Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents





Abstract
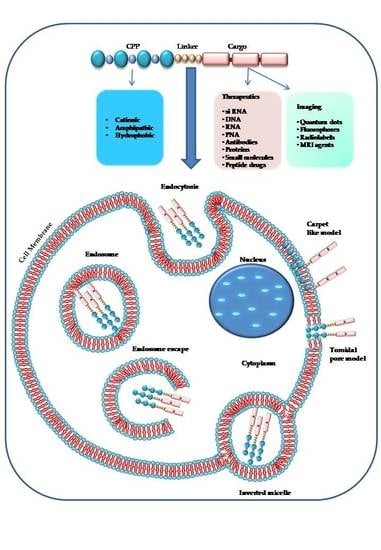
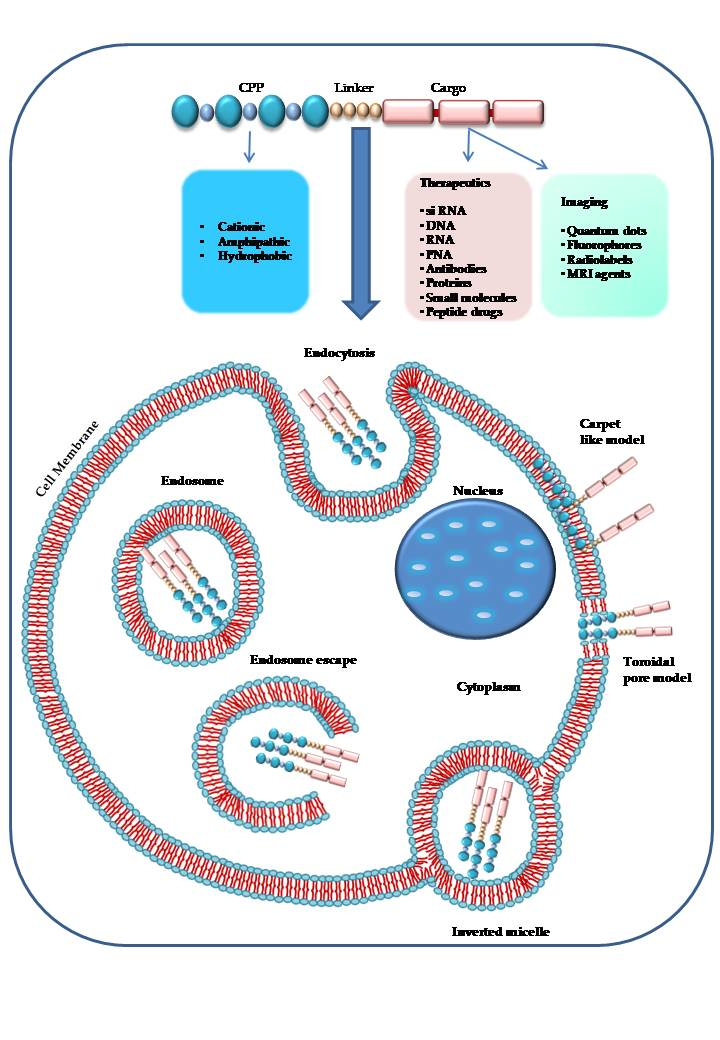
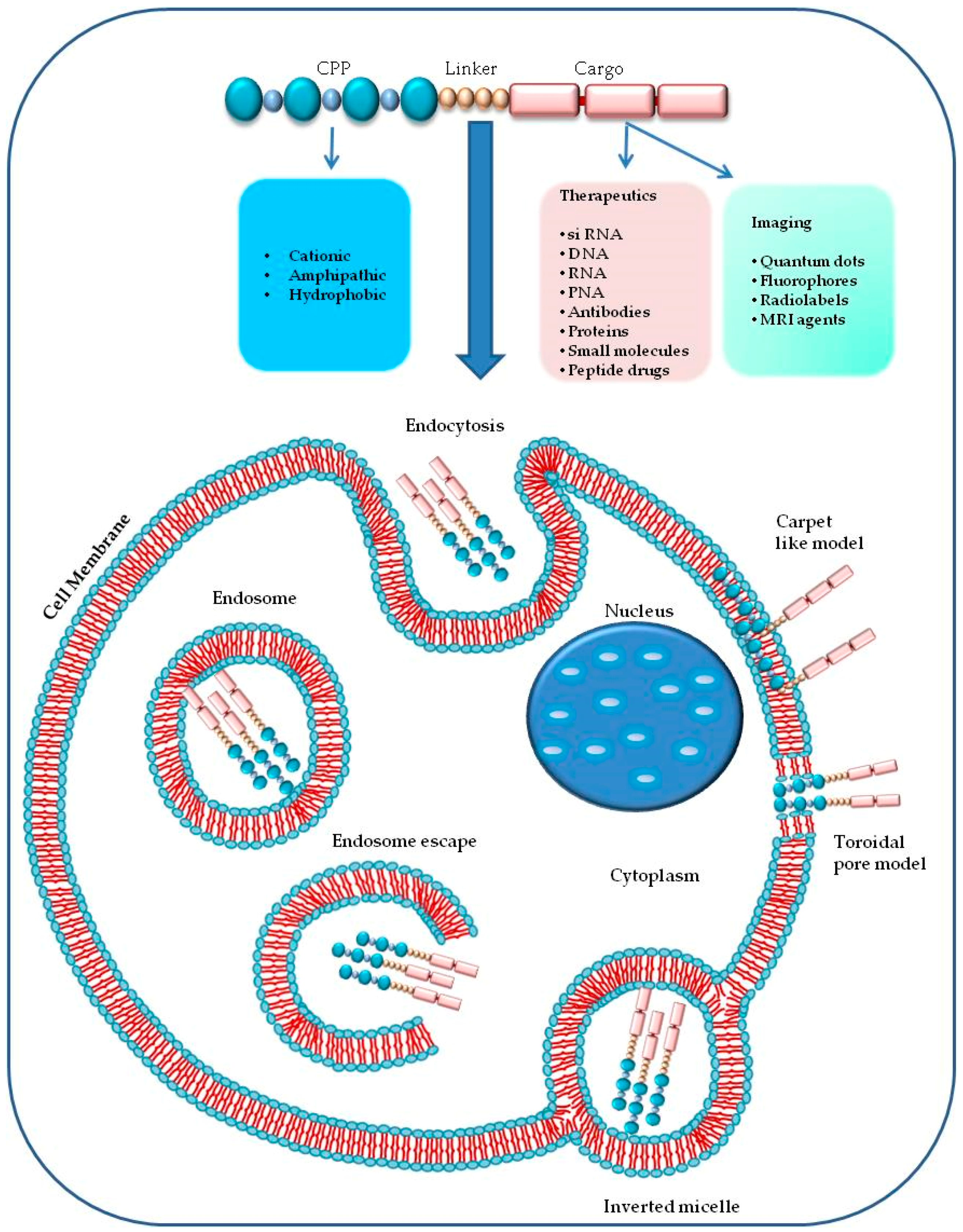
1. Introduction
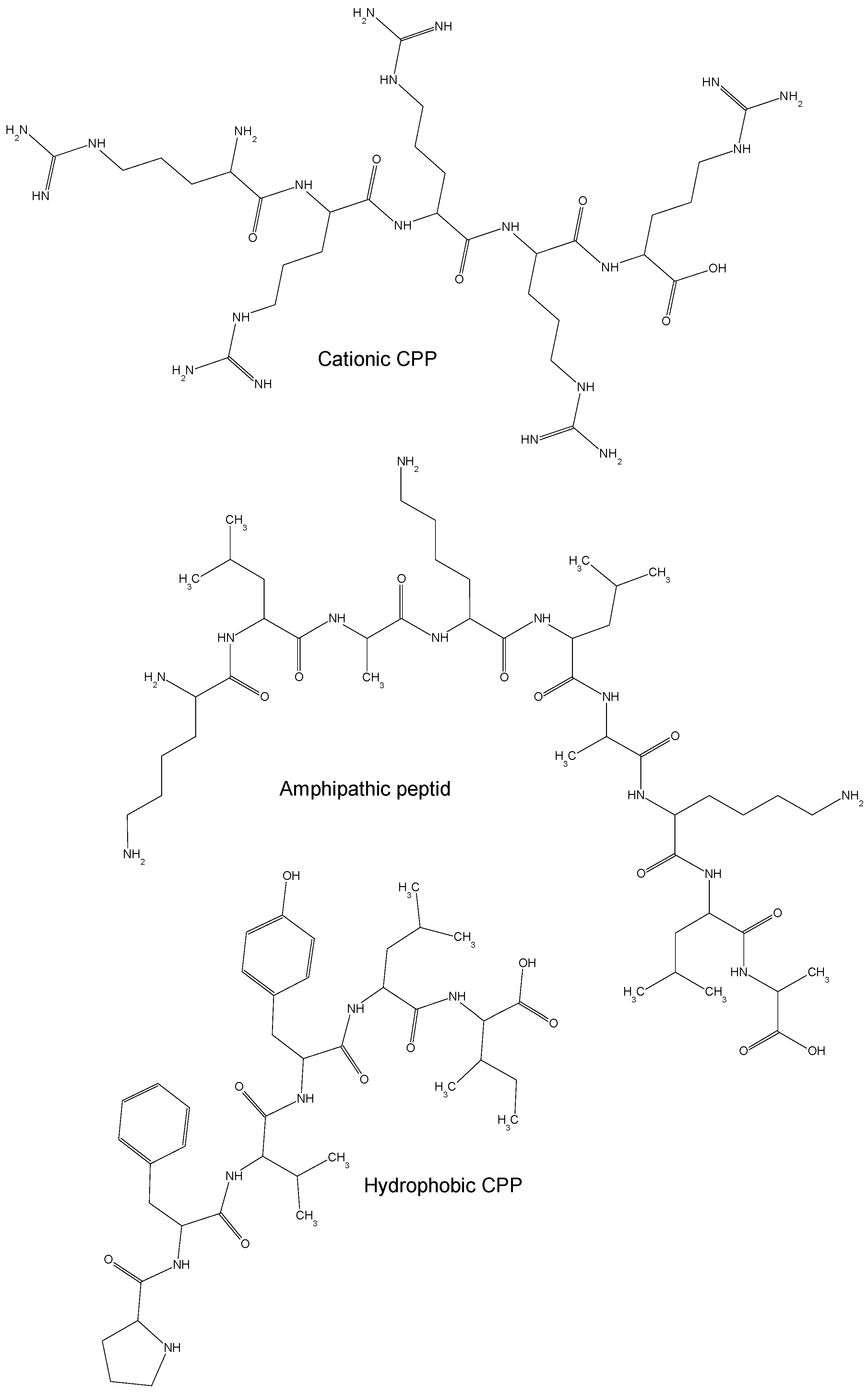
2. Chemical Properties of Cell Penetrating Peptides
Chemical Modifications of CPPs to Enhance Therapeutic Delivery
3. Mechanisms of Cell Uptake
3.1. Direct Penetration
3.2. Endocytosis
3.3. Escape from Endosomes
3.4. Chimeric and Synthetic CPPs
4. CPPs and Anti-Cancer Drug Delivery
4.1. CPPs for Delivery of Chemotherapeutic Agents
4.2. CPP and Nuclear Acids Delivery for Anti-Cancer Therapy
4.3. CPP and Protein Delivery for Anti-Cancer Therapy
4.4. “Smart” Intracellular Drug Delivery Systems for CPP-Mediated Cancer Therapy
4.5. Increasing Cell Specificity Systems for CPP-Mediated Cancer Therapy and Diagnosis
4.6. CPP and Organelle-Specific Delivery for Anti-Cancer Therapy: Mitochondrial Delivery
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Ruben, S.; Perkins, A.; Purcell, R.; Joung, K.; Sia, R.; Burghoff, R.; Haseltine, W.A.; Rosen, C.A. Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol. 1989, 63, 1–8. [Google Scholar] [PubMed]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ryu, J.; Kim, K.A.; Lee, H.J.; Bahn, J.H.; Han, K.; Choi, E.Y.; Lee, K.S.; Kwon, H.Y.; Choi, S.Y. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol. 2002, 83, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Vasconcelos, L.; Parn, K.; Langel, U. Therapeutic potential of cell-penetrating peptides. Ther. Deliv. 2013, 4, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.; Torchilin, V. Intracellular transduction using cell-penetrating peptides. Mol. Biosyst. 2010, 6, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Langel, U. Classes of Cell-Penetrating Peptides. Methods Mol. Biol. 2015, 1324, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Takechi, Y.; Tanaka, H.; Kitayama, H.; Yoshii, H.; Tanaka, M.; Saito, H. Comparative study on the interaction of cell-penetrating polycationic polymers with lipid membranes. Chem. Phys. Lipids 2012, 165, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Berkland, C.J. Polyarginine molecular weight determines transfection efficiency of calcium condensed complexes. Mol. Pharm. 2013, 10, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, H.A.; Matson, M.; Amand, H.L.; Esbjorner, E.K.; Norden, B. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry 2012, 51, 5531–5539. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Decaffmeyer, M.; Brasseur, R.; Thomas, A. Structural polymorphism of two CPP: An important parameter of activity. Biochim. Biophys. Acta 2008, 1778, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Ragin, A.D.; Morgan, R.A.; Chmielewski, J. Cellular import mediated by nuclear localization signal Peptide sequences. Chem. Biol. 2002, 9, 943–948. [Google Scholar] [CrossRef]
- Robbins, J.; Dilworth, S.M.; Laskey, R.A.; Dingwall, C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: Identification of a class of bipartite nuclear targeting sequence. Cell 1991, 64, 615–623. [Google Scholar] [CrossRef]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Eiriksdottir, E.; Konate, K.; Langel, U.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta 2010, 1798, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, C.J.; Nilsson, B.L. Self-assembly of amphipathic beta-sheet peptides: Insights and applications. Biopolymers 2012, 98, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.R.; Placone, J.; Hristova, K.; Wimley, W.C. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Davis, P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J. Biol. Chem. 2006, 281, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Mao, S.; Ditzel, H.J.; Farnaes, L.; Wirsching, P.; Lerner, R.A.; Janda, K.D. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorg. Med. Chem. 2002, 10, 4057–4065. [Google Scholar] [CrossRef]
- Dietrich, L.; Rathmer, B.; Ewan, K.; Bange, T.; Heinrichs, S.; Dale, T.C.; Schade, D.; Grossmann, T.N. Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets beta-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Palsuledesai, C.C.; Distefano, M.D. Protein prenylation: Enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Andaloussi, S.E.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.F.; Arzumanov, A.; Abes, R.; Owen, D.; Lebleu, B.; Gait, M.J. Synthesis and splice-redirecting activity of branched, arginine-rich peptide dendrimer conjugates of peptide nucleic acid oligonucleotides. Bioconj. Chem. 2010, 21, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Nasrolahi, S.A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. 2011, 50, 9633–9637. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Adjobo-Hermans, M.J.; Kohze, R.; Enderle, T.; Brock, R.; Milletti, F. Identification of Short Hydrophobic Cell-Penetrating Peptides for Cytosolic Peptide Delivery by Rational Design. Bioconj. Chem. 2017, 28, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Arukuusk, P.; Parnaste, L.; Oskolkov, N.; Copolovici, D.M.; Margus, H.; Padari, K.; Moll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim. Biophys. Acta 2013, 1828, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. 2009, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Perillo, E.; Allard-Vannier, E.; Falanga, A.; Stiuso, P.; Vitiello, M.T.; Galdiero, M.; Galdiero, S.; Chourpa, I. Quantitative and qualitative effect of gH625 on the nanoliposome-mediated delivery of mitoxantrone anticancer drug to HeLa cells. Int. J. Pharm. 2015, 488, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.H.; Niu, J.; Zhang, C.Z.; Yu, W.; Wu, J.H.; Shan, Y.H.; Wang, X.R.; Shen, Y.Q.; Mao, Z.W.; Liang, W.Q.; et al. TAT conjugated cationic noble metal nanoparticles for gene delivery to epidermal stem cells. Biomaterials 2014, 35, 5605–5618. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.P.; Pitingolo, G.; Celentano, M.; Cosentino, A.; Melone, P.; Vecchione, R.; Guarnieri, D.; Netti, P.A. Shuttle-mediated nanoparticle transport across an in vitro brain endothelium under flow conditions. Biotechnol. Bioeng. 2017, 114, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Perillo, E.; Herve-Aubert, K.; Allard-Vannier, E.; Falanga, A.; Galdiero, S.; Chourpa, I. Synthesis and in vitro evaluation of fluorescent and magnetic nanoparticles functionalized with a cell penetrating peptide for cancer theranosis. J. Colloid Interface Sci. 2017, 499, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Rosenthal-Aizman, K.; Saar, K.; Eiriksdottir, E.; Jiang, Y.; Sassian, M.; Ostlund, P.; Hallbrink, M.; Langel, U. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem. Pharmacol. 2006, 71, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Konishi, Y.; Ueda, M.; Saji, H.; Futaki, S. Accumulation of arginine-rich cell-penetrating peptides in tumors and the potential for anticancer drug delivery in vivo. J. Control. Release 2012, 159, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Komin, A.; Russell, L.M.; Hristova, K.A.; Searson, P.C. Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: Mechanisms and challenges. Adv. Drug Deliv. Rev. 2017, 110–111, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.; Shen, Q. Folic acid and cell-penetrating peptide conjugated PLGA-PEG bifunctional nanoparticles for vincristine sulfate delivery. Eur. J. Pharm. Sci. 2012, 47, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Husain, A.; Donnelly, R.J.; Barlos, D.; Riaz, S.; Ginjupalli, K.; Shodeinde, A.; Barton, B.E. Creation of a novel peptide with enhanced nuclear localization in prostate and pancreatic cancer cell lines. BMC Biotechnol. 2010, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.L.; Wang, S. Evaluation of the use of amphipathic peptide-based protein carrier for in vitro cancer research. Biochem. Biophys. Res. Commun. 2012, 419, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xi, L.; Luo, D.; Ma, X.; Yang, W.; Xi, Y.; Wang, H.; Qian, M.; Fan, L.; Xia, X.; et al. Enhanced targeted anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide TMTP1-TAT-NBD. J. Control. Release 2012, 161, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Y.; Guo, Q.; Wang, Z.; Wang, H.; Yang, Y.; Huang, Y. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials 2012, 33, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.A.; Oliveira, E.B.; Kerkis, I.; Karpel, R.L. Crotamine: A novel cell-penetrating polypeptide nanocarrier with potential anti-cancer and biotechnological applications. Methods Mol. Biol. 2012, 906, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Saito, K.; Tashiro, Y.; Kamide, K.; Uno, S.; Furuya, T.; Mashita, M.; Nakajima, K.; Tsumuraya, T.; Kobayashi, N.; et al. Tumour lineage-homing cell-penetrating peptides as anticancer molecular delivery systems. Nat. Commun. 2012, 3, 951. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Raina, D.; Kharbanda, S.; Kufe, D. Inhibition of the MUC1-C oncoprotein is synergistic with cytotoxic agents in the treatment of breast cancer cells. Cancer Biol. Ther. 2013, 14, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Braun, G.B.; Luo, X.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. Application of a proapoptotic peptide to intratumorally spreading cancer therapy. Cancer Res. 2013, 73, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Lemeshko, V.V. Electrical potentiation of the membrane permeabilization by new peptides with anticancer properties. Biochim. Biophys. Acta 2013, 1828, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Xu, X.D.; Yang, S.; Yang, J.; Zhuo, R.X.; Zhang, X.Z. Self-assembled BolA-like amphiphilic peptides as viral-mimetic gene vectors for cancer cell targeted gene delivery. Macromol. Biosci. 2013, 13, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, H.; Wan, L.; Cheng, J.; Lu, X. Penetratin-mediated delivery enhances the antitumor activity of the cationic antimicrobial peptide Magainin II. Cancer Biother. Radiopharm. 2013, 28, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Liu, J.; Ma, H.; Cheng, Q.; Huang, Y.; Zhao, J.; Huo, S.; Xue, X.; Liang, Z.; Liang, X.J. Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo. Nano Lett. 2013, 13, 2528–2534. [Google Scholar] [CrossRef] [PubMed]
- Weyland, M.; Griveau, A.; Bejaud, J.; Benoit, J.P.; Coursaget, P.; Garcion, E. Lipid nanocapsule functionalization by lipopeptides derived from human papillomavirus type-16 capsid for nucleic acid delivery into cancer cells. Int. J. Pharm. 2013, 454, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Dong, D.W.; Shi, N.Q.; Gao, W.; Yang, Z.Z.; Cui, Y.; Cao, D.Y.; Qi, X.R. PSA-responsive and PSMA-mediated multifunctional liposomes for targeted therapy of prostate cancer. Biomaterials 2013, 34, 6976–6991. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.H.; Yung, L.Y. Synergistic co-delivery of doxorubicin and paclitaxel using multi-functional micelles for cancer treatment. Int. J. Pharm. 2013, 454, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamazaki, D.; Yamauchi, J.; Harashima, H. The nanoparticulation by octaarginine-modified liposome improves alpha-galactosylceramide-mediated antitumor therapy via systemic administration. J. Control. Release 2013, 171, 216–224. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, C.B.; Russo, L.C.; Castro, L.M.; Forti, F.L.; do Monte, E.R.; Rioli, V.; Gozzo, F.C.; Colquhoun, A.; Ferro, E.S. A novel intracellular peptide derived from g1/s cyclin d2 induces cell death. J. Biol. Chem. 2014, 289, 16711–16726. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Raucher, D. Anti-tumor efficacy of a therapeutic peptide based on thermo-responsive elastin-like polypeptide in combination with gemcitabine. Cancer Lett. 2014, 348, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Tints, K.; Prink, M.; Neuman, T.; Palm, K. LXXLL peptide converts transportan 10 to a potent inducer of apoptosis in breast cancer cells. Int. J. Mol. Sci. 2014, 15, 5680–5698. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Raucher, D. Elastin-like polypeptides: The influence of its molecular weight on local hyperthermia-induced tumor accumulation. Eur. J. Pharm. Biopharm. 2014, 88, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Wang, H.; Gong, J.; He, H.; Shin, M.C.; Yang, V.C.; Huang, Y. Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. J. Control. Release 2014, 192, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhang, J.; Ah, M.K.; Lee, K.; Moon, C.; Balthasar, J.P.; Yang, V.C. Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. J. Control. Release 2014, 194, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Orzechowska, E.J.; Kozlowska, E.; Czubaty, A.; Kozlowski, P.; Staron, K.; Trzcinska-Danielewicz, J. Controlled delivery of BID protein fused with TAT peptide sensitizes cancer cells to apoptosis. BMC Cancer 2014, 14, 771. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Shi, K.; Hu, G.; Yang, Y.; Kuang, Q.; Lu, L.; Zhang, L.; Chen, W.; Dong, M.; Chen, Y.; et al. Tumor-targeted paclitaxel delivery and enhanced penetration using TAT-decorated liposomes comprising redox-responsive poly(ethylene glycol). J. Pharm. Sci. 2015, 104, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Qiu, Q.; Yang, B.; Wang, X.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of novel peptides with anti-cancer and drug resistance-reversing activities. Eur. J. Med. Chem. 2015, 89, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Ruczynski, J.; Siedlecka-Kroplewska, K.; Kaszubowska, L.; Rybarczyk, A.; Alenowicz, M.; Rekowski, P.; Kmiec, Z. Protein and siRNA delivery by transportan and transportan 10 into colorectal cancer cell lines. Folia Histochem. Cytobiol. 2014, 52, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Higa, M.; Katagiri, C.; Shimizu-Okabe, C.; Tsumuraya, T.; Sunagawa, M.; Nakamura, M.; Ishiuchi, S.; Takayama, C.; Kondo, E.; Matsushita, M. Identification of a novel cell-penetrating peptide targeting human glioblastoma cell lines as a cancer-homing transporter. Biochem. Biophys. Res. Commun. 2015, 457, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. p28, an anionic cell-penetrating peptide, increases the activity of wild type and mutated p53 without altering its conformation. Mol. Pharm. 2013, 10, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.H.; Cole, G.; et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J. Control. Release 2014, 189, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Kim, K.; Lim, D.; Jeong, K.; Hong, Y.; Nguyen, V.H.; Kim, T.H.; Ryu, S.; Lim, J.A.; Kim, J.I.; et al. Anti-tumoral effect of the mitochondrial target domain of Noxa delivered by an engineered Salmonella typhimurium. PLoS ONE 2014, 9, e80050. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.R.; Lin, M.D.; Chiang, H.J.; Lee, H.J. Arginine-rich cell-penetrating peptides deliver gene into living human cells. Gene 2012, 505, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Veiman, K.L.; Mager, I.; Ezzat, K.; Margus, H.; Lehto, T.; Langel, K.; Kurrikoff, K.; Arukuusk, P.; Suhorutsenko, J.; Padari, K.; et al. PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol. Pharm. 2013, 10, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Mevel, M.; Haudebourg, T.; Colombani, T.; Peuziat, P.; Dallet, L.; Chatin, B.; Lambert, O.; Berchel, M.; Montier, T.; Jaffres, P.A.; et al. Important role of phosphoramido linkage in imidazole-based dioleyl helper lipids for liposome stability and primary cell transfection. J. Gene Med. 2016, 18, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, P.; Cheetham, A.G.; Moon, J.H.; Moxley, J.W., Jr.; Lin, Y.A.; Cui, H. Controlled release of free doxorubicin from peptide-drug conjugates by drug loading. J. Control. Release 2014, 191, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Ma, Y.; Zhang, W.; Li, L.; Zhang, Y.; Zhang, L.; Liu, H.; Ni, J.; Wang, R. Design of new acid-activated cell-penetrating peptides for tumor drug delivery. PeerJ 2017, 5, e3429. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Hsu, C.Y.; Tsai, C.F.; Chiu, C.C.; Liang, S.S.; Wang, T.N.; Kuo, P.L.; Long, C.Y.; Tsai, E.M. A novel cell-penetrating peptide suppresses breast tumorigenesis by inhibiting beta-catenin/LEF-1 signaling. Sci. Rep. 2016, 6, 19156. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Kim, Y.H.; Hwangbo, S.; Cho, H. Photodynamic therapy by conjugation of cell-penetrating peptide with fluorochrome. Int. J. Nanomed. 2017, 12, 8185–8196. [Google Scholar] [CrossRef] [PubMed]
- Favretto, M.E.; Wallbrecher, R.; Schmidt, S.; van de Putte, R.; Brock, R. Glycosaminoglycans in the cellular uptake of drug delivery vectors—Bystanders or active players? J. Control. Release 2014, 180, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Simon Davis, D.A.; Parish, C.R. Heparan sulfate: A ubiquitous glycosaminoglycan with multiple roles in immunity. Front. Immunol. 2013, 4, 470. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Eitzen, G. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 2003, 1641, 175–181. [Google Scholar] [CrossRef]
- Gerbal-Chaloin, S.; Gondeau, C.; Aldrian-Herrada, G.; Heitz, F.; Gauthier-Rouviere, C.; Divita, G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol. Cell 2007, 99, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.F.; Pokorny, A. Mechanisms of antimicrobial, cytolytic, and cell-penetrating peptides: From kinetics to thermodynamics. Biochemistry 2009, 48, 8083–8093. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1998, 1376, 339–352. [Google Scholar] [CrossRef]
- Magzoub, M.; Graslund, A. Cell-penetrating peptides: [corrected] from inception to application. Q. Rev. Biophys. 2004, 37, 147–195. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Plenat, T.; Aldrian-Herrada, G.; Divita, G.; Le, G.C.; Heitz, F. Primary amphipathic cell-penetrating peptides: Structural requirements and interactions with model membranes. Biochemistry 2004, 43, 7698–7706. [Google Scholar] [CrossRef] [PubMed]
- Esbjorner, E.K.; Graslung, A.; Norden, P. Membrane interactions of cell penetrating peptide. In Cell-Penetrating Peptides; CRC Press: Boca Raton, FL, USA, 2007; pp. 109–138. [Google Scholar]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Futaki, S.; Niwa, M.; Tanaka, S.; Ueda, K.; Sugiura, Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 2002, 277, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Nielsen, H.M. Cell-penetrating peptides as tools to enhance non-injectable delivery of biopharmaceuticals. Tissue Barriers 2016, 4, e1178369. [Google Scholar] [CrossRef] [PubMed]
- Verdurmen, W.P.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hallbrink, M.; van Kuppevelt, T.H.; Brock, R. Preferential uptake of l- versus d-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Jarver, P.; Mager, I.; Langel, U. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol. Sci. 2010, 31, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Trehin, R.; Krauss, U.; Beck-Sickinger, A.G.; Merkle, H.P.; Nielsen, H.M. Cellular uptake but low permeation of human calcitonin-derived cell penetrating peptides and Tat(47–57) through well-differentiated epithelial models. Pharm. Res. 2004, 21, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Foerg, C.; Ziegler, U.; Fernandez-Carneado, J.; Giralt, E.; Merkle, H.P. Differentiation restricted endocytosis of cell penetrating peptides in MDCK cells corresponds with activities of Rho-GTPases. Pharm. Res. 2007, 24, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, E.; Yu, Z.; Pei, X.; Chen, S.; Zhang, P.; Shin, M.C.; Gong, J.; He, H.; Yang, V.C. CPP-Assisted Intracellular Drug Delivery, What Is Next? Int. J. Mol. Sci. 2016, 17, 1892. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Rinaldi, A.; Rufo, A.; Kinnunen, P.; Di Giulio, A. Structural and Change Requirements for Antimicrobial Peptide Insertion into Biological and Model Membranes. In Pore-Forming Peptides and Protein Toxins; Harwood Academic Publishers: Reading, UK, 2003; pp. 151–177. [Google Scholar]
- Lopez-Meza, J.E.; Ochoa-Zarzosa, A.; Barboza-Corona, J.E.; Bideshi, D.K. Antimicrobial peptides: Current and potential applications in biomedical therapies. Biomed. Res. Int. 2015, 2015, 367243. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Vives, E.; Richard, J.P.; Rispal, C.; Lebleu, B. TAT peptide internalization: Seeking the mechanism of entry. Curr. Protein Pept. Sci. 2003, 4, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppe, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Malerod, L.; Berg, T.; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J. 2004, 377, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Heuser, J. Three-dimensional visualization of coated vesicle formation in fibroblasts. J. Cell Biol. 1980, 84, 560–583. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Helenius, A. Endocytosis via caveolae. Traffic 2002, 3, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Medina-Kauwe, L.K. “Alternative” endocytic mechanisms exploited by pathogens: New avenues for therapeutic delivery? Adv. Drug Deliv. Rev. 2007, 59, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Hitz, T.; Iten, R.; Gardiner, J.; Namoto, K.; Walde, P.; Seebach, D. Interaction of alpha-and beta-oligoarginine-acids and amides with anionic lipid vesicles: A mechanistic and thermodynamic study. Biochemistry 2006, 45, 5817–5829. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Dinca, A.; Chien, W.M.; Chin, M.T. Intracellular Delivery of Proteins with Cell-Penetrating Peptides for Therapeutic Uses in Human Disease. Int. J. Mol. Sci. 2016, 17, 263. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Masuda, T.; Nishio, T.; Niikura, K.; Ijiro, K.; Harashima, H. Improving in vivo hepatic transfection activity by controlling intracellular trafficking: The function of GALA and maltotriose. Mol. Pharm. 2011, 8, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Akita, H.; Takayama, K.; Kobayashi, S.; Futaki, S.; Harashima, H. Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials 2011, 32, 5733–5742. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Cardarelli, F.; Di, L.M.; Boccardi, C.; Nifosi, R.; Bardi, G.; Di, B.L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.; Lehto, T.; Mager, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, S.; Munoz-Alarcon, A.; Helmfors, H.; Mosqueira, D.; Gyllborg, D.; Tudoran, O.; Langel, U. PepFect15, a novel endosomolytic cell-penetrating peptide for oligonucleotide delivery via scavenger receptors. Int. J. Pharm. 2013, 441, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dubikovskaya, E.A.; Thorne, S.H.; Pillow, T.H.; Contag, C.H.; Wender, P.A. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc. Natl. Acad. Sci. USA 2008, 105, 12128–12133. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; De, W.M.; Breard, J.; Kenani, A. Efficient induction of apoptosis by doxorubicin coupled to cell-penetrating peptides compared to unconjugated doxorubicin in the human breast cancer cell line MDA-MB 231. Cancer Lett. 2009, 285, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Dawidczyk, C.M.; Russell, L.M.; Searson, P.C. Recommendations for Benchmarking Preclinical Studies of Nanomedicines. Cancer Res. 2015, 75, 4016–4020. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; Waard, M.D.; Kenani, A. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: A comparative study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liu, J.; He, Q.; Wang, L.; Shi, J. Overcoming multidrug resistance of cancer cells by direct intranuclear drug delivery using TAT-conjugated mesoporous silica nanoparticles. Biomaterials 2013, 34, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; Perkins, E.; Kratz, F.; Raucher, D. Cell penetrating peptides fused to a thermally targeted biopolymer drug carrier improve the delivery and antitumor efficacy of an acid-sensitive doxorubicin derivative. Int. J. Pharm. 2012, 436, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Nasrolahi, S.A.; Mandal, D.; Tiwari, R.K.; Guo, L.; Lu, W.; Parang, K. Cyclic peptide-capped gold nanoparticles as drug delivery systems. Mol. Pharm. 2013, 10, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kanazawa, T.; Shibata, Y.; Suda, Y.; Fukuda, T.; Takashima, Y.; Okada, H. Development of cell-penetrating peptide-modified MPEG-PCL diblock copolymeric nanoparticles for systemic gene delivery. Int. J. Pharm. 2010, 396, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Li, J.F.; Liou, J.S.; Charng, Y.C.; Huang, Y.W.; Lee, H.J. A gene delivery system for human cells mediated by both a cell-penetrating peptide and a piggyBac transposase. Biomaterials 2011, 32, 6264–6276. [Google Scholar] [CrossRef] [PubMed]
- Rajpal; Mann, A.; Khanduri, R.; Naik, R.J.; Ganguli, M. Structural rearrangements and chemical modifications in known cell penetrating peptide strongly enhance DNA delivery efficiency. J. Control. Release 2012, 157, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003, 31, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; Sivakumar, J.; Garimidi, P.; Rangaraj, N.; Kumar, J.M.; Rao, N.M.; Gopal, V. Targeting human epidermal growth factor receptor 2 by a cell-penetrating peptide-affibody bioconjugate. Biomaterials 2012, 33, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, E.J. Assembly and function of RNA silencing complexes. Nat. Rev. Mol. Cell Biol. 2005, 6, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeong, E.J.; Lee, J.; Rhim, T.; Lee, S.K.; Lee, K.Y. Preparation and characterization of nonaarginine-modified chitosan nanoparticles for siRNA delivery. Carbohydr. Polym. 2013, 92, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Baoum, A.; Ovcharenko, D.; Berkland, C. Calcium condensed cell penetrating peptide complexes offer highly efficient, low toxicity gene silencing. Int. J. Pharm. 2012, 427, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Jiang, L.; Zhang, M.; Ren, F.Z. A novel cell-penetrating peptide TAT-A1 delivers siRNA into tumor cells selectively. Biochimie 2013, 95, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Van Asbeck, A.H.; Beyerle, A.; McNeill, H.; Bovee-Geurts, P.H.; Lindberg, S.; Verdurmen, W.P.; Hallbrink, M.; Langel, U.; Heidenreich, O.; Brock, R. Molecular parameters of siRNA—Cell penetrating peptide nanocomplexes for efficient cellular delivery. ACS Nano 2013, 7, 3797–3807. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc, M.G.; Coll, J.L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotechnol. 2009, 27, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Michiue, H.; Eguchi, A.; Scadeng, M.; Dowdy, S.F. Induction of in vivo synthetic lethal RNAi responses to treat glioblastoma. Cancer Biol. Ther. 2009, 8, 2306–2313. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Yang, X.Z.; Sun, C.Y.; Mao, C.Q.; Zhu, Y.H.; Wang, J. Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials 2014, 35, 7622–7634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.T.; Kang, T.H.; Ma, B.; Xu, Y.; Hung, C.F.; Wu, T.C. LAH4 enhances CD8+ T cell immunity of protein/peptide-based vaccines. Vaccine 2012, 30, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mehta, R.R.; Lekmine, F.; Christov, K.; King, M.L.; Majumdar, D.; Shilkaitis, A.; Green, A.; Bratescu, L.; Beattie, C.W.; et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol. Cancer Ther. 2009, 8, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L.; et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae, B.L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.M.; Hurley, J.P.; Salmaso, S.; Kale, A.; Tolcheva, E.; Levchenko, T.S.; Torchilin, V.P. “SMART” drug delivery systems: Double-targeted pH-responsive pharmaceutical nanocarriers. Bioconj. Chem. 2006, 17, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Yuan, W.; Li, W.; Qin, Y.; Tang, J.; Yuan, M.; Fu, L.; Ran, R.; Zhang, Z.; He, Q. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol. Pharm. 2011, 8, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shin, M.C.; David, A.E.; Zhou, J.; Lee, K.; He, H.; Yang, V.C. Long-circulating heparin-functionalized magnetic nanoparticles for potential application as a protein drug delivery platform. Mol. Pharm. 2013, 10, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; David, A.E.; Yang, V.C. Magnetic tumor targeting of beta-glucosidase immobilized iron oxide nanoparticles. Nanotechnology 2013, 24, 375102. [Google Scholar] [CrossRef] [PubMed]
- He, H.; David, A.; Chertok, B.; Cole, A.; Lee, K.; Zhang, J.; Wang, J.; Huang, Y.; Yang, V.C. Magnetic nanoparticles for tumor imaging and therapy: A so-called theranostic system. Pharm. Res. 2013, 30, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiong, M.; Gong, J.; Zhang, Y.; Bai, N.; Luo, Y.; Li, L.; Wei, Y.; Liu, Y.; Tan, X.; et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat. Commun. 2014, 5, 4280. [Google Scholar] [CrossRef] [PubMed]
- Savariar, E.N.; Felsen, C.N.; Nashi, N.; Jiang, T.; Ellies, L.G.; Steinbach, P.; Tsien, R.Y.; Nguyen, Q.T. Real-time in vivo molecular detection of primary tumors and metastases with ratiometric activatable cell-penetrating peptides. Cancer Res. 2013, 73, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Lan, K.H.; Yao, J.; Lu, C.H.; Sun, M.; Neal, C.L.; Lu, J.; Yu, D. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res. 2006, 66, 3764–3772. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Olson, E.S.; Aguilera, T.A.; Jiang, T.; Scadeng, M.; Ellies, L.G.; Tsien, R.Y. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc. Natl. Acad. Sci. USA 2010, 107, 4317–4322. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Torchilin, V.P. Nanopreparations for organelle-specific delivery in cancer. Adv. Drug Deliv. Rev. 2014, 66, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, A.; Schiattarella, A.; Bonelli, P.; Tuccillo, F.M.; Buonaguro, F.M.; Mancini, A. The functional role of MnSOD as a biomarker of human diseases and therapeutic potential of a new isoform of a human recombinant MnSOD. Biomed. Res. Int. 2014, 2014, 476789. [Google Scholar] [CrossRef] [PubMed]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, U. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; McCormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M.; Prabhakar, B.; Ganesh, B.; et al. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother. Pharmacol. 2011, 68, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.F.; Figg, W.D.; Sparreboom, A. Pharmacogenetics of irinotecan metabolism and transport: An update. Toxicol. In Vitro 2006, 20, 163–175. [Google Scholar] [CrossRef] [PubMed]
- De Coupade, C.; Fittipaldi, A.; Chagnas, V.; Michel, M.; Carlier, S.; Tasciotti, E.; Darmon, A.; Ravel, D.; Kearsey, J.; Giacca, M.; et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem. J. 2005, 390, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Losic, F.; Nicolazzi, C.; Quinonero, J.; Ribes, F.; Michel, M.; Dubois, V.; De, C.C.; Boukaissi, M.; Chene, A.S.; Tranchant, I.; et al. DTS-108, a novel peptidic prodrug of SN38: In vivo efficacy and toxicokinetic studies. Clin. Cancer Res. 2008, 14, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Faivre, S.J.; Mir, O.; Dreyer, C.; Ropert, S.; Bouattour, M.; Desjardins, R.; Goldwasser, F.; Raymond, E. Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: A Phase I study. Int. J. Nanomed. 2016, 11, 6207–6216. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Walton, J.G.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Name | Peptide Sequence * | Activity | Cells/Tumors | Cargo | Ref. |
|---|---|---|---|---|---|
| YTA2 YTA4 | YTAIAWVKAFIRKLRK IAWVKAFIRKLRKGPLG | CPP conjugated to the methotrexate (MTX) as therapeutic for drug resistant tumor cells. | Breast cancer cells MDA-MB-231 | MTX | [47] |
| TAT Pen R12 R16 r8 r12 R5 R7 | GRKKRRQRRRPPQC RQIKIWFQNRRMKWKKGC RRRRRRRRRRRRGC RRRRRRRRRRRRRRRRGC RrrrrrrrGC rrrrrrrrrrrrGC Fl-ahx-RRRRR RRRRRRR | Accumulation of oligoarginine peptides in tumor-xenografted mice. | Nude mice implanted with HeLa and CHO-K1 cells | Doxorubicin Paclitaxel | [40,48,49,50] |
| Glu-Oct-6 Glu-Lys 6-Oct Phe-Oct-6 Asn-Oct-6 Tyr-Oct-6 | EEEAAGRKRKKRT EEEAAKKK GRKRKKRT FFFAAGRKRKKRT NNNAAGRKRKKRT YYYAAGRKRKKRT | CPP with enhanced nuclear localization in prostate cancer cells. | Prostate cancer cells DU-145 and LNCaP | Nucleic acid | [51] |
| RV24 | RRRRRRRRRGPGVTWTPQAWFQWV | Amphipathic peptide-carrier for targeting cancer cells. | T98G, HepG2 and HeLa cells | β-galactosidase and eGFP | [52] |
| TAT-NBD TMTP1-TAT-NBD | YGRKKRRQRRRGTALDWSWLQTE CGNVVRQGC-G-YGRK-KRRQRRR-G-TALDWSWLQTE | Anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide. | BALB/c nu/nu mice; PC-3M-1E8, MDA-MB-231, MCF-7 and PC-3M-2B4 cells | Tumor molecular targeted peptide 1 (TMTP1) | [53] |
| AgNP-TAT | CGGGYGRKKRRQRRR | TAT-modified nanosilver for multidrug-resistant cancer. | Nude mice implanted with B16 melanoma cells; Caco-2 cells | Nanosilver Nanoparticles | [54] |
| Crotamine | YKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSG | Crotamine as carrier for anti-cancer molecules. | B16-F10, HCT116, 3T3 cells; C57BL6 or nude mice | Nucleic acid | [55] |
| 1 (TAT) 2 7 10 28 30 33 44 45 47 48 | YGRKKRPQRRR DSLKSYWYLQKFSWR KLWMRWWSPTTRRYG RLWMRWYSPWTRRWG RLIMRIYAPTTRRYG RLYMRYYSPTTRRYG RLWMRWYSPRTRAYG KRPTMRFRYTWNPMK WKCRRQCFRVLHHWN WKCRRQAFRVLHHWN WKARRQCFRVLHHWN | -TAT derived CPP as anticancer molecular delivery systems. -Co-delivery of doxorubicin and paclitaxel using multi-functional micelles. | NOD-SCID mice model of xenograft human tumor cells; Many Human neoplastic cells including HeLa, Lovo, A549, MCF-7,MKN45,HepG2, LNCap, KPK,U2OS, RC15,RDES,H28,K562,U251,NHDF KB cells | Doxorubicin and Paclitaxel | [56] |
| R9-GO-203 | rrrrrrrrrcqcrrkn | Inhibition of the MUC1-C oncoprotein and delivery of cytotoxic agents in breast cancer cell lines. | MCF-7 and ZR-75-1 cell lines | Taxol and Doxorubicin | [57] |
| iRGD-CDD | CRGDKGPDC | Proapoptotic peptide to intratumorally spreading cancer therapy. | Athymic nude mice and Balb/C mice; Human embryonic kidney (HEK) 293T, prostate cancer PPC1, mouse breast cancer 4T1, Human tumor cell line M21, Human breast cancer cell line MCF-10CA1a. | Bit1 (a pro-apoptotic mitochondrial protein) | [58] |
| P7-4 P7-5 P7-6 P7-7 R7-KLA KLA-R7 | RRRRRRRGGIYLATALAKWALKQGF IYLATALAKWALKQGFGGRRRRRRR RRRRRRRGGIYLATALAKWALKQ IYLATALAKWALKQGGRRRRRRR RRRRRRRGGKLAKLAKKLAKLAK KLAKLAKKLAKLAKGGRRRRRRR | Membrane permeabilization by peptides with anticancer properties. | Male white rats; Jurkat and CHO | Peptide P7–27 | [59] |
| P1 P2 P3 P4 | RGD-ADDA-RRRRRRRR RGD-Ahx-RRRRRRRR RGD-RRRRRRRR RRRRRRRR | Self-assembled BolA-like amphiphilic peptides as viral-mimetic gene vectors. | 293T and HeLa | Plasmid DNA | [60] |
| MG2A | GIGKFLHSAKKFGKAFVGEIMNSGGKKWKMRRNQFWVKVQRG | Penetratin-mediated delivery for antitumor activity of the cationic antimicrobial peptide Magainin II. | HeLa and A549 cells | Antimicrobial peptide MG2A | [61] |
| CRGDK | CRGDK | Functionalized micelles for delivery of anticancer drugs. | Breast MDA-MB-231 and prostatic PC3 cancer cell lines | Doxorubicin | [62] |
| L1 | CTSTTAKRKKRKLK | Lipopeptides derived from human papillomavirus type-16 capsid for gene delivery. | Malignant human glioma cells U87MG and COS-7 cells | Plasmid DNA and siRNA | [63] |
| oligoarginine | rrrrrrrr | Multifunctional liposomes for targeted therapy of prostate cancer. | 22Rv1 xenograft murine model; PC-3 cells | Folate | [64] |
| GC/R8-Lip | RRRRRRRR | Octaarginine-modified liposome as carriers of alpha-galactosylceramide. | C57BL/6 (H-2b) female mice; JAWSII cells | GC (-galactosylceramide), ovalbumin | [65] |
| p21-ELP1-Bac Bac-ELP43 Bac-ELP63 Bac-ELP122 | RRIRPRPPRLPRPRPRPLPFPRPG | Therapeutic peptide based on thermo-responsive elastin-like polypeptide. | Female athymic nude mice (Ncr-nu/nu); S2013, Mia PaCa-2 and Panc-1. | p21 peptide | [66,67] |
| TP10-SRC1LXXLL R7-SRC1LXXLL TP10-SRC1(1222–1245) R7-SRC1(1222–1245) | PKKKRKV-AGYLLGKINLKALAALAKKIL- PQMQQNVFQYPGAGMVPQGEANF PKKKRKV-RRRRRRR-YSQTSHKLVQLLTTAEQQ PKKKRKV-AGYLLGKINLKALAALAKKIL- PQMQQNVFQYPGAGMVPQGEANF PKKKRKV-RRRRRRR-PQMQQNVFQYPGAGMVPQGEANF | LXXLL peptide to convert transportan 10 to a potent inducer of apoptosis in breast cancer cells. | MCF-7 cells | Peptide (LXXLL) | [68] |
| pep5-cpp N-pep5-cpp N2-pep5-cpp N3-pep5-cpp C2-pep5-cpp C3-pep5-cpp * C4-pep5-cpp C5-pep5-cpp C6-pep5-cpp C7-pep5-cpp A B C Ac-pep5-cpp | WELVVLGKL-YGRKKRRQRRR ELVVLGKL-YGRKKRRQRRR LVVLGKL-YGRKKRRQRRR VVLGKL-YGRKKRRQRRR WELVVLG-YGRKKRRQRRR WELVVL-YGRKKRRQRRR WELVV-YGRKKRRQRRR WELV-YGRKKRRQRRR WEL-YGRKKRRQRRR WE-YGRKKRRQRRR WELVVA-YGRKKRRQRRR WEAVVL-YGRKKRRQRRR WEAVVA-YGRKKRRQRRR Ac-WELVVL-YGRKKRRQRRR | Peptide derived from g1/s cyclin d2 that induces cell death. | C6 rat; HeLa cells | Pep-5 derivatives | [69] |
| C24-LMWP | VSRRRRRRGGRRRR | Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. | BALB/c-nu nude mice; A549/T and MCF-7/ADR | LMWP/PLGA nanoparticles and doxorubicin | [70] |
| TAT-gelonin | YGRKKRRQRRR | Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer. | C57BL/6 mice; LS174T and HCT116, MDCK and 293 HEK | Gelonin | [71] |
| TAT-BID | YGRKKRRQRRR | Controlled delivery of BID protein fused with TAT peptide sensitizes cancer cells to apoptosis. | PC3, LNCaP, A549, and HeLa | BID protein | [72] |
| PTX-TAT-LP PTX-C-TAT-LP PTX-N-TAT-LP | CAYGRKKRRQRRR CAYGRKKRRQRRR CYGRKKRRQRRR | Tumor-targeted paclitaxel delivery and enhanced penetration using TAT-decorated liposomes comprising redox-responsive poly(ethylene glycol). | B16F1 tumor-bearing C57 mice; Murine B16F1 melanoma tumor cells | Paclitaxel (PTX) | [73] |
| B1 B1-Leu B1-Lys | VKRFKKFFRKLKKSV VKRFKKFFRKLKKLV VKRFKKFFRKLKKKV | Design, synthesis and biological evaluation of novel peptides with anti-cancer and drug resistance-reversing activities. | MCF-7 cells | B1 peptides | [74] |
| TAT-LP-PTX T7/TAT-LP-PTX T7-LP | CAYGRKKRRQRRR CAYGRKKRRQRRR CHAIYPRH | Efficacy of dual-functional liposomes containing paclitaxel for treatment of lung cancer. | BALB/c male athymic nude mice; A549 | Paclitaxel (PTX) | [75] |
| TP TP-biot1 TP-biot13 TP-10 TP10-biot1 | GWTLNSAGYLLGKINLKALAALAKKIL GWTLNSAGYLLGKINLKALAALAKKIL GWTLNSAGYLLGKINLKALAALAKKIL AGYLLGKINLKALAALAKKIL AGYLLGKINLKALAALAKKIL | Protein and siRNA delivery by transportan and transportan 10 into colorectal cancer cell lines. | HT29 and HCT116 | siRNA | [76] |
| Peptide 1 Peptide 2 Peptide 3 Peptide 4 Peptide 5 Peptide 6 Peptide 7 Peptide 8 Peptide 9 Peptide 10 R9 Peptide 1-C3G Peptide 1-NΔ Peptide 1-SΔ Peptide 1-NSΔ Peptide 1-NTSΔ Peptide 1-NTCSΔ Peptide 1-NTHSΔ | NTCTWLKYHS CASGQQGLLKLC YNNFAYSVFL ECYPKKGQDP RHVYHVLLSQ HATKSQNINF YRDRFAFQPH IWRYSLASQQ YQKQAKIMCS VQLRRRWC RRRRRRRRR NTGTWLKYHS TCTWLKYHS NTCTWLKYH TCTWLKYH CTWLKYH TWLKYH CTWLKY | Novel cell-penetrating peptide targeting human glioblastoma cell lines. | U87MG cells | p16(INK4a) functional peptide | [40,49,77] |
| P28 | LSTAADMQGVVTDGMASGLDKDYLKPDD | p28, an anionic cell-penetrating peptide, increases the activity of wild type and mutated p53. | MCF-7, MDA-MB-231, and T47D, HCT116 and HT29, HT1080, (HTB-88), osteosarcoma (TE85), rhabdomyosarcoma (RD), glioblastoma (U87 and LN229), neuroblastoma (SK-N-BE2), prostate cancer (DU145), pancreatic cancer (MIA-Paca2) and ovarian cancer (ES-2)]. Melanoma lines (UISO-Mel-23, 29 | P28 | [78] |
| RALA peptide | WEARLARALARALARHLARALARA | Readily traversed the plasma membrane of both cancer and fibroblast cell lines and elicited reporter-gene expression following intravenous delivery in vivo. | ZR-75-1 human breast cancer, PC-3 human prostate cancerand NCTC-929 murine fibroblast cell lines | Plasmid DNA | [79] |
| TAT(47–57) Penetratin PEP-1 DS4.3 | YGRKKRRQRRR RQIKIWFQNRRMKWKK KETWWETWWTEWSQPKKKRKV RIMRILRILKLAR | Anti-tumoral effect of the mitochondrial target domain of Noxa delivered by an engineered Salmonella typhimurium. | Male Balb/c mice; CT26 mouse colon cancer cells, HeLa and Hep3B cells | Mitochondrial Target Domain of NOXA | [80] |
| SR9 HR9 PR9 | SRRRRRRRRR CHHHHHRRRRRRRRRHHHHHC FFLIPKGRRRRRRRRR | Direct membrane traslocation. Enhance the gene expression intensity. | A549 cells | Plasmid DNA | [81] |
| PF14 | AGYLLGKLLOOLAAAALOOLL | Delivery pDNA forming stable nanoparticles that improve the transfection efficiency. | HeLa pLuc705 cells | Nucleic acids | [82,83] |
| d-NTD q-NTD | KGRKKRRQRRRPPQ KGRKKRRQRRRPPQ | d-NTD is the most potent conjugate against HepG2 human liver cancer cells. | HepG2 | Doxorubicin | [84] |
| TH | AGYLLGHINLHHLAHL(Aib)HHIL-NH2 | Acid-activated pH response for targeting delivery of antitumor drugs. | Hela cells | pH-responsive | [85] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. https://doi.org/10.3390/molecules23020295
Borrelli A, Tornesello AL, Tornesello ML, Buonaguro FM. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules. 2018; 23(2):295. https://doi.org/10.3390/molecules23020295
Chicago/Turabian StyleBorrelli, Antonella, Anna Lucia Tornesello, Maria Lina Tornesello, and Franco M. Buonaguro. 2018. "Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents" Molecules 23, no. 2: 295. https://doi.org/10.3390/molecules23020295
APA StyleBorrelli, A., Tornesello, A. L., Tornesello, M. L., & Buonaguro, F. M. (2018). Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules, 23(2), 295. https://doi.org/10.3390/molecules23020295