Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes



Abstract
:1. Introduction
2. Results and Discussion
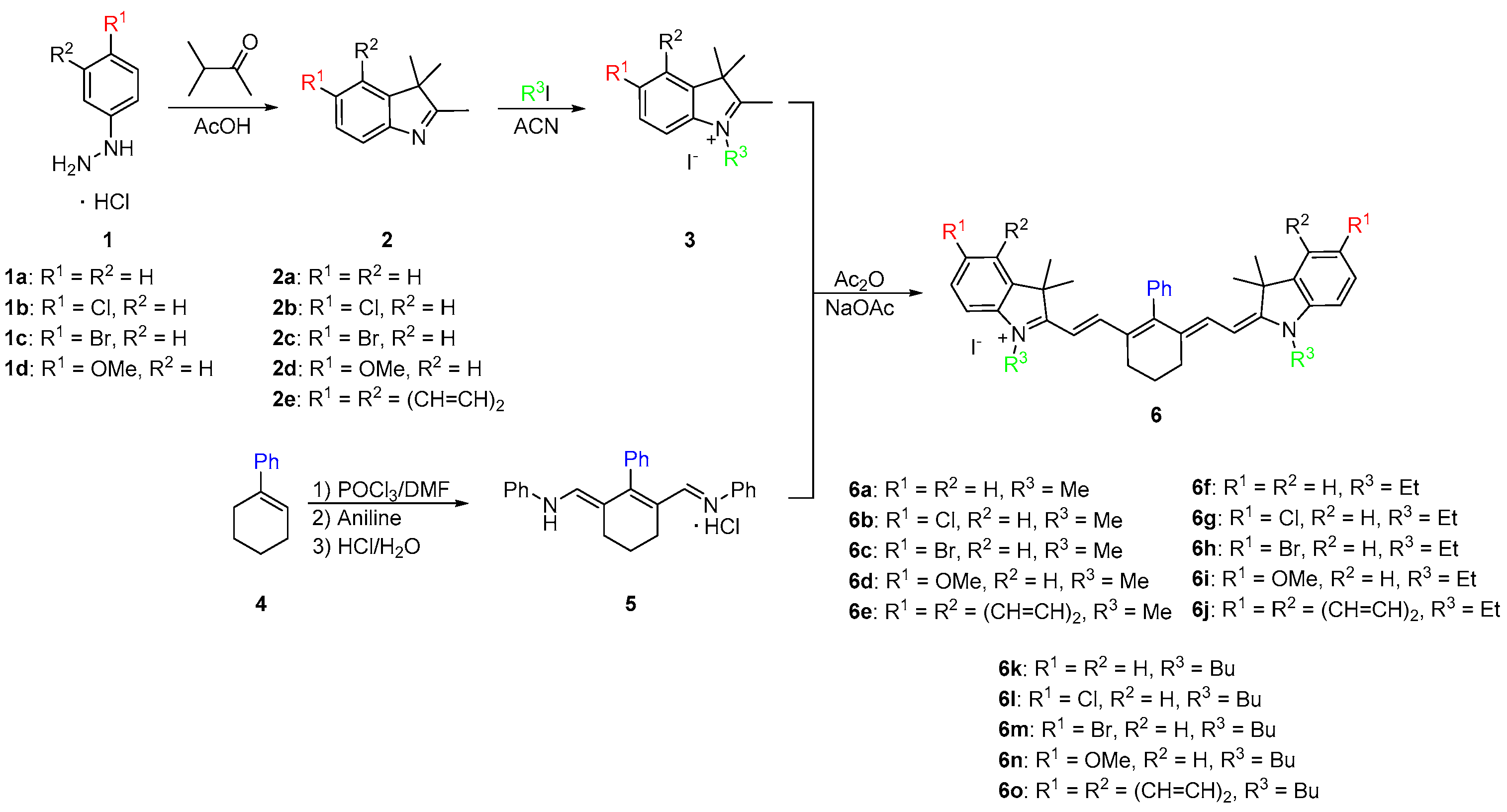
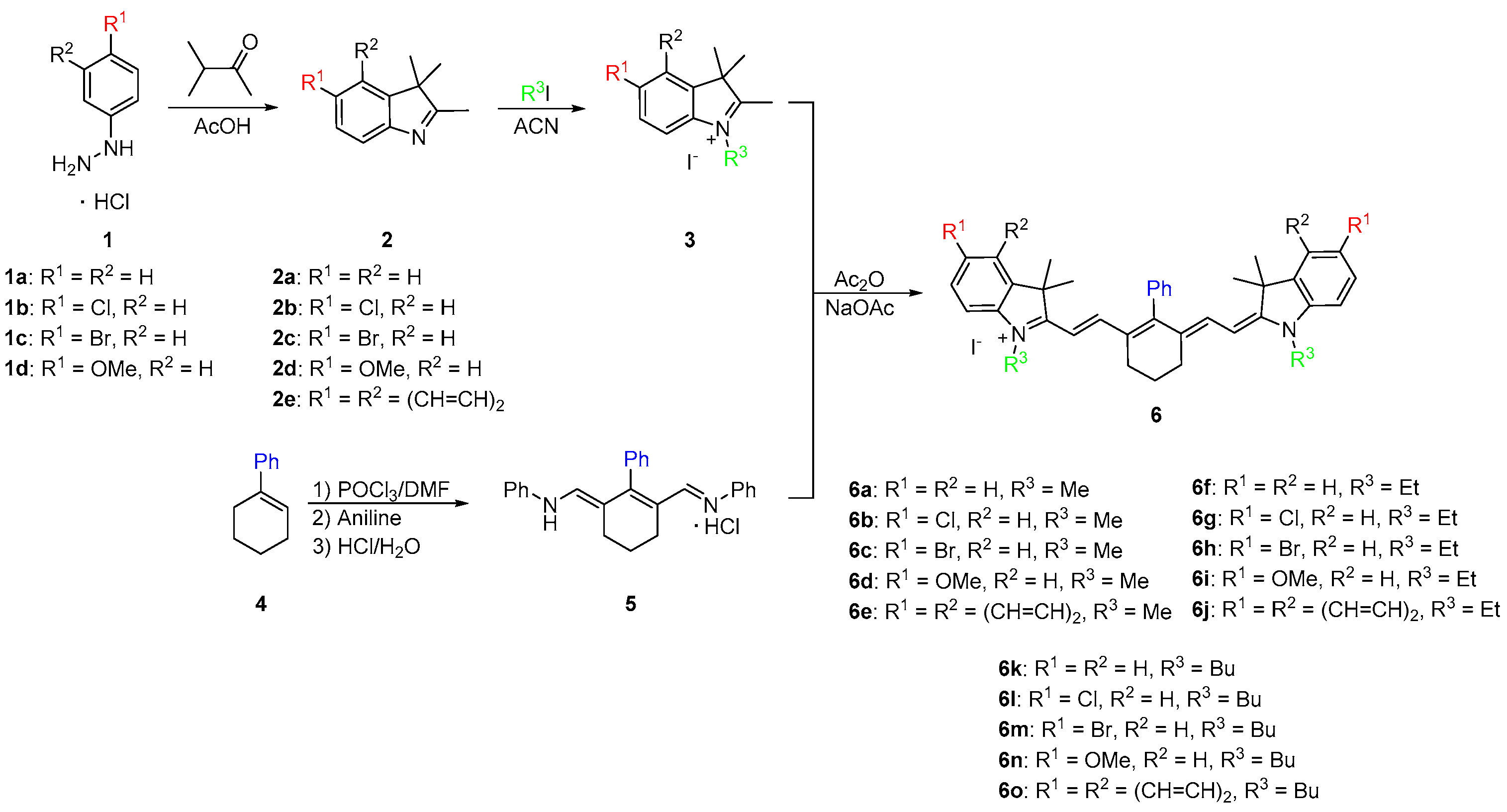
2.1. Synthesis
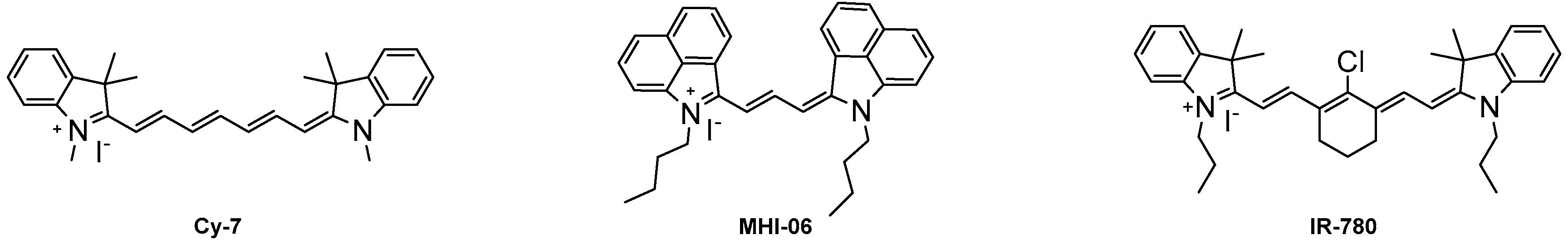
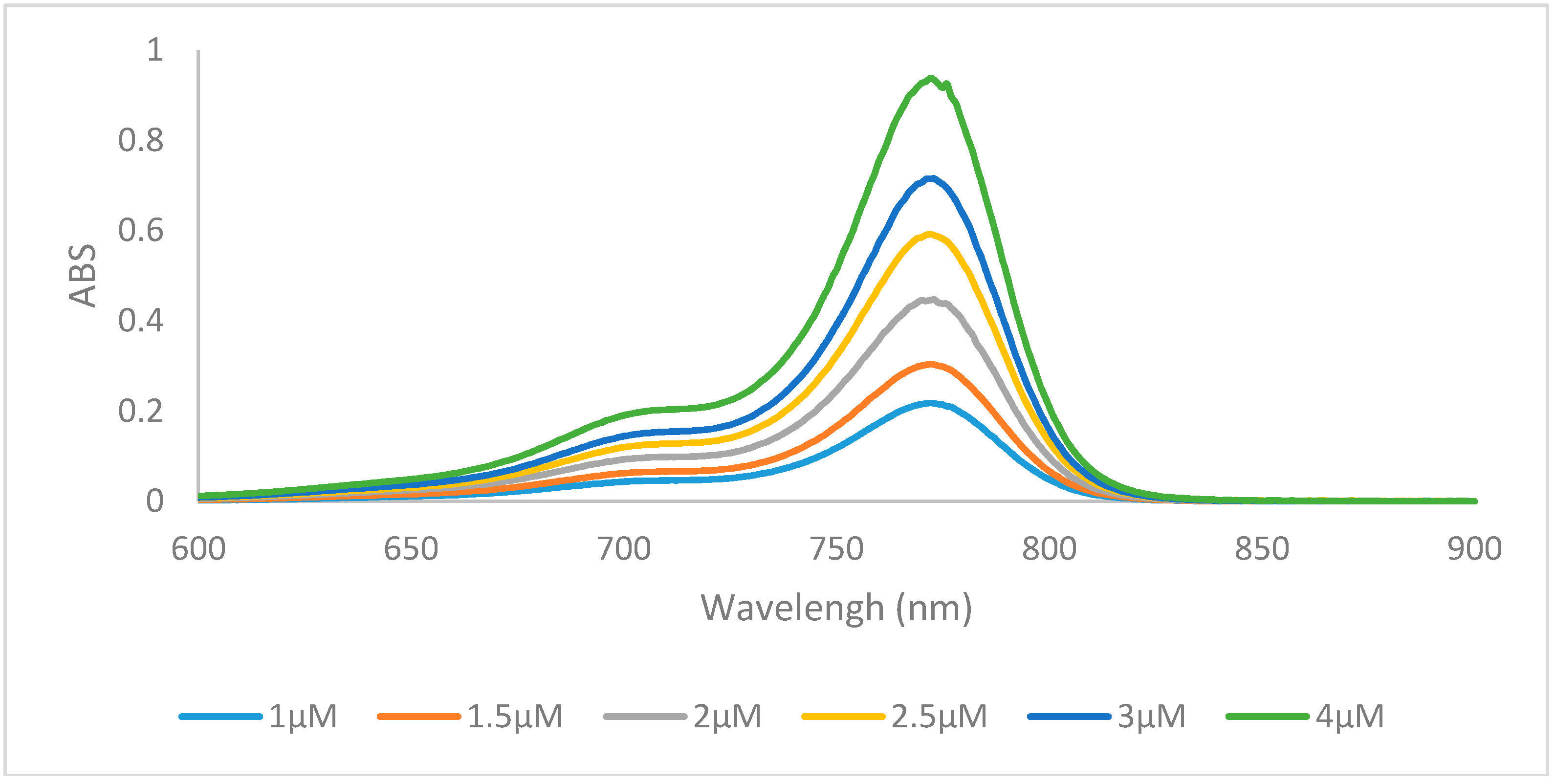
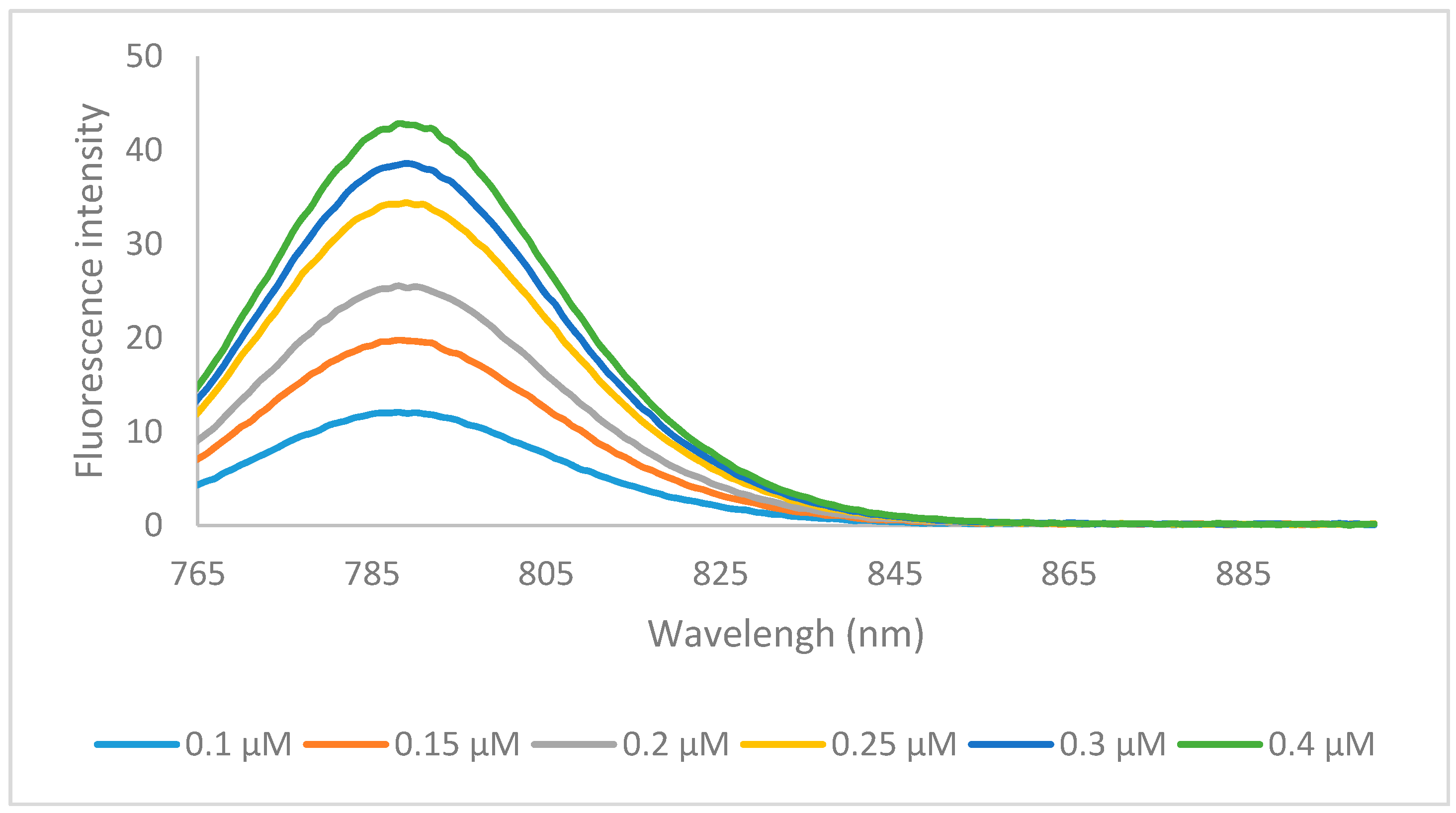
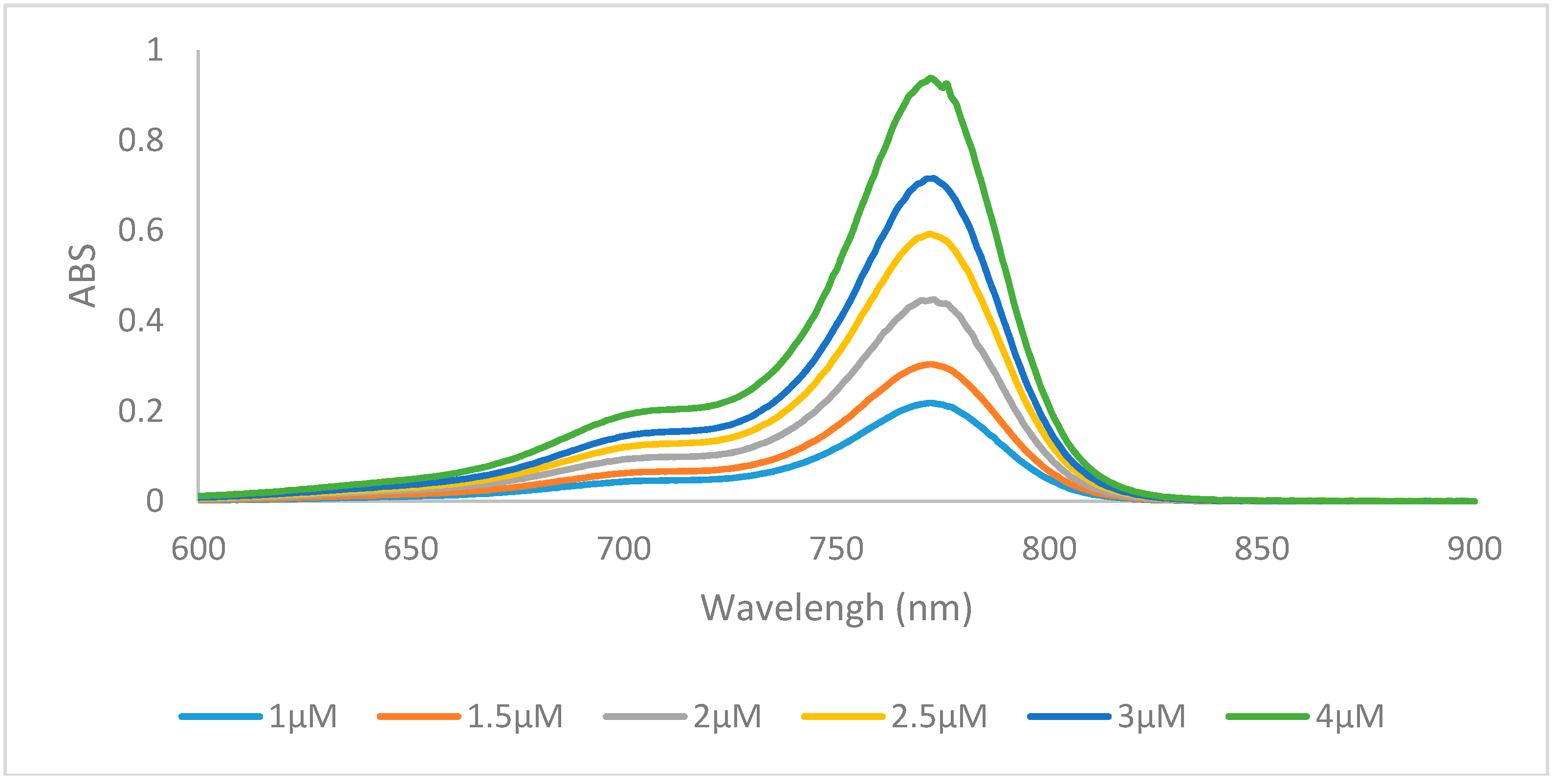
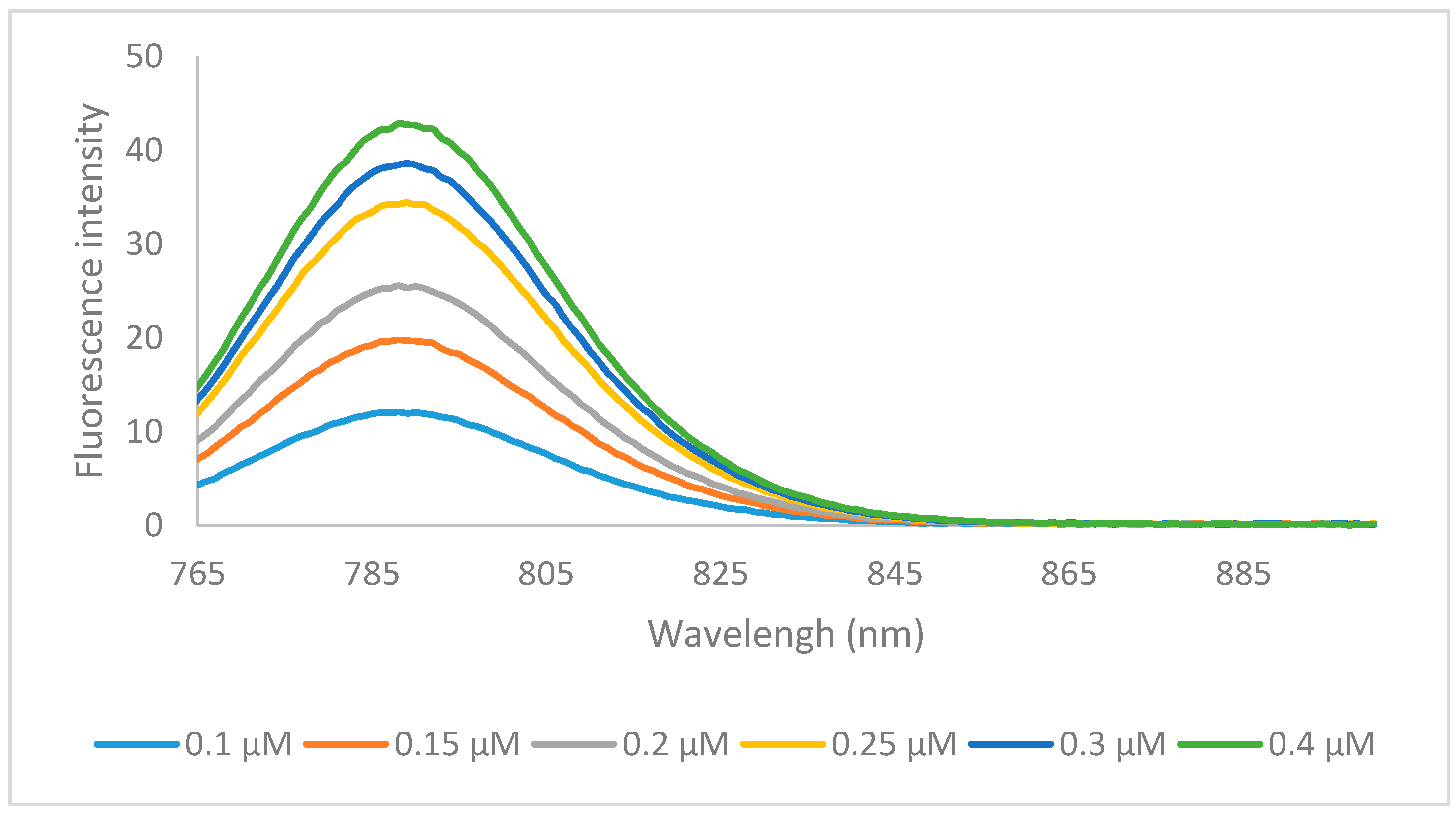
2.2. Optical Properties
2.3. Physiochemical Properties
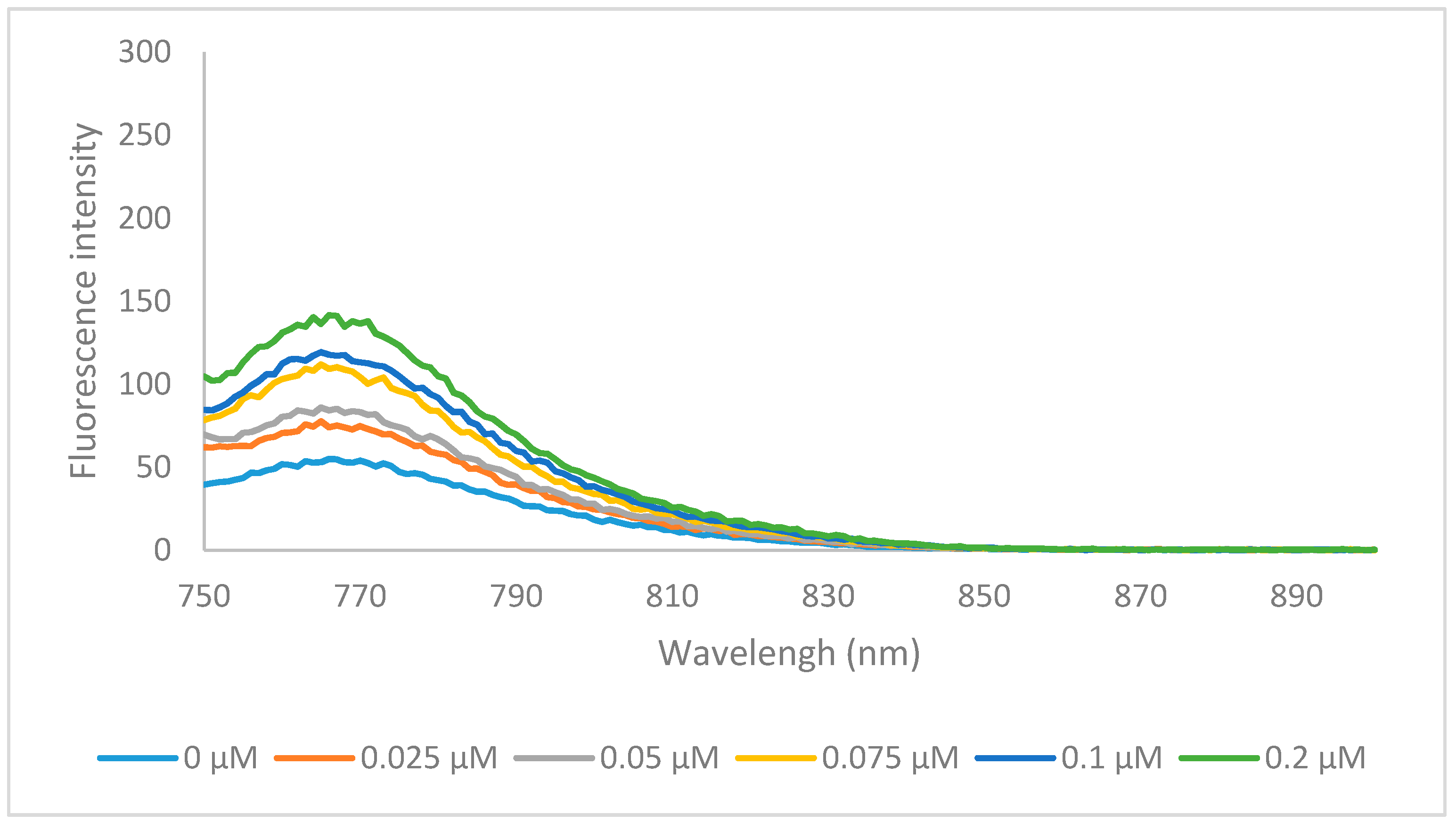
2.4. HSA Binding
3. Experimental Section
3.1. General Information
3.2. Characterization
3.3. Stock Solutions
3.4. Method of Determining Molar Absorptivity and Fluorescence Quantum Yield
3.5. HSA Binding Study
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flanagan, J.H., Jr.; Khan, S.H.; Menchen, S.; Soper, S.A.; Hammer, R.P. Functionalized tricarbocyanine dyes as near-infrared fluorescent probes for biomolecules. Bioconjug. Chem. 1997, 8, 751–756. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Patonay, G. Fluorescence studies of carbocyanines using AOTF. Talanta 1997, 45, 285–289. [Google Scholar] [CrossRef]
- Patonay, G.; Salon, J.; Sowell, J.; Strekowski, L. Noncovalent labeling of biomolecules with red and near-infrared dyes. Molecules 2004, 9, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shi, C.; Tong, R.; Qian, W.; Zhau, H.E.; Wang, R.; Zhu, G.; Cheng, J.; Yang, V.W.; Cheng, T.; et al. Near IR heptamethine cyanine dye-mediated cancer imaging. Clin. Cancer Res. 2010, 16, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wu, J.B.; Pan, D. Review on near-infrared heptamethine cyanine dyes as theranostic agents for tumor imaging, targeting, and photodynamic therapy. J. Biomed. Opt. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Li, Y.L.M.; Chen, H.; Li, H. Near-infrared chemodosimetric probes based on heptamethine cyanine dyes for the “naked-eye” detection of cyanide in aqueous media. J. Lumin. 2017, 185, 286–291. [Google Scholar] [CrossRef]
- Soriano, E.; Holder, C.; Levitz, A.; Henary, M. Benz[c,d]indolium-containing monomethine cyanine dyes: Synthesis and photophysical properties. Molecules 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Kurutosa, A.; Tarabara, O.R.U.; Trusovab, V.; Gorbenko, G.; Gadjev, N.; Deligeorgiev, T. Novel synthetic approach to near-infrared heptamethine cyanine dyes and spectroscopic characterization in presence of biological molecules. J. Photochem. Photobiol. A 2016, 328, 87–96. [Google Scholar] [CrossRef]
- Ernst, L.A.; Gupta, R.K.; Mujumdar, R.B.; Waggoner, A.S. Cyanine dye labeling reagents for sulfhydryl groups. Cytometry 1989, 10, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Honig, B.; Greenberg, A.D.; Dinur, U.; Ebrey, T.G. Visual-pigment spectra: Implications of the protonation of the retinal Schiff base. Biochemistry 1976, 15, 4593–4599. [Google Scholar] [CrossRef] [PubMed]
- Ashitate, Y.; Levitz, A.; Park, M.H.; Hyun, H.; Venugopal, V.; Park, G.; El Fakhri, G.; Henary, M.; Gioux, S.; Frangioni, J.V.; et al. Endocrine-specific NIR fluorophores for adrenal gland targeting. Chem. Commun. 2016, 52, 10305–10308. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Owens, E.A.; Wada, H.; Levitz, A.; Park, G.; Park, M.H.; Frangioni, J.V.; Henary, M.; Choi, H.S. Cartilage-specific near-infrared fluorophores for biomedical imaging. Angew. Chem. Int. Ed. Engl. 2015, 54, 8648–8652. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Park, M.H.; Owens, E.A.; Wada, H.; Henary, M.; Handgraaf, H.J.; Vahrmeijer, A.L.; Frangioni, J.V.; Choi, H.S. Structure-inherent targeting of near-infrared fluorophores for parathyroid and thyroid gland imaging. Nat. Med. 2015, 21, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Wada, H.; Bao, K.; Gravier, J.; Yadav, Y.; Laramie, M.; Henary, M.; Frangioni, J.V.; Choi, H.S. Phosphonated near-infrared fluorophores for biomedical imaging of bone. Angew. Chem. Int. Ed. Engl. 2014, 53, 10668–10672. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, G.; Hyun, H.; Lee, J.H.; Ashitate, Y.; Choi, J.; Hong, G.H.; Owens, E.A.; Henary, M.; Choi, H.S. Near-infrared lipophilic fluorophores for tracing tissue growth. Biomed. Mater. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Owens, E.A.; Henary, M.; El Fakhri, G.; Choi, H.S. Tissue-specific near-infrared fluorescence imaging. Acc. Chem. Res. 2016, 49, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Shershov, V.E.; Spitsyn, M.A.; Kuznetsova, V.E.; Timofeev, E.N.; Ivashkina, O.A.; Abramov, I.S.; Nasedkina, T.V.; Zasedatelev, A.S.; Chudinov, A.V. Near-infrared heptamethine cyanine dyes. Synthesis, spectroscopic characterization, thermal properties and photostability. Dyes Pigment. 2013, 97, 353–360. [Google Scholar] [CrossRef]
- Lee, H.; Berezin, M.Y.; Tang, R.; Zhegalova, N.; Achilefu, S. Pyrazole-substituted near-infrared cyanine dyes exhibit pH-dependent fluorescence lifetime properties. Photochem. Photobiol. 2013, 89, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mason, J.C.; Achilefu, S. Heptamethine cyanine dyes with a robust C-C bond at the central position of the chromophore. J. Org. Chem. 2006, 71, 7862–7865. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mason, J.C.; Achilefu, S. Synthesis and spectral properties of near-infrared aminophenyl-, hydroxyphenyl-, and phenyl-substituted heptamethine cyanines. J. Org. Chem. 2008, 73, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Stanford, C.; Morton, M.D.; Zhu, Q.; Smith, M.B. Structurally modified indocyanine green dyes. Modification of the polyene linker. Dyes Pigment. 2013, 99, 275–283. [Google Scholar] [CrossRef]
- Jozef Salon, E.W.; Raszkiewicz, A.; Patonay, G.; Strekowski, L. Synthesis of Benz[e]indolium Heptamethine Cyanines Containing C-Substituents at the Central Portion of the Heptamethine Moiety. J. Heterocycl. Chem. 2005, 42, 959–961. [Google Scholar] [CrossRef]
- Theodore Peters, J. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic: San Diego, CA, USA, 1995. [Google Scholar]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Maggioli, C.; Zaccherini, G. Human albumin in the management of complications of liver cirrhosis. Crit. Care 2012, 16. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Beckford, G.; Owens, E.; Henary, M.; Patonay, G. The solvatochromic effects of side chain substitution on the binding interaction of novel tricarbocyanine dyes with human serum albumin. Talanta 2012, 92, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rurack, K.; Spieles, M. Fluorescence quantum yields of a series of red and near-infrared dyes emitting at 600–1000 nm. Anal. Chem. 2011, 83, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.; Henary, M.; Patonay, G. The effect of varying short-chain alkyl substitution on the molar absorptivity and quantum yield of cyanine dyes. Anal. Chem. Insights 2011, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.A.; Jacobs, L.D.; Furlani, T.R.; Nalley, E.A.; Laboy, J. Conformationally dependent heavy atom effect of chlorine on alkene triplet lifetimes. experimental and ab initio calculations. J. Am. Chem. Soc. 1991, 144, 1623–1625. [Google Scholar]
- Dost, T.L.; Gressel, M.T.; Henary, M. Synthesis and optical properties of pentamethine cyanine dyes with carboxylic acid moieties. Anal. Chem. Insights 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Toseland, C.P. Fluorescent labeling and modification of proteins. J. Chem. Biol. 2013, 6, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, A. Covalent labeling of proteins and nucleic acids with fluorophores. Methods Enzymol. 1995, 246, 362–373. [Google Scholar] [PubMed]
- Patonay, G.; Kim, J.S.; Kodagahally, R.; Strekowski, L. Spectroscopic study of a novel bis(heptamethine cyanine) dye and its interaction with human serum albumin. Appl. Spectrosc. 2005, 59, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, H.; Tang, Y.; Xu, G.; Yan, W. Spectroscopic investigation on the interaction of J-aggregate with human serum albumin. Biophys Chem. 2007, 128, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Berezin, M.Y.; Lee, H.; Akers, W.; Nikiforovich, G.; Achilefu, S. Ratiometric analysis of fluorescence lifetime for probing binding sites in albumin with near-infrared fluorescent molecular probes. Photochem. Photobiol. 2007, 83, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kodagahally, R.; Strekowski, L.; Patonay, G. A study of intramolecular H-complexes of novel bis(heptamethine cyanine) dyes. Talanta 2005, 67, 947–954. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
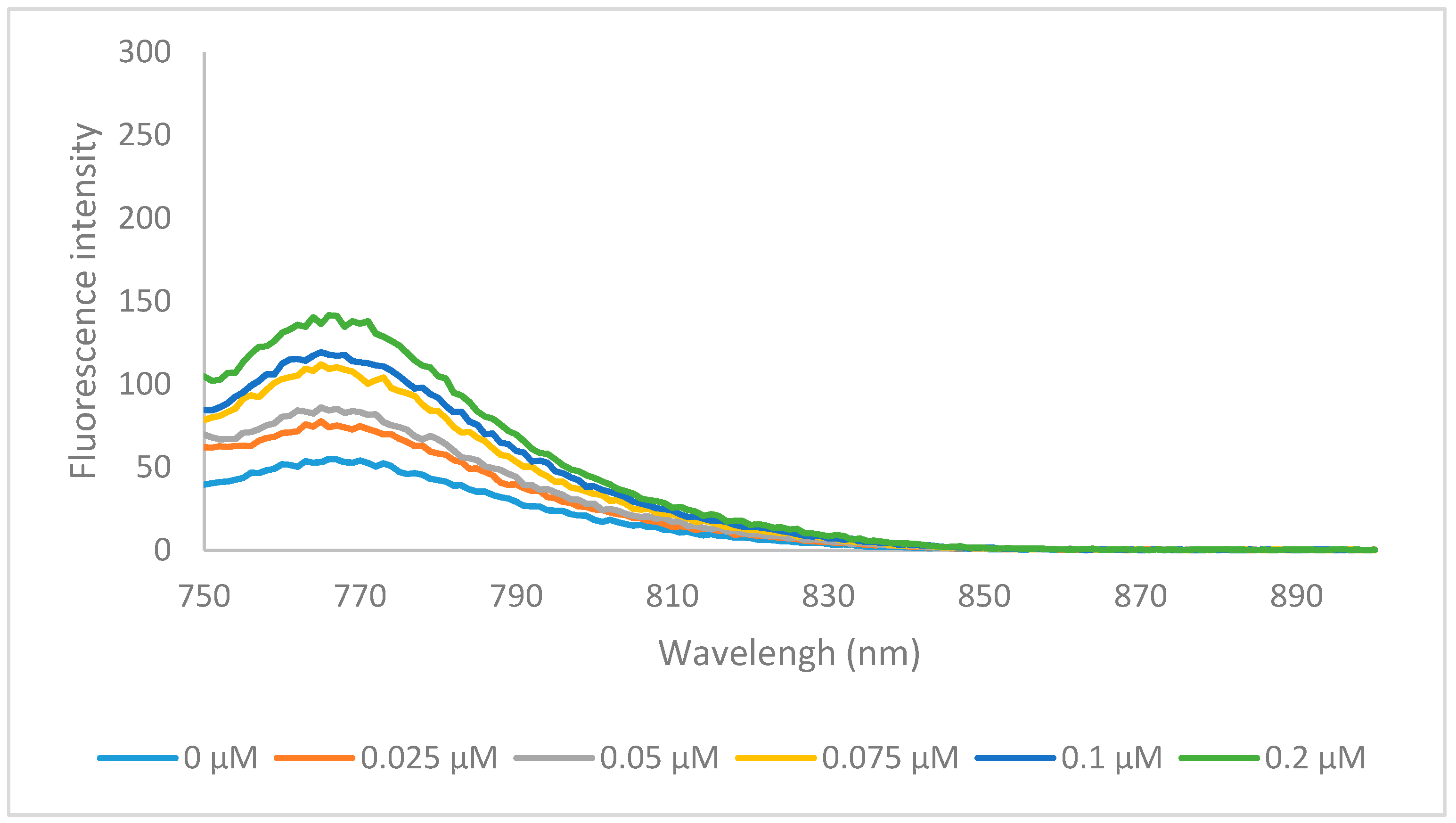

{kind=link}
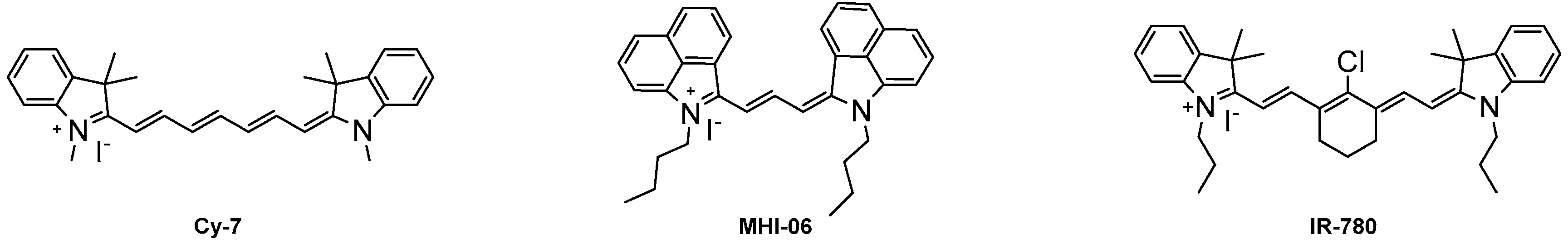
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | λmax (nm) | λemission (nm) | Stokes Shift (nm) | ε (L·mol−1·cm−1) | Φ (%) | Molecular Brightness (M−1·cm−1) |
|---|---|---|---|---|---|---|
| Cy-7 | 753 | 775 | 22 | 200,000 | 28 | 56,000 |
| IR-780 | 779 | 799 | 20 | 274,000 | 8.0 | 20,800 |
| 6a | 759 | 774 | 15 | 265,700 | 31 | 82,000 |
| 6b | 765 | 780 | 15 | 261,000 | 34 | 88,700 |
| 6c | 767 | 783 | 16 | 275,600 | 35 | 96,500 |
| 6d | 782 | 802 | 20 | 249,500 | 10 | 25,000 |
| 6e | 798 | 810 | 12 | 255,400 | 16 | 40,900 |
| 6f | 760 | 781 | 21 | 263,900 | 39 | 102,300 |
| 6g | 769 | 785 | 16 | 286,600 | 38 | 109,300 |
| 6h | 770 | 786 | 16 | 282,900 | 42 | 119,200 |
| 6i | 786 | 804 | 18 | 143,500 | 12 | 17,200 |
| 6j | 797 | 810 | 13 | 231,800 | 17 | 39,400 |
| 6k | 763 | 780 | 17 | 198,500 | 45 | 89,300 |
| 6l | 772 | 787 | 15 | 123,400 | 47 | 58,000 |
| 6m | 773 | 788 | 15 | 239,200 | 48 | 113,600 |
| 6n | 789 | 805 | 16 | 249,900 | 11 | 27,100 |
| 6o | 800 | 812 | 12 | 226,600 | 17 | 38,500 |
| Dye | logD | Polarizability | Dipole Moment | Rot. Bonds | Volume | Molec. Surface Area | Molar Mass |
|---|---|---|---|---|---|---|---|
| MHI-06 | 4.97 | 59.45 | 2.48 | 8 | 441.44 | 693.216 | 584.54 |
| 6a | 6.07 | 65.57 | 8.35 | 4 | 519.85 | 814.001 | 652.66 |
| 6b | 7.28 | 69.15 | 12.3 | 4 | 547.45 | 845.982 | 721.55 |
| 6c | 7.61 | 70.67 | 13.02 | 4 | 556.15 | 854.732 | 810.46 |
| 6d | 5.75 | 70.51 | 27.66 | 6 | 569.71 | 907.778 | 712.72 |
| 6e | 8.05 | 80.82 | 11.87 | 4 | 604.27 | 936.392 | 752.78 |
| 6f | 6.75 | 69.26 | 3.2 | 6 | 552.14 | 874.685 | 680.72 |
| 6g | 7.99 | 72.84 | 4.81 | 6 | 579.48 | 906.491 | 749.6 |
| 6h | 8.32 | 74.35 | 4.97 | 6 | 588.47 | 915.629 | 838.51 |
| 6i | 6.47 | 74.2 | 25.29 | 8 | 603.83 | 969.908 | 740.77 |
| 6j | 8.76 | 84.51 | 4.59 | 6 | 638.63 | 999.272 | 7803.84 |
| 6k | 8.72 | 76.65 | 2.85 | 10 | 620.01 | 996.744 | 736.81 |
| 6l | 9.92 | 80.23 | 4.63 | 10 | 647.91 | 1029.597 | 805.71 |
| 6m | 10.25 | 81.71 | 4.96 | 10 | 656.64 | 1038.143 | 894.62 |
| 6n | 8.40 | 81.59 | 26.66 | 12 | 672.63 | 1094.66 | 796.88 |
| 6o | 10.69 | 91.9 | 3.96 | 10 | 708.29 | 1124.8 | 836.95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levitz, A.; Marmarchi, F.; Henary, M. Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes. Molecules 2018, 23, 226. https://doi.org/10.3390/molecules23020226
Levitz A, Marmarchi F, Henary M. Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes. Molecules. 2018; 23(2):226. https://doi.org/10.3390/molecules23020226
Chicago/Turabian StyleLevitz, Andrew, Fahad Marmarchi, and Maged Henary. 2018. "Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes" Molecules 23, no. 2: 226. https://doi.org/10.3390/molecules23020226
APA StyleLevitz, A., Marmarchi, F., & Henary, M. (2018). Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes. Molecules, 23(2), 226. https://doi.org/10.3390/molecules23020226