal injection or intracerebral infusion are the preferred drug delivery routes to study the effect of anti-AD peptides in animal models [350,351,352,353,354]. For example, intraperitoneal injection of TFP5, an inhibitor Cdk5/p25 activity, attenuated tau hyperphosphorylation, neurofilament formation, Aβp accumulation and inflammation in mice brains [355]. In human, intraperitoneal injection or intracerebral infusion are not practical administration routes for long term treatment. An alternative to both, oral administration, was tested in animals with the anti-AD peptide D3; however, the peptide only elicited an improvement in cognitive behavior and a reduction in amyloid deposition with the use of very large doses (0.5–1 mg/mouse/day) [356]. Experiments with radioactively labeled exendin (9–39) imply that intranasal delivery is the only non-invasive drug delivery route, studied in an animal model, that could be used as a testing model for long term treatment in human; it has also a considerably higher peptide delivery to the brain than intravenous administration [357]. The use of polyethylenimine (PEI) conjugated peptides (e.g., V24P(10-40)-PEI), or peptides with a cell-penetrating peptide segment with multiple positively charged residues (e.g., wtNBD), further increase peptide brain delivery efficiency, to more than 17% of the intranasally administered dose [358,359].
Intranasal application of potential anti-AD peptides for human treatment (e.g., wtNBD, PACAP38 and NAP) reduced amyloidopathy in animal models by targeting diverse amyloidogenic pathways. For example: (a) wtNBD inhibits the induction of NF-κB activation, suppresses microglial activation, attenuates Aβ plaque deposition and improves cognition [358]; (b) PACAP38 increases α-secretase activity and improves cognition [360]; and (c) NAP promotes microtubule assembly, attenuates Aβ peptide accumulation and tau hyperphosphorylation [273,361]. The efficiency of intranasally delivered antiamyloidogenic peptides could be further enhanced by increasing their protease resistance [169]. The strategy to treat AD in human, with intranasal application of peptides, is also vindicated by phase 2 clinical trials on AD patients with intranasal insulin application that improved cognition [181,183,189,191,362,363,364,365].
3.5. Summary of Presented AD Modifying Peptides
A summary of AD modifying peptides, including references, is presented in Table 1.

Table 1.
AD modifying peptides with brief description. Abbreviations: act. (activity); agg. (aggregation); att. (attenuates); hyperph. (hyperphosphorylation); infl. (inflammation); NFF (neurofilament formation); pII. c.t. (phase II clinical trials); pl. form. (plaque formation); ref. (reference); stim. (stimulates); α-S (α-secretase); ● (observed effect).
3.6. Development of a Personalized Therapy for AD Patients
Several research groups have recently articulated the need for a personalized approach to the treatment of patients with AD, a multifactorial disease due to a combination of genes and environmental factors [8,164,169]. Peng et al. proposed a stage-specific strategy for a comprehensive and personalized treatment of patients with AD. This strategy includes genome editing of AD associated mutations, physical activity, brain stimulation, adequate social communication, appropriate diet, multiple drug therapy targeting amyloidogenesis and inflammation or stimulating brain blood flow and neuronal regeneration with stem cell technology [164].
4. Conclusions
AD is a progressive neurodegenerative disorder, where the rate of disease progression varies considerably from person to person due to life style, genetic and environmental factors. Chronic inflammation, vascular pathology and accumulation of toxic Aβ peptide products tend to sustain and re-inforce each other overtime, and these processes are accompanied by changes in brain structure, connectivity and function. Therefore, an effective peptide based AD therapy has to be personalized and stage specific. Chemical kinetics and aggregation prediction algorithms are essential tools for the development of peptide based AD modifying drugs, and intranasal application is the preferred delivery route for the development and treatment with anti-amyloidogenic peptides.
Acknowledgments
This work was supported by ARRS grant number P3-0171.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Pahnke, J.; Walker, L.C.; Scheffler, K.; Krohn, M. Alzheimer’s disease and blood-brain barrier function—Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci. Biobehav. Rev. 2009, 33, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future; Alzheimer’s Disease International (ADI): London, UK, 2016. [Google Scholar]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Vandal, M.; Bourassa, P.; Calon, F. Can insulin signaling pathways be targeted to transport abeta out of the brain? Front. Aging Neurosci. 2015, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein e type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic alzheimer disease: Therapeutic implications. Front. Clin. Neurosci. 2004, 135–152. [Google Scholar] [CrossRef]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for alzheimer disease in persons with type 2 diabetes and apoe epsilon4: The cardiovascular health study cognition study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M. Apolipoprotein e ε4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life alzheimer disease. Ann. Intern. Med. 2002, 137, 149. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.-X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The rotterdam study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Pellegrini, S.; Siciliano, G.; Murri, L. Causative and susceptibility genes for Alzheimer’s disease: A review. Brain Res. Bull. 2003, 61, 1–24. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B.; Whishaw, I.Q. Plasticity in the neocortex: Mechanisms underlying recovery from early brain damage. Prog. Neurobiol. 1989, 32, 235–276. [Google Scholar] [CrossRef]
- Li, N.; Daie, K.; Svoboda, K.; Druckmann, S. Robust neuronal dynamics in premotor cortex during motor planning. Nature 2016, 532, 459–464. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Retamal, C.; Cuitiño, L.; Caruano-Yzermans, A.; Shin, J.-E.; van Kerkhof, P.; Marzolo, M.-P.; Bu, G. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 2008, 283, 11501–11508. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [PubMed]
- Kinoshita, A. Demonstration by fret of bace interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, A.; Okochi, M.; Tagami, S.; Jiang, J.; Itoh, N.; Nakayama, T.; Yanagida, K.; Ishizuka-Katsura, Y.; Morihara, T.; Kamino, K.; et al. Presenilin-dependent gamma-secretase on plasma membrane and endosomes is functionally distinct†. Biochemistry 2006, 45, 4907–4914. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Cleavage of alzheimer’s amyloid Precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry 1999, 38, 9728–9734. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.W.; Harmon, A.D. Microglial activation by alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997, 388, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Dodart, J.C.; Mathis, C.; Little, S.; Clemens, J.; Paul, S.M.; Ungerer, A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12683–12688. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, G.L.; Gandy, S.E.; Buxbaum, J.D.; Ramabhadran, T.V.; Greengard, P. Protein phosphorylation regulates secretion of alzheimer beta/a4 amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1992, 89, 3055–3059. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Basun, H.; Wahlund, L.-O.; Rowe, B.A.; Wagner, S.L. Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for alzheimer’s diseas. Nat. Med. 1995, 1, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Trusko, S.P.; Howland, D.S.; Pinsker, L.R.; Mistretta, S.; Reaume, A.G.; Greenberg, B.D.; Siman, R.; Scott, R.W. Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 1998, 18, 1743–1752. [Google Scholar] [PubMed]
- Van Nostrand, W.E.; Wagner, S.L.; Shankle, W.R.; Farrow, J.S.; Dick, M.; Rozemuller, J.M.; Kuiper, M.A.; Wolters, E.C.; Zimmerman, J.; Cotman, C.W. Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live alzheimer disease patients. Proc. Natl. Acad. Sci. USA 1992, 89, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Characterization of detergent-insoluble complexes containing the familial Alzheimer’s disease-associated presenilins. J. Neurochem. 2001, 72, 1534–1543. [Google Scholar] [CrossRef]
- Parkin, E.T.; Turner, A.J.; Hooper, N.M. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem. J. 1999, 344, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.M. Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem. Soc. Trans. 2005, 33, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides abeta 42 and abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase adam 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Abramov, E.; Dolev, I.; Fogel, H.; Ciccotosto, G.D.; Ruff, E.; Slutsky, I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009, 12, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ohyagi, Y.; Miyoshi, K.; Sakae, N.; Motomura, K.; Taniwaki, T.; Furuya, H.; Takeda, K.; Tabira, T.; Kira, J. Increase in p53 protein levels by presenilin 1 gene mutations and its inhibition by secretase inhibitors. J. Alzheimer Dis. 2009, 16, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y. Intracellular amyloid beta-protein as a therapeutic target for treating Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S. The role of cerebral amyloid beta accumulation in common forms of alzheimer disease. J. Clin. Investig. 2005, 115, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Kuret, J. Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer’s disease pathology. Int. J. Alzheimers Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting nmda receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of nmda receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, W.; Yan, Z. β-amyloid impairs ampa receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase ii synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef] [PubMed]
- Tyszkiewicz, J.P. β-amyloid peptides impair pkc-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophysiol. 2005, 93, 3102–3111. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Zamponi, G.W.; Ferguson, S.S.G. Glutamate receptors function as scaffolds for the regulation of beta-amyloid and cellular prion protein signaling complexes. Mol. Brain 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A.; et al. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2004, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Poling, A.; Morgan-Paisley, K.; Panos, J.J.; Kim, E.-M.; O’Hare, E.; Cleary, J.P.; Lesné, S.; Ashe, K.H.; Porritt, M.; Baker, L.E. Oligomers of the amyloid-beta protein disrupt working memory: Confirmation with two behavioral procedures. Behav. Brain Res. 2008, 193, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the alzheimer amyloid-beta protein induce reversible synapse loss by modulating an nmda-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Galvan, V.; Lange, M.B.; Tang, H.; Sowell, R.A.; Spilman, P.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Sultana, R.; et al. In vivo oxidative stress in brain of alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of app. Free Radic. Biol. Med. 2010, 48, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.; Morris, K.L.; Sada, A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of amyloid-beta in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J. Neurosci. 2005, 25, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Devi, L. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of a beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2001, 80, 91–100. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-M.; Ou, H.-C.; Xu, H.; Chen, H.-L.; Fowler, C.; Gibson, G.E. Inhibition of α-ketoglutarate dehydrogenase complex promotes cytochromec release from mitochondria, caspase-3 activation, and necrotic cell death. J. Neurosci. Res. 2003, 74, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Inhibition of krebs cycle enzymes by hydrogen peroxide: A key role of α-ketoglutarate dehydrogenase in limiting nadh production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [PubMed]
- Li, F.; Calingasan, N.Y.; Yu, F.; Mauck, W.M.; Toidze, M.; Almeida, C.G.; Takahashi, R.H.; Carlson, G.A.; Flint Beal, M.; Lin, M.T.; et al. Increased plaque burden in brains of app mutant mnsod heterozygous knockout mice. J. Neurochem. 2004, 89, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P.; Davis, R.E.; Parker, W.D. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. Abad directly links abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Tamaoka, A.; Araki, W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J. Neurosci. Res. 2010, 88, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Baez, R.; Rojas, E.; Arias, C. Oxidative stress promotes jnk-dependent amyloidogenic processing of normally expressed human app by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem. Int. 2009, 55, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes abeta production through jnk-dependent activation of gamma-secretase. J. Biol. Chem. 2008, 283, 17721–17730. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates bace1 gene expression and abeta generation. J. Neural Transm. 2004, 112, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [PubMed]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic tg2576 mice with alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced maillard reaction end products are associated with alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.D.; Bonay, P.; Colaco, C.; Avila, J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 1994, 269, 21614–21619. [Google Scholar] [PubMed]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M. Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer’s disease. A meta-analysis. Neurosciences (Riyadh) 2012, 17, 321–326. [Google Scholar] [PubMed]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Apolipoprotein e alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. Apoe promotes the proteolytic degradation of abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein e genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme a: Cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Kovacs, D.M. Acat inhibition and amyloid beta reduction. Biochim. Biophys. Acta 2010, 1801, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Hutter-Paier, B.; Huttunen, H.J.; Puglielli, L.; Eckman, C.B.; Kim, D.Y.; Hofmeister, A.; Moir, R.D.; Domnitz, S.B.; Frosch, M.P.; Windisch, M.; et al. The acat inhibitor cp-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 2010, 68, 1014. [Google Scholar] [CrossRef]
- Shibuya, Y.; Niu, Z.; Bryleva, E.Y.; Harris, B.T.; Murphy, S.R.; Kheirollah, A.; Bowen, Z.D.; Chang, C.C.Y.; Chang, T.-Y. Acyl-coenzyme a:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human p301l-tau content at the presymptomatic stage. Neurobiol. Aging 2015, 36, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L.; Berg, J.D.; Schafer, K.; Campbell, K.; Thomas, R.G.; Weiner, M.F.; Farlow, M.R.; Sano, M.; Grundman, M.; et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s disease cooperative study. Neurology 2000, 54, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J.; et al. Effects of rofecoxib or naproxen vs placebo on alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Blalock, E.M.; Chen, K.-C.; Stromberg, A.J.; Norris, C.M.; Kadish, I.; Kraner, S.D.; Porter, N.M.; Landfield, P.W. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer’s disease: Statistical reliability and functional correlation. Res. Rev. 2005, 4, 481–512. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial activation coincides with increased bace1 activation and precedes amyloid plaque deposition in app[v717i] transgenic mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.L.; Davis, K.L.; Haroutunian, V. Large-scale microarray studies of gene expression in multiple regions of the brain in schizophrenia and Alzheimer’s disease. Int. Rev. Neurobiol. 2005, 41–82. [Google Scholar] [CrossRef]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Gordon, M.N.; Tan, J.; Wilcock, D.; Rojiani, A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: Implications for alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 2005, 64, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Reines, S.A.; Block, G.A.; Morris, J.C.; Liu, G.; Nessly, M.L.; Lines, C.R.; Norman, B.A.; Baranak, C.C.; Rofecoxib Protocol 091 Study Group. Rofecoxib: No effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 2004, 62, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.S.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, W.A.; Weinstein, H.C.; Scheltens, P.K.; Walstra, G.J. Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: An 18-month randomised, double-blind, placebo-controlled study. Lancet 2001, 358, 455–460. [Google Scholar] [CrossRef]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S. Csf and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 2013, 3, 128. [Google Scholar] [CrossRef] [PubMed]
- Capetillo-Zarate, E.; Gracia, L.; Yu, F.; Banfelder, J.R.; Lin, M.T.; Tampellini, D.; Gouras, G.K. High-resolution 3d reconstruction reveals intra-synaptic amyloid fibrils. Am. J. Pathol. 2011, 179, 2551–2558. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Bogdanovic, N.; Alafuzoff, I.; Ekman, R.; Davidsson, P. Synaptic pathology in Alzheimer’s disease: Relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the apoe4 allele. J. Neural Transm. 1996, 103, 603–618. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, S.; Parra, M.A.; Fabi, K.; Luzzi, S.; Abrahams, S. Short-term memory binding is impaired in ad but not in non-ad dementias. Neuropsychologia 2012, 50, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Levy, M.; Dickson, D.W.; Potter, H. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer’s disease brain, induces tau phosphorylation in neurons. Brain 2006, 129, 3020–3034. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chinnici, C.; Tang, H.; Trojanowski, J.Q.; Lee, V.M.; Pratico, D. Brain inflammation and oxidative stress in a transgenic mouse model of alzheimer-like brain amyloidosis. J. Neuroinflamm. 2004, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild-Bode, C.; Yamazaki, T.; Capell, A.; Leimer, U.; Steiner, H.; Ihara, Y.; Haass, C. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J. Biol. Chem. 1997, 272, 16085–16088. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.J.; Chandswangbhuvana, D.; Shu, T.; Henschen, A.; Glabe, C.G. Intracellular accumulation of insoluble, newly synthesized a n-42 in amyloid precursor protein-transfected cells that have been treated with a 1-42. J. Biol. Chem. 1999, 274, 20650–20656. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J. Mol. Neurosci. 2001, 17, 137–145. [Google Scholar] [CrossRef]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J. Astrocyte regulation of synaptic behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in ca1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.-A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. Nfκb-activated astroglial release of complement c3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the cns. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Shin, R.W.; Iwaki, T.; Kitamoto, T.; Tateishi, J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Investig. 1991, 64, 693–702. [Google Scholar] [PubMed]
- Mawal-Dewan, M.; Henley, J.; Van de Voorde, A.; Trojanowski, J.Q.; Lee, V.M. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J. Biol. Chem. 1994, 269, 30981–30987. [Google Scholar] [PubMed]
- Taniguchi, T.; Kawamata, T.; Mukai, H.; Hasegawa, H.; Isagawa, T.; Yasuda, M.; Hashimoto, T.; Terashima, A.; Nakai, M.; Mori, H.; et al. Phosphorylation of tau is regulated by pkn. J. Biol. Chem. 2001, 276, 10025–10031. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Jung, H.-J.; Kim, Y.-J.; Eggert, S.; Chung, K.C.; Choi, K.S.; Park, S.A. Age-dependent increases in tau phosphorylation in the brains of type 2 diabetic rats correlate with a reduced expression of p62. Exp. Neurol. 2013, 248, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, B.; Moskophidis, D.; Mivechi, N.F. Loss of hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol. Cell. Biol. 2010, 30, 4626–4643. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Lauretti, E.; Pratico, D. Caspase-3-dependent cleavage of akt modulates tau phosphorylation via gsk3beta kinase: Implications for Alzheimer’s disease. Mol. Psychiatry 2017, 22, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Tokutake, T.; Kasuga, K.; Yajima, R.; Sekine, Y.; Tezuka, T.; Nishizawa, M.; Ikeuchi, T. Hyperphosphorylation of tau induced by naturally secreted amyloid-beta at nanomolar concentrations is modulated by insulin-dependent akt-gsk3beta signaling pathway. J. Biol. Chem. 2012, 287, 35222–35233. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. Gsk3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ramos, A.; Díaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 2002, 71, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in m17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R.; Zhou, X.Z.; Lu, K.P.; Butterfield, D.A. Oxidative modification and down-regulation of pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol. Aging 2006, 27, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Okamura, N.; Furukawa, K.; Furumoto, S.; Harada, R.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Ishikawa, Y.; Tago, T.; et al. Longitudinal assessment of tau pathology in patients with Alzheimer’s disease using [18f]thk-5117 positron emission tomography. PLoS ONE 2015, 10, e0140311. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Marzloff, K.; Arriagada, P.V. The lack of accumulation of senile plaques or amyloid burden in Alzheimer’s disease suggests a dynamic balance between amyloid deposition and resolution. J. Neuropathol. Exp. Neurol. 1993, 52, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Carrillo, M.C.; Schenk, D.; Cacace, A.; Desanti, S.; Fremeau, R.; Bhat, R.; Glicksman, M.; May, P.; Swerdlow, R.; et al. Beyond amyloid: Getting real about nonamyloid targets in Alzheimer’s disease. Alzheimers Dement 2013, 9, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Hattori, C.; Iwata, N.; Saido, T.C.; Sasagawa, N.; Szabó, B.; Hashimoto, Y.; Maruyama, K.; Tanuma, S.; Kiso, Y.; et al. The novel beta-secretase inhibitor kmi-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice. J. Neurochem. 2006, 96, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A. Acute gamma-secretase inhibition improves contextual fear conditioning in the tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates alzheimer-disease-like pathology in the pdapp mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gravitz, L. Drugs: A tangled web of targets. Nature 2011, 475, S9–S11. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase i trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of abeta immunization (an1792) in patients with ad in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with ad after a 42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.; Cache County Study Group. Reduced risk of alzheimer disease in users of antioxidant vitamin supplements: The cache county study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Saumier, D.; Briand, R.; Laurin, J.; Gervais, F.; Tremblay, P.; Garceau, D. A phase ii study targeting amyloid-beta with 3aps in mild-to-moderate alzheimer disease. Neurology 2006, 67, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Xing, P.; Li, X.; Qian, Y.; Song, F.; Bai, Z.; Han, G.; Lei, H. Towards personalized intervention for Alzheimer’s disease. Genom. Proteom. Bioinf. 2016, 14, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Pawar, A.P.; Campioni, S.; Dobson, C.M.; Chiti, F.; Vendruscolo, M. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 2008, 380, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Trainor, K.; Broom, A.; Meiering, E.M. Exploring the relationships between protein sequence, structure and solubility. Curr. Opin. Struct. Biol. 2017, 42, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 2014, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally designed peptides and peptidomimetics as inhibitors of amyloid-beta (abeta) aggregation: Potential therapeutics of Alzheimer’s disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.P.Y. From nose to brain: The promise of peptide therapy for Alzheimer’s disease and other neurodegenerative diseases. J. Alzheimers Dis. Parkinsonism 2017, 7. [Google Scholar] [CrossRef]
- Unger, J.; Livingston, J.; Moss, A. Insulin receptors in the central nervous system: Localization, signalling mechanisms and functional aspects. Prog. Neurobiol. 1991, 36, 343–362. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Gluck, M.A.; Mercado, E.; Myers, C.E. Learning and Memory: From Brain to Behavior, 2nd ed.; Worth Publishers: New York, NY, USA, 2014; pp. xxiii, 497. [Google Scholar]
- Ito, R.; Lee, A.C.H. The role of the hippocampus in approach-avoidance conflict decision-making: Evidence from rodent and human studies. Behav. Brain Res. 2016, 313, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Moser, E.I.; Kropff, E.; Moser, M.B. Place cells, grid cells, and the brain’s spatial representation system. Annu. Rev. Neurosci. 2008, 31, 69–89. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2007, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.J.; Martinez, J.A.; Liu, W.Q.; Xu, K.; Ayer, A.; Fine, J.; Tuor, U.I.; Glazner, G.; Hanson, L.R.; Frey, W.H., 2nd; et al. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain 2008, 131, 3311–3334. [Google Scholar] [CrossRef] [PubMed]
- Haj-ali, V.; Mohaddes, G.; Babri, S.H. Intracerebroventricular insulin improves spatial learning and memory in male wistar rats. Behav. Neurosci. 2009, 123, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.; Benedict, C.; Born, J.; Hallschmid, M. Comparable sensitivity of postmenopausal and young women to the effects of intranasal insulin on food intake and working memory. J. Clin. Endocrinol. Metab. 2010, 95, E468–E472. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Frey, W.H.; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by apoe genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans: Superiority of insulin aspart. Neuropsychopharmacology 2006, 32, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 2013, 154, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.-H.; Kar, S.; Doré, S.; Quirion, R. Insulin-like growth factor-1 (igf-1): A neuroprotective trophic factor acting via the akt kinase pathway. Adv. Res. Neurodegener. 2000, 261–272. [Google Scholar] [CrossRef]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases csf abeta42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Teutenberg, K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005, 12, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., 2nd; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Bernhardt, T.; Reger, M.A.; Cholerton, B.A.; Baker, L.D.; Peskind, E.R.; Asthana, S.; Plymate, S.R.; Frolich, L.; Craft, S. Insulin effects on csf norepinephrine and cognition in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D. Insulin and Alzheimer’s disease: An amyloid connection. Neurology 2003, 60, 1886–1887. [Google Scholar] [CrossRef] [PubMed]
- Tarassishin, L.; Yin, Y.I.; Bassit, B.; Li, Y.M. Processing of notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc. Natl. Acad. Sci. USA 2004, 101, 17050–17055. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Opar, A. Mixed results for disease-modification strategies for Alzheimer’s disease. Nat. Rev. Drug Discov. 2008, 7, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Menting, K.W.; Claassen, J.A. Beta-secretase inhibitor; a promising novel therapeutic drug in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S. Gamma-secretase as a target for Alzheimer’s disease. Adv. Pharmacol. 2012, 64, 127–153. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Lessene, G.; Wilkins, S. Bace inhibitors as potential drugs for the treatment of Alzheimer’s disease: Focus on bioactivity. Recent Pat. CNS Drug Discov. 2011, 6, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Govitrapong, P. Activation of the alpha-secretase processing of abetapp as a therapeutic approach in Alzheimer’s disease. J. Alzheimers Dis. 2011, 24 (Suppl. 2), 75–94. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Salloway, S.; van Dyck, C.; Kerselaers, W.; Kaplita, S.; Curtis, C.; Ross, J.; Richter, R.W.; Andreasen, N.; Brody, M.; et al. A phase ii study of the gamma-secretase inhibitor avagacestat (bms-708163) in predementia Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2013, 9, 283. [Google Scholar] [CrossRef]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients with Mild Alzheimer Disease: A Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent bace1 inhibitor ly2886721 elicits robust central abeta pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wei, W.; Shen, Z. Drug-like chelating agents: A potential lead for Alzheimer’s disease. RSC Adv. 2014, 4, 52088–52099. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of pbt2 in targeting abeta as a modifying therapy for Alzheimer’s disease: A phase iia, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.-X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting abeta amyloid deposition and toxicity in alzheimer disease. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Wisniewski, T. Tau-based therapeutic approaches for Alzheimer’s disease—A mini-review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Ittner, A.; Ittner, L.M. Tau-targeted treatment strategies in Alzheimer’s disease. Br. J. Pharmacol. 2012, 165, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, L.P.; Perez, P.; Morales, I.; Maccioni, R.B. Novel drugs affecting tau behavior in the treatment of Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2011, 8, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy Study Evaluating TRx0237 in Subjects with Mild Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01689233 (accessed on 29 January 2018).
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.; Bhattacharya, M.; Perchiacca, J.M.; Cao, P.; Raleigh, D.P.; Abedini, A.; Schmidt, A.M.; Varkey, J.; Langen, R.; Tessier, P.M. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 19965–19970. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Schmidt, M.E.; Margolin, R.; Sperling, R.; Koeppe, R.; Mason, N.S.; Klunk, W.E.; Mathis, C.A.; Salloway, S.; Fox, N.C.; et al. Amyloid-beta 11 c-pib-pet imaging results from 2 randomized bapineuzumab phase 3 ad trials. Neurology 2015, 85, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, X.; Li, L.; Zheng, J. Inhibition of amyloid-beta aggregation in Alzheimer’s disease. Curr. Pharm. Des. 2014, 20, 1223–1243. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Soto, C. Disrupting beta-amyloid aggregation for alzheimer disease treatment. Curr. Top. Med. Chem. 2007, 7, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Su, R.; Liang, M.; Huang, R.; Wang, M.; Qi, W.; He, Z. Physicochemical strategies for inhibition of amyloid fibril formation: An overview of recent advances. Curr. Med. Chem. 2012, 19, 4157–4174. [Google Scholar] [CrossRef] [PubMed]
- Dasilva, K.A.; Shaw, J.E.; McLaurin, J. Amyloid-beta fibrillogenesis: Structural insight and therapeutic intervention. Exp. Neurol. 2010, 223, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Ayyannan, S.R.; Panda, G. Perspectives on inhibiting beta-amyloid aggregation through structure-based drug design. Chem. Med. Chem. 2015, 10, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of Protein−Protein Interactions: What are the Preferred Ways for Proteins to Interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.; Hu, L.; Falkner, J.A.; Benson, M.L.; Nerothin, J.P.; Carlson, H.A. Exploring protein–ligand recognition with binding moad. J. Mol. Graph. Model. 2006, 24, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Hambly, K.; Danzer, J.; Muskal, S.; Debe, D.A. Interrogating the druggable genome with structural informatics. Mol. Divers. 2006, 10, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Crabtree, G.R.; Graef, I.A. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004, 306, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Gardberg, A.S.; Dice, L.T.; Ou, S.; Rich, R.L.; Helmbrecht, E.; Ko, J.; Wetzel, R.; Myszka, D.G.; Patterson, P.H.; Dealwis, C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 15659–15664. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Cecal, R.; Kierstead, M.E.; Tian, X.; Phinney, A.L.; Manea, M.; French, J.E.; Lambermon, M.H.L.; Darabie, A.A.; Brown, M.E.; et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat. Med. 2002, 8, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-H.; Ke, S.-C.; Lin, T.-H.; Huang, H.-B.; Chen, Y.-C. Effect of c-terminal residues of abeta on copper binding affinity, structural conversion and aggregation. PLoS ONE 2014, 9, e90385. [Google Scholar] [CrossRef]
- Lazo, N.D.; Grant, M.A.; Condron, M.C.; Rigby, A.C.; Teplow, D.B. On the nucleation of amyloid beta-protein monomer folding. Protein Sci. 2009, 14, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state nmr. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant abeta(1–42) amyloid fibril. Proc. Nat. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the het-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Xu, S. Aggregation drives “misfolding” in protein amyloid fiber formation. Amyloid 2007, 14, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lei, M.; Yordanov, B.; Huo, S. Peptide plane can flip in two opposite directions: Implication in amyloid formation of transthyretin. J. Phys. Chem. B 2006, 110, 5829–5833. [Google Scholar] [CrossRef] [PubMed]
- Milner-White, J.E.; Watson, J.D.; Qi, G.; Hayward, S. Amyloid formation may involve α- to beta sheet interconversion via peptide plane flipping. Structure 2006, 14, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; Milner-White, E.J. The geometry of alpha-sheet: Implications for its possible function as amyloid precursor in proteins. Proteins 2008, 71, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Grillo-Bosch, D.; Carulla, N.; Cruz, M.; Sánchez, L.; Pujol-Pina, R.; Madurga, S.; Rabanal, F.; Giralt, E. Retro-enantio N-methylated peptides as beta-amyloid aggregation inhibitors. Chem. Med. Chem. 2009, 4, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Derreumaux, P. Understanding amyloid fibril nucleation and abeta oligomer/drug interactions from computer simulations. Acc. Chem. Res. 2014, 47, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Ronicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Lasagna, C.; Soto, C. Design of inhibitors of amyloid-beta misfolding and aggregation for alzheimer’s therapy. In Pharmacological Mechanisms in Alzheimer’s Therapeutics; Springer: New York, NY, USA, 2007; pp. 238–254. [Google Scholar]
- Amijee, H.; Madine, J.; Middleton, D.A.; Doig, A.J. Inhibitors of protein aggregation and toxicity. Biochem. Soc. Trans. 2009, 37, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Nerelius, C.; Sandegren, A.; Sargsyan, H.; Raunak, R.; Leijonmarck, H.; Chatterjee, U.; Fisahn, A.; Imarisio, S.; Lomas, D.A.; Crowther, D.C.; et al. Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9191–9196. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-M.; Velkova, A.; Tatarek-Nossol, M.; Rammes, G.; Sibaev, A.; Andreetto, E.; Kracklauer, M.; Bakou, M.; Malideli, E.; Göke, B.; et al. Selectively N-methylated soluble iapp mimics as potent iapp receptor agonists and nanomolar inhibitors of cytotoxic self-assembly of both iapp and abeta40. Angew. Chem. Int. Ed. 2013, 52, 10378–10383. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Crespo, R.; Börner, H.; Martins, P.M.; Rocha, F.A.; Coelho, M.; Pereira, M.C.; Rocha, S. Fluorinated beta-sheet breaker peptides. J. Mater. Chem. B 2014, 2, 2259–2264. [Google Scholar] [CrossRef]
- Francioso, A.; Punzi, P.; Boffi, A.; Lori, C.; Martire, S.; Giordano, C.; D’Erme, M.; Mosca, L. beta-sheet interfering molecules acting against beta-amyloid aggregation and fibrillogenesis. Biorgan. Med. Chem. 2015, 23, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Jagota, S.; Rajadas, J. Synthesis of d-amino acid peptides and their effect on beta-amyloid aggregation and toxicity in transgenic caenorhabditis elegans. Med. Chem. Res. 2012, 22, 3991–4000. [Google Scholar] [CrossRef]
- Chorev, M.; Goodman, M. Recent developments in retro peptides and proteins—An ongoing topochemical exploration. Trends Biotechnol. 1995, 13, 438–445. [Google Scholar] [CrossRef]
- Fletcher, M.D.; Campbell, M.M. Partially modified retro-inverso peptides: Development, synthesis, and conformational behavior. Chem. Rev. 1998, 98, 763–796. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M. Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med. Chem. 2010, 2, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Matharu, B.; El-Agnaf, O.; Razvi, A.; Austen, B.M. Development of retro-inverso peptides as anti-aggregation drugs for beta-amyloid in Alzheimer’s disease. Peptides 2010, 31, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Parthsarathy, V.; McClean, P.L.; Hölscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M.; et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the appswe/ps1Δe9 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e54769. [Google Scholar] [CrossRef]
- Taylor, M.; Moore, S.; Mayes, J.; Parkin, E.; Beeg, M.; Canovi, M.; Gobbi, M.; Mann, D.M.A.; Allsop, D. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry 2010, 49, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of d-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1-42 by mirror image phage display. ChemBioChem 2003, 4, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Araya, T.; Sasaki, D.; Taniguchi, A.; Sato, T.; Sohma, Y.; Kanai, M. Rational design and identification of a non-peptidic aggregation inhibitor of amyloid-beta based on a pharmacophore motif obtained from cyclo[-lys-leu-val-phe-phe-]. Angew. Chem. Int. Ed. Engl. 2014, 53, 8236–8239. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Abrahams, J.P. Cyclic peptides as inhibitors of amyloid fibrillation. Chemistry 2014, 20, 2410–2419. [Google Scholar] [CrossRef] [PubMed]
- Amijee, H.; Bate, C.; Williams, A.; Virdee, J.; Jeggo, R.; Spanswick, D.; Scopes, D.I.C.; Treherne, J.M.; Mazzitelli, S.; Chawner, R.; et al. The N-methylated peptide sen304 powerfully inhibits abeta(1–42) toxicity by perturbing oligomer formation. Biochemistry 2012, 51, 8338–8352. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, E.; Scopes, D.I.C.; Kim, E.-M.; Palmer, P.; Spanswick, D.; McMahon, B.; Amijee, H.; Nerou, E.; Treherne, J.M.; Jeggo, R. Novel 5-aryloxypyrimidine sen1576 as a candidate for the treatment of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2013, 17, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hopping, G.; Kellock, J.; Barnwal, R.P.; Law, P.; Bryers, J.; Varani, G.; Caughey, B.; Daggett, V. Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. Elife 2014, 3, e01681. [Google Scholar] [CrossRef] [PubMed]
- Ashur-Fabian, O.; Segal-Ruder, Y.; Skutelsky, E.; Brenneman, D.E.; Steingart, R.A.; Giladi, E.; Gozes, I. The neuroprotective peptide nap inhibits the aggregation of the beta-amyloid peptide. Peptides 2003, 24, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, A.; Barca, A.; Romano, A.; Guerrieri, S.; Storelli, C.; Rinaldi, R.; Verri, T. Anti-aggregating effect of the naturally occurring dipeptide carnosine on abeta1-42 fibril formation. PLoS ONE 2013, 8, e68159. [Google Scholar] [CrossRef] [PubMed]
- Herculano, B.; Tamura, M.; Ohba, A.; Shimatani, M.; Kutsuna, N.; Hisatsune, T. Beta-alanyl-l-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, G.; Zhang, M.; Zhao, J.; Patel, K.; Yu, X.; Zhao, C.; Ding, B.; Zhang, G.; Zhou, F.; et al. De novo design of self-assembled hexapeptides as beta-amyloid (abeta) peptide inhibitors. ACS Chem. Neurosci. 2014, 5, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Morimoto, B.H.; Tiong, J.; Fox, A.; Sutherland, K.; Dangoor, D.; Holser-Cochav, M.; Vered, K.; Newton, P.; Aisen, P.S.; et al. Nap: Research and development of a peptide derived from activity-dependent neuroprotective protein (adnp). CNS Drug Rev. 2006, 11, 353–368. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Jouroukhin, Y.; Gray, A.J.; Ma, L.; Hirata-Fukae, C.; Li, H.-F.; Feng, L.; Lecanu, L.; Walker, B.R.; Planel, E.; et al. A neuronal microtubule-interacting agent, napvsipq, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 325, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; van Groen, T.; Kadish, I.; Nayyar, G.; Cao, D.; Palgunachari, M.N.; Li, L.; Anantharamaiah, G.M. Oral apolipoprotein a-i mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yamin, G.; Ruchala, P.; Teplow, D.B. A peptide hairpin inhibitor of amyloid beta-protein oligomerization and fibrillogenesis. Biochemistry 2009, 48, 11329–11331. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- DuBay, K.F.; Pawar, A.P.; Chiti, F.; Zurdo, J.; Dobson, C.M.; Vendruscolo, M. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 2004, 341, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Taddei, N.; Baroni, F.; Capanni, C.; Stefani, M.; Ramponi, G.; Dobson, C.M. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 2002, 9, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Lindquist, S.L. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005, 435, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Tartaglia, G.G.; Pawar, A.; Christopeit, T.; Hortschansky, P.; Schroeckh, V.; Dobson, C.M.; Vendruscolo, M.; Fändrich, M. Similarities in the thermodynamics and kinetics of aggregation of disease-related abeta(1-40) peptides. Protein Sci. 2007, 16, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Sanchez de Groot, N.; Pallares, I.; Aviles, F.X.; Vendrell, J.; Ventura, S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct. Biol. 2005, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.D.; Portelius, E.; Kheterpal, I.; Guo, J.; Cook, K.D.; Xu, Y.; Wetzel, R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 2004, 335, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Chalifour, R.J.; McLaughlin, R.W.; Lavoie, L.; Morissette, C.; Tremblay, N.; Boule, M.; Sarazin, P.; Stea, D.; Lacombe, D.; Tremblay, P.; et al. Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide. J. Biol. Chem. 2003, 278, 34874–34881. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Asok Kumar, R.; Castaño, E.M.; Frangione, B. beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Tatarek-Nossol, M.; Yan, L.-M.; Schmauder, A.; Tenidis, K.; Westermark, G.; Kapurniotu, A. Inhibition of hiapp amyloid-fibril formation and apoptotic cell death by a designed hiapp amyloid- core-containing hexapeptide. Chem. Biol. 2005, 12, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of alzheimer’s amyloid-β (1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.-M.; Tycko, R. Experimental constraints on quaternary structure in alzheimer’s beta-amyloid fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Vendruscolo, M.; Tartaglia, G.G. Sequence-based prediction of protein solubility. J. Mol. Biol. 2012, 421, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Charoenkwan, P.; Kao, T.F.; Lee, H.C.; Chang, F.L.; Huang, W.L.; Ho, S.J.; Shu, L.S.; Chen, W.L.; Ho, S.Y. Prediction and analysis of protein solubility using a novel scoring card method with dipeptide composition. BMC Bioinform. 2012, 13 (Suppl. 17), S3. [Google Scholar] [CrossRef]
- Niu, X.H.; Shi, F.; Hu, X.H.; Xia, J.B.; Li, N.N. Predicting the protein solubility by integrating chaos games representation and entropy in information theory. Exp. Syst. Appl. 2014, 41, 1672–1679. [Google Scholar] [CrossRef]
- Samak, T.; Gunter, D.; Wang, Z. Prediction of protein solubility in E. coli. In Proceedings of the 2012 IEEE 8th International Conference on E-Science (e-Science), Chicago, IL, USA, 8–12 October 2012; Curran Associates: Red Hook, NY, USA, 2013; pp. 178–185. [Google Scholar]
- Smialowski, P.; Doose, G.; Torkler, P.; Kaufmann, S.; Frishman, D. Proso ii—A new method for protein solubility prediction. FEBS J. 2012, 279, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niroula, A.; Shen, B.; Vihinen, M. Pon-sol: Prediction of effects of amino acid substitutions on protein solubility. Bioinformatics 2016, 32, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. Aggrescan: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Cho, P.Y.; Yang, D.T.; Murphy, R.M. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng. Des. Sel. 2012, 25, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Siekierska, A.; Beerten, J.; Brams, M.; Van Durme, J.; De Baets, G.; Van der Kant, R.; Gallardo, R.; Ramakers, M.; Langenberg, T.; et al. Structural hot spots for the solubility of globular proteins. Nat. Commun. 2016, 7, 10816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. Foldamyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2009, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Gasior, P.; Kotulska, M. Fish amyloid—A new method for finding amyloidogenic segments in proteins based on site specific co-occurence of aminoacids. BMC Bioinform. 2014, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Noguchi, T. Espresso: A system for estimating protein expression and solubility in protein expression systems. Proteomics 2013, 13, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; de la Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Corrigendum: Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 855. [Google Scholar] [CrossRef]
- Navarro, S.; Villar-Pique, A.; Ventura, S. Selection against toxic aggregation-prone protein sequences in bacteria. BBA-Mol. Cell Res. 2014, 1843, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Robustelli, P.; Cavalli, A.; Walsh, P.; Lundstrom, P.; Zarrine-Afsar, A.; Sharpe, S.; Vendruscolo, M.; Kay, L.E. Structure of an intermediate state in protein folding and aggregation. Science 2012, 336, 362–366. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.W.; Waldispuhl, J.; Lis, M.; Halfmann, R.; Devadas, S.; Lindquist, S.; Berger, B. A method for probing the mutational landscape of amyloid structure. Bioinformatics 2011, 27, I34–I42. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Bhattacharya, M.; Tessier, P.M. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins Struct. Funct. Bioinform. 2011, 79, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Jordan, J.L.; Spatara, M.L.; Naranjo, A.; Costanzo, J.A.; Weiss, W.F.; Robinson, A.S.; Fernandez, E.J.; Roberts, C.J. Computational design and biophysical characterization of aggregation-resistant point mutations for gamma d crystallin illustrate a balance of conformational stability and intrinsic aggregation propensity. Biochemistry 2011, 50, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The camsol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Sievers, S.A.; Karanicolas, J.; Ivanova, M.I.; Baker, D.; Eisenberg, D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4074–4078. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The pasta server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Villar-Pique, A.; de Groot, N.S.; Sabaté, R.; Acebrón, S.P.; Celaya, G.; Fernàndez-Busquets, X.; Muga, A.; Ventura, S. The effect of amyloidogenic peptides on bacterial aging correlates with their intrinsic aggregation propensity. J. Mol. Biol. 2012, 421, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Seno, F.; Tosatto, S.C.E.; Trovato, A. Pasta 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.W.; O’Donnell, C.W.; Menke, M.; Cowen, L.J.; Lindquist, S.; Berger, B. Stitcher: Dynamic assembly of likely amyloid and prion beta-structures from secondary structure predictions. Proteins Struct. Funct. Bioinform. 2011, 80, 410–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Trainor, K.; Gingras, Z.; Shillingford, C.; Malakian, H.; Gosselin, M.; Lipovsek, D.; Meiering, E.M. Ensemble modeling and intracellular aggregation of an engineered immunoglobulin-like domain. J. Mol. Biol. 2016, 428, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 1994, 12, 193–198. [Google Scholar] [CrossRef]
- Chan, P.; Curtis, R.A.; Warwicker, J. Soluble expression of proteins correlates with a lack of positively-charged surface. Sci. Rep. 2013, 3, 3333. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Schaller, A.; Connors, N.K.; Oelmeier, S.A.; Hubbuch, J.; Middelberg, A.P.J. Predicting recombinant protein expression experiments using molecular dynamics simulation. Chem. Eng. Sci. 2015, 121, 340–350. [Google Scholar] [CrossRef]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. Aggrescan3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.K.; Kepp, K.P. Modeling the aggregation propensity and toxicity of amyloid-beta variants. J. Alzheimers Dis. 2015, 47, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The physical basis of how prion conformations determine strain phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated polymerization with secondary pathways. III. Equilibrium behavior and oligomer populations. J. Chem. Phys. 2011, 135, 065107. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.F.; Ferrone, F.A. Kinetics of nucleation-controlled polymerization. A perturbation treatment for use with a secondary pathway. Biophys. J. 1984, 46, 631–644. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Toyama, B.H.; Wille, H.; Colby, D.W.; Collins, S.R.; May, B.C.H.; Prusiner, S.B.; Weissman, J.; Shoichet, B.K. Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 2008, 4, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Lendel, C.; Bolognesi, B.; Wahlström, A.; Dobson, C.M.; Gräslund, A. Detergent-like interaction of congo red with the amyloid beta peptide. Biochemistry 2010, 49, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Lang, L.; Lendel, C.; Gräslund, A.; Danielsson, J. Transient small molecule interactions kinetically modulate amyloid beta peptide self-assembly. FEBS Lett. 2012, 586, 3991–3995. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Dobson, C.M.; Knowles, T.P.J.; Welland, M.E. Interactions between amyloidophilic dyes and their relevance to studies of amyloid inhibitors. Biophys. J. 2010, 99, 3492–3497. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Esbjorner, E.K.; Riss, P.J.; White, D.A.; Aigbirhio, F.I.; Toth, G.; Welland, M.E.; Dobson, C.M.; Knowles, T.P. Probing small molecule binding to amyloid fibrils. Phys. Chem. Chem. Phys. 2011, 13, 20044–20052. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro-Lago, C.; Quinlan-Pluck, F.; Lynch, I.; Lindman, S.; Minogue, A.M.; Thulin, E.; Walsh, D.M.; Dawson, K.A.; Linse, S. Inhibition of amyloid beta protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc. 2008, 130, 15437–15443. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Waudby, C.A.; Wang, S.; Buell, A.K.; Knowles, T.P.J.; Ecroyd, H.; Welland, M.E.; Carver, J.A.; Dobson, C.M.; Meehan, S. Binding of the molecular chaperone αb-crystallin to abeta amyloid fibrils inhibits fibril elongation. Biophys. J. 2011, 101, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.A.; Platt, G.W.; Hellewell, A.L.; Hewitt, E.W.; Homans, S.W.; Ashcroft, A.E.; Radford, S.E. Ligand binding to distinct states diverts aggregation of an amyloid-forming protein. Nat. Chem. Biol. 2011, 7, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Q.; Wu, S.; Buell, A.K.; Cohen, S.I.; Chen, L.J.; Hu, W.H.; Cusack, S.A.; Itzhaki, L.S.; Zhang, H.; Knowles, T.P.; et al. Influence of specific hsp70 domains on fibril formation of the yeast prion protein Ure2. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110410. [Google Scholar] [CrossRef] [PubMed]
- Cukalevski, R.; Lundqvist, M.; Oslakovic, C.; Dahlbäck, B.; Linse, S.; Cedervall, T. Structural changes in apolipoproteins bound to nanoparticles. Langmuir 2011, 27, 14360–14369. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Molecular mechanisms underlying the flavonoid-induced inhibition of α-synuclein fibrillation. Biochemistry 2009, 48, 8206–8224. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. Egcg redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.A.; Dordick, J.S.; Tessier, P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. J. Biol. Chem. 2010, 286, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Moody, L.; Olaivar, J.F.; Lewis, N.A.; Khade, R.L.; Holder, A.A.; Zhang, Y.; Rangachari, V. Inhibition of abeta42 peptide aggregation by a binuclear ruthenium(ii)−platinum(ii) complex: Potential for multimetal organometallics as anti-amyloid agents. ACS Chem. Neurosci. 2010, 1, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.I.; Yang, M.; Brender, J.R.; Subramanian, V.; Sun, K.; Joo, N.E.; Jeong, S.-H.; Ramamoorthy, A.; Kotov, N.A. Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: Functional similarities with proteins. Angew. Chem. Int. Ed. 2011, 50, 5110–5115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, L.-Q.; Perrett, S. Studying the effects of chaperones on amyloid fibril formation. Methods 2011, 53, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Marki, A.; Vass, G.; Fulop, L.; Budai, D.; Penke, B.; Falkay, G.; Szegedi, V. An intraperitoneally administered pentapeptide protects against abeta (1-42) induced neuronal excitation in vivo. J. Alzheimers Dis. 2009, 16, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. beta-sheet breaker peptide prevents abeta-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Frydman-Marom, A.; Rechter, M.; Shefler, I.; Bram, Y.; Shalev, D.E.; Gazit, E. Cognitive-performance recovery of Alzheimer’s disease model mice by modulation of early soluble amyloidal assemblies. Angew. Chem. 2009, 48, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Permanne, B.; Adessi, C.; Saborio, G.P.; Fraga, S.; Frossard, M.J.; Van Dorpe, J.; Dewachter, I.; Banks, W.A.; Van Leuven, F.; Soto, C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002, 16, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, T.; Kadish, I.; Funke, A.; Bartnik, D.; Willbold, D. Treatment with abeta42 binding d-amino acid peptides reduce amyloid deposition and inflammation in app/ps1 double transgenic mice. Adv. Protein Chem. Struct. Biol. 2012, 133–152. [Google Scholar] [CrossRef]
- Van Groen, T.; Wiesehan, K.; Funke, S.A.; Kadish, I.; Nagel-Steger, L.; Willbold, D. Reduction of Alzheimer’s disease amyloid plaque load in transgenic mice by D3, a D-enantiomeric peptide identified by mirror image phage display. Chem. Med. Chem. 2008, 3, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Zheng, Y.-L.; Mishra, S.K.; Amin, N.D.; Steiner, J.; Grant, P.; Kesavapany, S.; Pant, H.C. A truncated peptide from p35, a cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 2012, 27, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Aileen Funke, S.; van Groen, T.; Kadish, I.; Bartnik, D.; Nagel-Steger, L.; Brener, O.; Sehl, T.; Batra-Safferling, R.; Moriscot, C.; Schoehn, G.; et al. Oral treatment with thed-enantiomeric peptide D3 improves the pathology and behavior of Alzheimer’s disease transgenic mice. ACS Chem. Neurosci. 2010, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.E.; Salameh, T.S.; Banks, W.A. Intranasal delivery of proteins and peptides in the treatment of neurodegenerative diseases. AAPS J. 2015, 17, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.B.; Corbett, G.T.; Roy, A.; Modi, K.K.; Bennett, D.A.; Mufson, E.J.; Ghosh, S.; Pahan, K. Intranasal delivery of nemo-binding domain peptide prevents memory loss in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 47, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Chen, Z.T.; Liao, T.Y.; Lin, C.; Shen, H.C.; Wang, Y.H.; Chang, C.W.; Liu, R.S.; Chen, R.P.; Tu, P.H. An intranasally delivered peptide drug ameliorates cognitive decline in alzheimer transgenic mice. EMBO Mol. Med. 2017, 9, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Rat, D.; Schmitt, U.; Tippmann, F.; Dewachter, I.; Theunis, C.; Wieczerzak, E.; Postina, R.; van Leuven, F.; Fahrenholz, F.; Kojro, E. Neuropeptide pituitary adenylate cyclase-activating polypeptide (pacap) slows down Alzheimer’s disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 2011, 25, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Gray, A.J.; Hirata-Fukae, C.; Minami, S.S.; Waterhouse, E.G.; Mattson, M.P.; LaFerla, F.M.; Gozes, I.; Aisen, P.S. Intranasal nap administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J. Mol. Neurosci. 2007, 31, 165–170. [Google Scholar] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early ad. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Dhamoon, M.S.; Noble, J.M.; Craft, S. Intranasal insulin improves cognition and modulates beta-amyloid in early ad. Neurology 2009, 72, 292–293; author reply 293–294. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Craft, S.; Brooks, S.J.; Frey, W.H.; Benedict, C. Brain insulin signaling and Alzheimer’s disease: Current evidence and future directions. Mol. Neurobiol. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Rudich, A.; Harman-Boehm, I.; Cukierman-Yaffe, T. Effect of intranasal insulin on cognitive function: A systematic review. J. Clin. Endocrinol. Metab. 2012, 97, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Kapurniotu, A.; Buck, A.; Weber, M.; Schmauder, A.; Hirsch, T.; Bernhagen, J.; Tatarek-Nossol, M. Conformational restriction via cyclization in beta-amyloid peptide abeta(1-28) leads to an inhibitor of abeta(1-28) amyloidogenesis and cytotoxicity. Chem. Biol. 2003, 10, 149–159. [Google Scholar] [CrossRef]
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).