Accurate Estimation of the Standard Binding Free Energy of Netropsin with DNA


 ,
, 
Abstract
1. Introduction
2. Results and Discussion
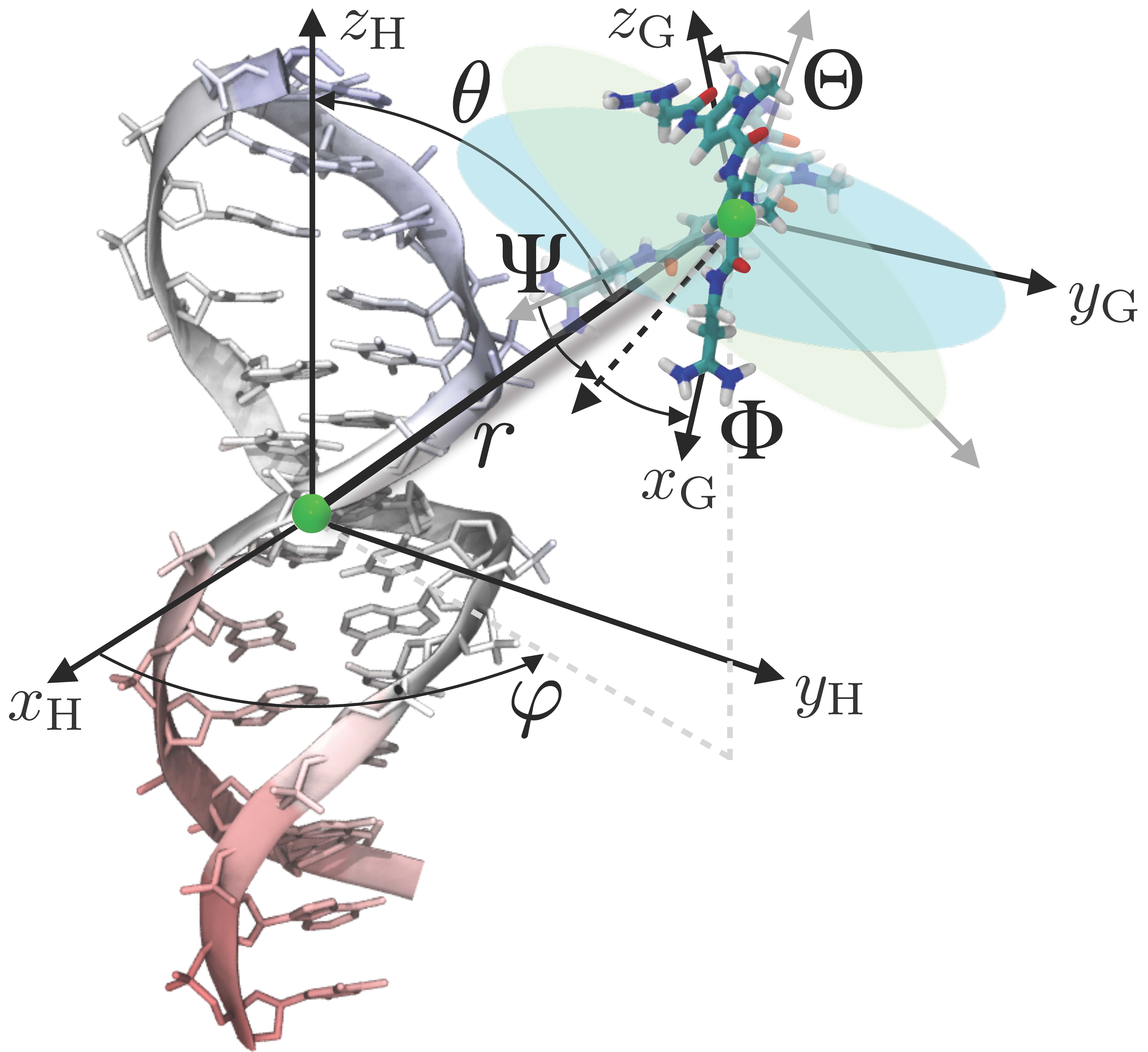
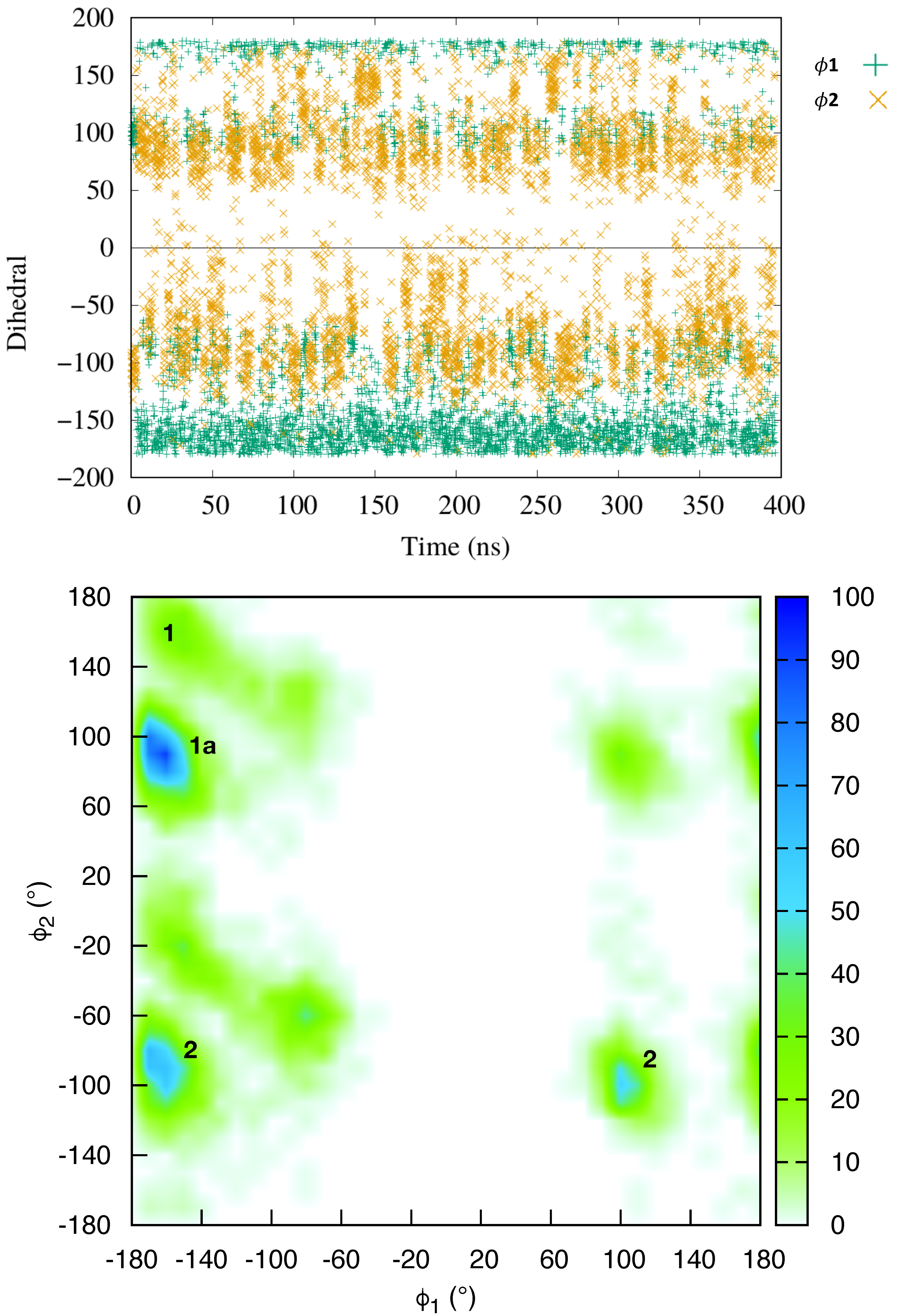
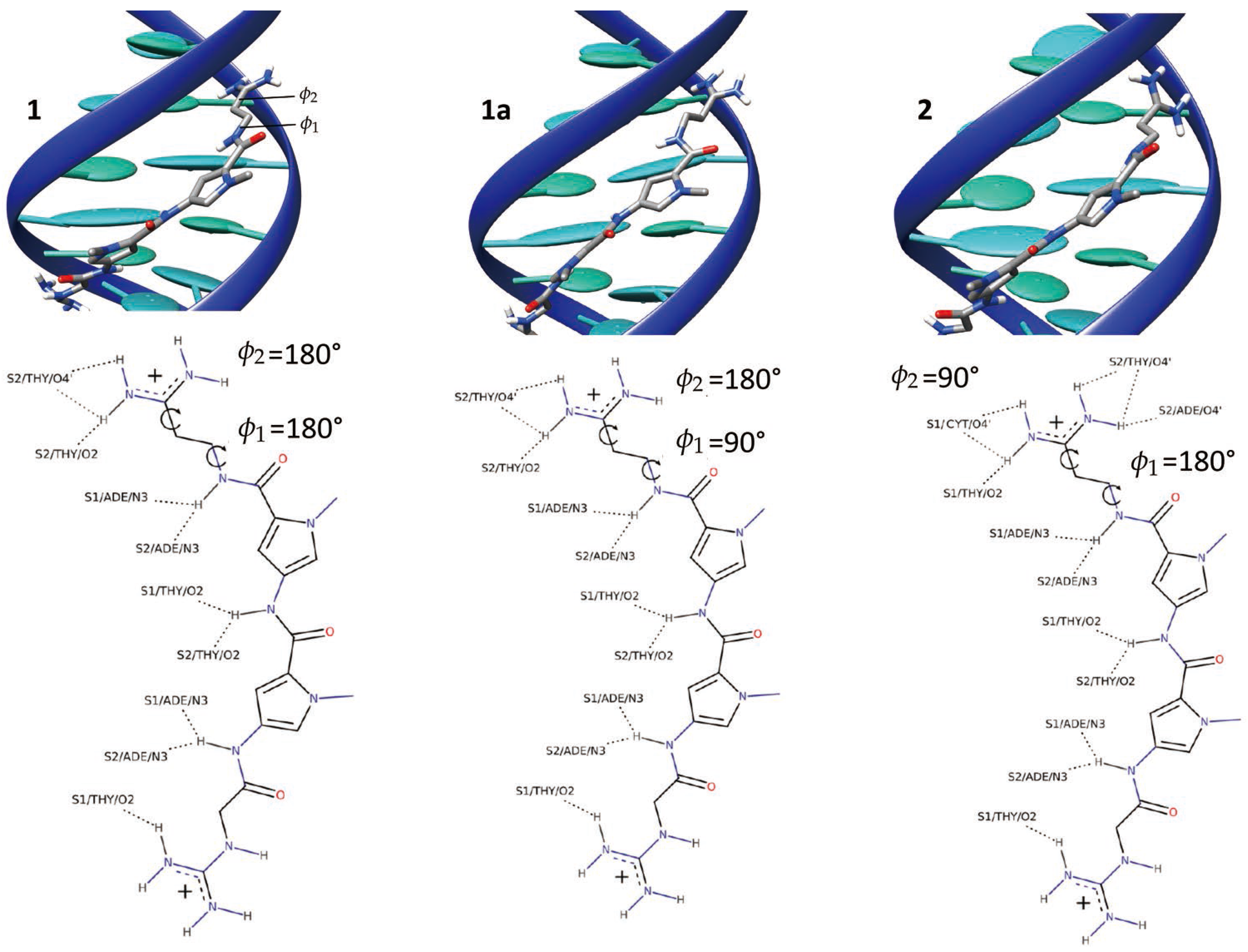
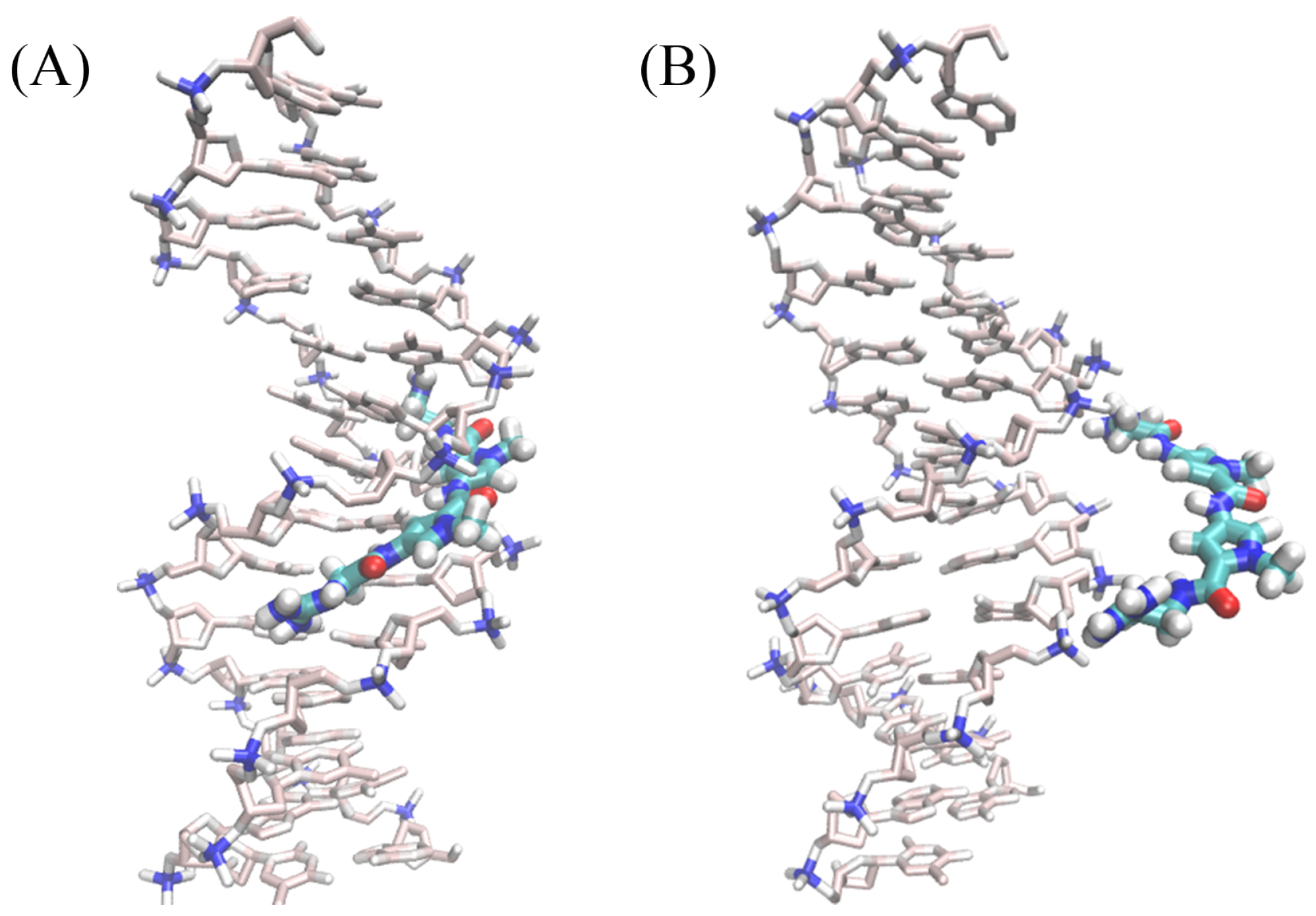
2.1. Dynamics of the Netropsin/DNA Complex
2.2. Absolute Binding Free-Energy of Netropsin to DNA
3. Conclusions
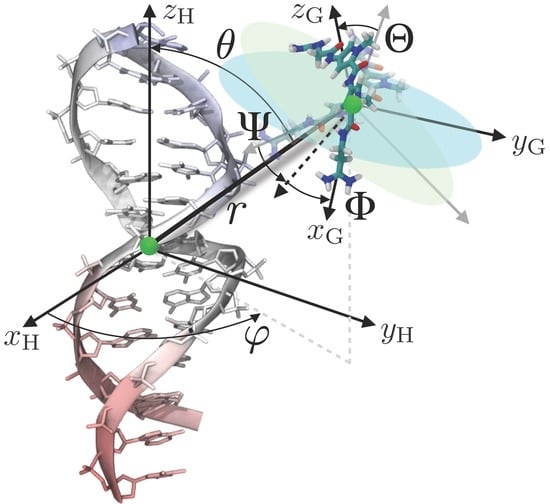
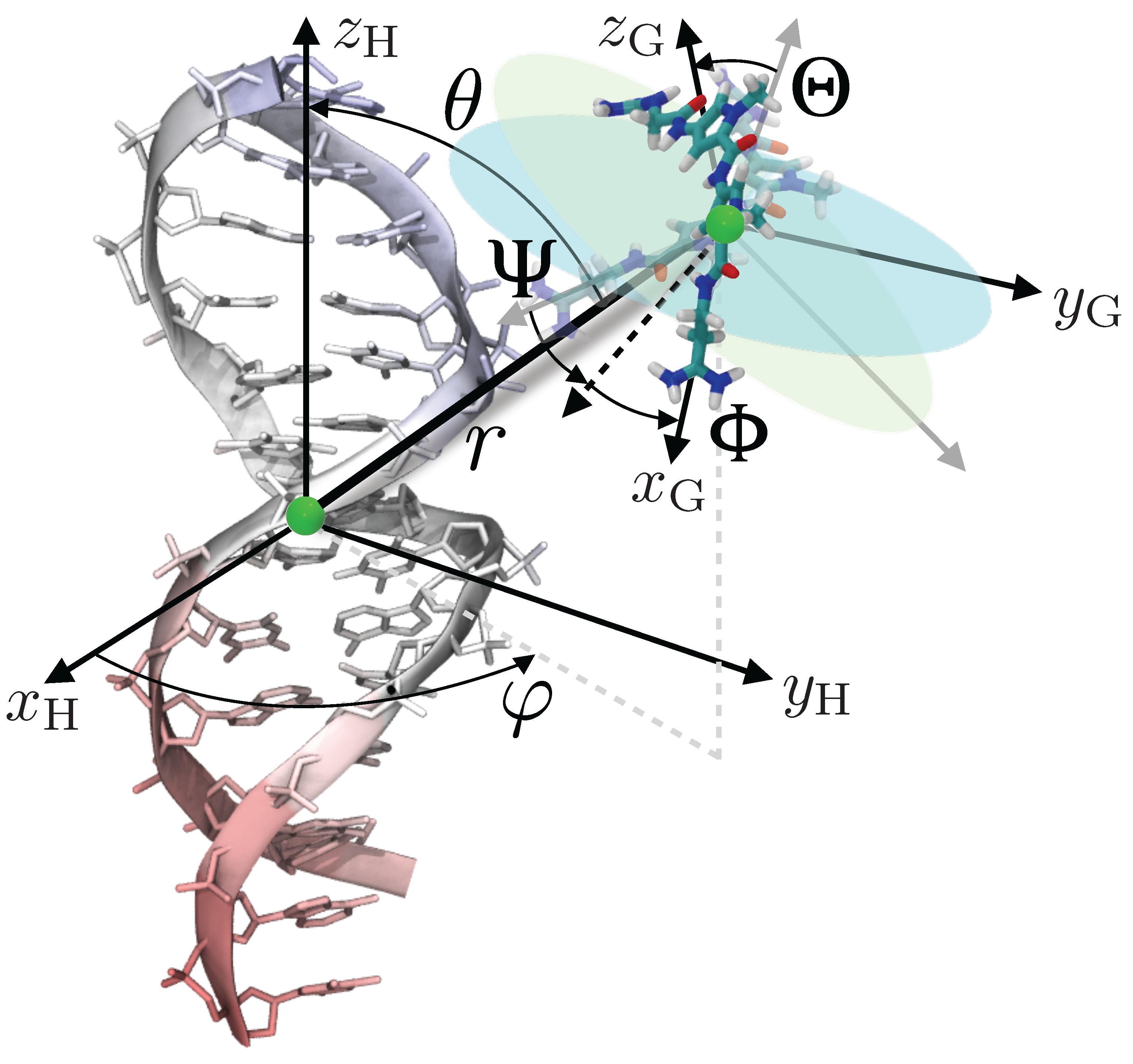
4. Theoretical Background
5. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Cauet, E.; Bogatko, S.A.; Lievin, J.; De Proft, F.; Geerlings, P. Electron attachment-induced DNA damage: Instantaneous strand breaks. J. Phys. Chem. B 2013, 117, 9669–9676. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Delatour, T.; Douki, T.; Gasparutto, D.; Pouget, J.P.; Ravanat, J.L.; Sauvaigo, S. Hydroxyl radicals and DNA base damage. Mutat. Res., Fundam. Mol. Mech. Mutagen. 1999, 424, 9–21. [Google Scholar] [CrossRef]
- Cadet, J.; Mouret, S.; Ravanat, J.L.; Douki, T. Photoinduced damage to cellular DNA: Direct and photosensitized reactions. Photochem. Photobiol. 2012, 88, 1048–1065. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Shikazono, N.; Noguchi, M.; Fujii, K.; Urushibara, A.; Yokoya, A. The yield, processing, and biological consequences of clustered DNA damage induced by ionizing radiation. J. Radiat. Res. 2009, 50, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Magnander, K.; Elmroth, K. Biological consequences of formation and repair of complex DNA damage. Cancer Lett. 2012, 327, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication fork collapse is a major cause of the high mutation frequency at three-base lesion clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, S.; Walker, A.; Stabler, R.; O’Neill, P.; Lomax, M.E. Increased mutability and decreased repairability of a three-lesion clustered DNA-damaged site comprised of an AP site and bi-stranded 8-oxoG lesions. Int. J. Radiat. Biol. 2014, 90, 468–479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Salmon, T.B. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 2004, 32, 3712–3723. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C. Mitochondrial DNA mutations , oxidative stress , and apoptosis in mammalian aging. Science 2007, 481, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.L.; Martinez, G.R.; Medeiros, M.H.G.; di Mascio, P. Singlet oxygen oxidation of isolated and cellular DNA: Product formation and mechanistic insights. Photochem. Photobiol. 2006, 82, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Sheu, C.; Kang, P.; Khan, S.; Foote, C.S. Low-temperature photosensitized oxidation of a guanosine derivative and formation of an imidazole ring-opened product. J. Am. Chem. Soc. 2002, 124, 3905–3913. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Mundy, C.J.; Colvin, M.E.; Car, R. On the mechanisms of OH radical induced DNA-base damage: A comparative quantum chemical and Car - Parrinello molecular dynamics study. J. Phys. Chem. A 2004, 108, 2922–2929. [Google Scholar] [CrossRef]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Improta, R.; Markovitsi, D. DNA/RNA: Building blocks of life under UV irradiation. J. Phys. Chem. Lett. 2010, 1, 2025–2030. [Google Scholar] [CrossRef]
- Rauer, C.; Nogueira, J.J.; Marquetand, P.; González, L. Cyclobutane thymine photodimerization mechanism revealed by nonadiabatic molecular dynamics. J. Am. Chem. Soc. 2016, 138, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Dehez, F.; Gattuso, H.; Bignon, E.; Morell, C.; Dumont, E.; Monari, A. Conformational polymorphism or structural invariance in DNA photoinduced lesions: Implications for repair rates. Nucleic Acids Res. 2017, 45, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Epe, B. DNA damage spectra induced by photosensitization. Photochem. Photobiol. Sci. 2012, 11, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Benzophenone and DNA: Evidence for a double insertion mode and its spectral signature. J. Phys. Chem. Lett. 2013, 4, 4119–4124. [Google Scholar] [CrossRef]
- Cuquerella, M.C.; Lhiaubet-Vallet, V.; Cadet, J.; Miranda, M.A. Benzophenone photosensitized DNA damage. Acc. Chem. Res. 2012, 45, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Bignon, E.; Marazzi, M.; Besancenot, V.; Gattuso, H.; Drouot, G.; Morell, C.; Eriksson, L.; Grandemange, S.; Dumont, E.; Monari, A. Ibuprofen and ketoprofen potentiate UVA-induced cell death by a photosensitization process. Sci. Rep. 2017, 7, 8885. [Google Scholar] [CrossRef] [PubMed]
- Bignon, E.; Gattuso, H.; Morell, C.; Dumont, E.; Monari, A. DNA photosensitization by an “insider”: Photophysics and triplet energy transfer of 5-Methyl-2-pyrimidone deoxyribonucleoside. Chem. Eur. J. 2015, 21, 11509–11516. [Google Scholar] [CrossRef] [PubMed]
- Véry, T.; Ambrosek, D.; Otsuka, M.; Gourlaouen, C.; Assfeld, X.; Monari, A.; Daniel, C. Photophysical properties of ruthenium (ii) polypyridyl dna intercalators: Effects of the molecular surroundings investigated by theory. Chem. Eur. J. 2014, 20, 12901–12909. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, M.; Wibowo, M.; Gattuso, H.; Dumont, E.; Roca-Sanjuán, D.; Monari, A. Hydrogen abstraction by photoexcited benzophenone: Consequences for DNA photosensitization. Phys. Chem. Chem. Phys. 2016, 18, 7829–7836. [Google Scholar] [CrossRef] [PubMed]
- Chantzis, A.; Very, T.; Daniel, C.; Monari, A.; Assfeld, X. Theoretical evidence of photo-induced charge transfer from DNA to intercalated ruthenium (II) organometallic complexes. Chem. Phys. Lett. 2013, 578, 133–137. [Google Scholar] [CrossRef]
- Very, T.; Despax, S.; Hebraud, P.; Monari, A.; Assfeld, X. Spectral properties of polypyridyl ruthenium complexes intercalated in DNA: Theoretical insights into the surrounding effects of [Ru(dppz)(bpy)2]2+. Phys. Chem. Chem. Phys. 2012, 14, 12496–12504. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Dumont, E.; Marazzi, M.; Monari, A. Two-photon-absorption DNA sensitization via solvated electrons production: Unraveling the photochemical pathways by molecular modeling and simulation. Phys. Chem. Chem. Phys. 2016, 18, 18598–18606. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.J.; Oppel, M.; González, L. Enhancing intersystem crossing in phenotiazinium dyes by intercalation into DNA. Angew. Chem. Int. Ed. 2015, 54, 4375–4378. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Wibowo, M.; Roca-Sanjuán, D.; Garavelli, M.; Assfeld, X.; Monari, A. Resolving the benzophenone DNA-photosensitization mechanism at QM/MM level. J. Phys. Chem. Lett. 2015, 6, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Vendrell-Criado, V.; Rodríguez-Muñiz, G.M.; Cuquerella, M.C.; Lhiaubet-Vallet, V.; Miranda, M.A. Photosensitization of DNA by 5-methyl-2-pyrimidone deoxyribonucleoside: (6-4) photoproduct as a possible trojan horse. Angew. Chem. Int. Ed. 2013, 52, 6476–6479. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.K.; Sinha, S.S.; Maiti, S.; Pal, S.K. Sequence dependent ultrafast electron transfer of nile blue in oligonucleotides. J. Fluoresc. 2009, 19, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Besancenot, V.; Grandemange, S.; Marazzi, M.; Monari, A. From non-covalent binding to irreversible DNA lesions: Nile blue and nile red as photosensitizing agents. Sci. Rep. 2016, 6, 28480. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zhao, H.; Zhao, Y.; Fang, Y.; Chen, D.; Ren, J.; Wang, X.; Wang, Y.; Gu, Y.; Wu, F. Effective two-photon excited photodynamic therapy of xenograft tumors sensitized by water-soluble bis(arylidene)cycloalkanone photosensitizers. J. Med. Chem. 2015, 58, 7949–7958. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Dumont, E.; Chipot, C.; Monari, A.; Dehez, F. Thermodynamics of DNA: sensitizer recognition. Characterizing binding motifs with all-atom simulations. Phys. Chem. Chem. Phys. 2016, 18, 33180–33186. [Google Scholar] [CrossRef] [PubMed]
- Monari, A.; Rivail, J.L.L.; Assfeld, X. Theoretical modeling of large molecular systems. Advances in the local self consistent field method for mixed quantum mechanics/molecular mechanics calculations. Acc. Chem. Res. 2013, 46, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Interaction of palmatine with DNA: An environmentally controlled phototherapy drug. J. Phys. Chem. B 2015, 119, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.; Van de Rijn, M.; Jeffrey, S.; et al. National Academy of Sciences | Proceedings of the National Academy of Sciences of the United States of America. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Dolenc, J.; Oostenbrink, C.; Koller, J.; van Gunsteren, W.F. Molecular dynamics simulations and free energy calculations of netropsin and distamycin binding to an AAAAA DNA binding site. Nucleic Acids Res. 2005, 33, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Duchanois, T.; Besancenot, V.; Barbieux, C.; Assfeld, X.; Becuwe, P.; Gros, P.C.; Grandemange, S.; Monari, A. Interaction of iron II complexes with B-DNA. Insights from molecular modeling, spectroscopy, and cellular biology. Front. Chem. 2015, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Treesuwan, W.; Wittayanarakul, K.; Anthony, N.G.; Huchet, G.; Alniss, H.; Hannongbua, S.; Khalaf, A.I.; Suckling, C.J.; Parkinson, J.A.; Mackay, S.P. A detailed binding free energy study of 2:1 ligand-DNA complex formation by experiment and simulation. Phys. Chem. Chem. Phys. 2009, 11, 10682–10693. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Schlick, T. Innovations in Biomolecular Modeling and Simulations; Royal Society of Chemistry: London, UK, 2012; pp. 129–155. [Google Scholar]
- Kirkwood, J.G. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-temperature equation of state by a perturbation method. I. nonpolar gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Gumbart, J.C.; Roux, B.; Chipot, C. Standard binding free energies from computer simulations: What is the best strategy? J. Chem. Theory Comput. 2013, 9, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Roux, B.; Chipot, C. Efficient determination of protein-protein standard binding free energies from first principles. J. Chem. Theory Comput. 2013, 9, 3789–3798. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cai, W.; Hénin, J.; Roux, B.; Chipot, C. New coarse variables for the accurate determination of standard binding free energies. J. Chem. Theory Comput. 2017, 13, 5173–5178. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.A.; Munde, M.; Wang, S.; Rettig, M.; Le, V.; MacHha, V.; Wilson, W.D. Complexity in the binding of minor groove agents: Netropsin has two thermodynamically different DNA binding modes at a single site. Nucleic Acids Res. 2011, 39, 9649–9658. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.; Germann, M.W.; Wang, S.; Wilson, W.D. Molecular basis for sequence-dependent induced DNA bending. ChemBioChem 2013, 14, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Marky, L.A.; Blumenfeld, K.S.; Breslauer, K.J. Calorimetric and spectroscopic investigation of drug-DNA interactions. 1. The binding of netropsin to polyd(AT). Nucleic Acids Res. 1983, 11, 2857–2870. [Google Scholar] [CrossRef] [PubMed]
- Kopka, M.L.; Yoon, C.; Goodsell, D.; Pjura, P.; Dickerson, R.E. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc. Natl. Acad. Sci. 1985, 82, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kollman, P.A. Calculating the absolute free energy of association of netropsin and DNA. J. Am. Chem. Soc. 1999, 121, 3267–3271. [Google Scholar] [CrossRef]
- Fu, H.; Shao, X.; Chipot, C.; Cai, W. Extended adaptive biasing force algorithm. An on–the–fly implementation for accurate free-energy calculations. J. Chem. Theory Comput. 2016, 12, 3506–3513. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Lelièvre, T.; Stoltz, G.; Hénin, J. Smoothed biasing forces yield unbiased free energies with the extended-system adaptive biasing force method. J. Phys. Chem. B 2017, 121, 3676–3685. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; Roux, B. Calculation of absolute protein–ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 6825–6830. [Google Scholar] [CrossRef] [PubMed]
- Hermans, J.; Wang, L. Inclusion of loss of translational and rotational freedom in theoretical estimates of free energies of binding. Application to a complex of benzene and mutant T4 lysozyme. J. Am. Chem. Soc. 1997, 119, 2707–2714. [Google Scholar] [CrossRef]
- Dixit, S.B.; Chipot, C. Can absolute free energies of association be estimated from molecular mechanical simulations? The biotin-streptavidin system revisited. J. Phys. Chem. A 2001, 105, 9795–9799. [Google Scholar] [CrossRef]
- Mobley, D.L.; Chodera, J.D.; Dill, K.A. Confine-and-release method: obtaining correct binding free energies in the presence of protein conformational change. J. Chem. Theory Comput. 2007, 3, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Velez-Vega, C.; Gilson, M.K. Overcoming dissipation in the calculation of standard binding free energies by ligand extraction. J. Comput. Chem. 2013, 34, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H. AMBER16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An nlog(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contribution | PMF (kcal/mol) | Simulation Time (ns) |
|---|---|---|
| 445 | ||
| 14 | ||
| 10 | ||
| 10 | ||
| 18 | ||
| 14 | ||
| ln(SIC) | 245 | |
| 215 | ||
| +7.9 | - | |
| 971 | ||
| (exp) [58] | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Gattuso, H.; Dumont, E.; Cai, W.; Monari, A.; Chipot, C.; Dehez, F. Accurate Estimation of the Standard Binding Free Energy of Netropsin with DNA. Molecules 2018, 23, 228. https://doi.org/10.3390/molecules23020228
Zhang H, Gattuso H, Dumont E, Cai W, Monari A, Chipot C, Dehez F. Accurate Estimation of the Standard Binding Free Energy of Netropsin with DNA. Molecules. 2018; 23(2):228. https://doi.org/10.3390/molecules23020228
Chicago/Turabian StyleZhang, Hong, Hugo Gattuso, Elise Dumont, Wensheng Cai, Antonio Monari, Christophe Chipot, and François Dehez. 2018. "Accurate Estimation of the Standard Binding Free Energy of Netropsin with DNA" Molecules 23, no. 2: 228. https://doi.org/10.3390/molecules23020228
APA StyleZhang, H., Gattuso, H., Dumont, E., Cai, W., Monari, A., Chipot, C., & Dehez, F. (2018). Accurate Estimation of the Standard Binding Free Energy of Netropsin with DNA. Molecules, 23(2), 228. https://doi.org/10.3390/molecules23020228