2. Results and Discussion
Compound
1 was obtained as a white, amorphous powder. Its (−)- and (+)-ESI-MS displayed quasi-molecular ion peaks at
m/
z 1087 [M − H]
− and 1111 [M + Na]
+, respectively, corresponding to the molecular formula of C
53H
84O
23, which was confirmed by further HR-ESI-MS (+) analysis ([M + Na]
+ m/
z 1111.5297, calcd. 1111.5296). The
1H- and
13C-NMR spectra of
1 exhibited four sugar anomeric protons at δ
H 6.22 (Glc-I-1), 4.95 (Glc-II-1), 5.87 (Rha-1), and 5.24 (Xyl-1) (
Table 1), and anomeric carbons at δ
C 95.5, 104.7, 102.2 and 107.1 (
Table 2), suggesting the existence of four sugar moieties in the molecule. Acid hydrolysis of
1 with 2N HCl afforded
d-glucose,
l-rhamnose and
d-xylose in the ratio of 2:1:1, which were identified by GC-MS analysis of their chiral derivatives (see experimental part). The
1H- and
13C-NMR assignments (
Table 1 and
Table 2) of the sugar moieties in
1 were established by interpretation of combined HSQC and HMBC data (See the
Supplementary Materials). After having excluded the resonances due to the sugar moieties, the remaining signals in the
1H-NMR spectrum for the aglycone unit of
1 were readily recognized for six tertiary methyls at δ
H 1.42 (3H, s), 1.20 (3H, s), 1.07 (3H, s), 1.04 (3H, s), 0.90 (3H, s) and 0.88 (3H, s), an olefinic proton at δ
H 5.40 (1H, t), two oxymethine protons at δ
H 4.21 (1H, m) and 4.04 (1H, d,
J = 10.1 Hz), and an aldehyde proton at δ
H 9.61 (1H, s). The
13C-NMR spectrum indicated, besides the signals for the sugar moieties, 30 carbons for the aglycone unit, including six methyls, nine methylenes, six methines (including an olefinic methine at δ
C 122.4, two oxygenated methines at δ
C 67.9 and 76.9 and an aldehyde carbon at δ
C 206.3), and nine quaternary carbons (including an olefinic carbon at δ
C 144.0 and a carbonyl carbon at δ
C 176.3). By comparison, it was found that the
1H- and
13C-NMR spectroscopic data (
Table 1 and
Table 2) of the aglycone of
1 were closely related to those of 2α,3β-dihydroxy-23-oxo-olean-12-en-28-oic acid [
11], a known triterpene which was also obtained in this study as
5, with the major difference of the chemical shift value of the carboxyl group at C-28 was shifted from δ
C 180.0 in
5 to δ
C 176.3 in
1. These findings supported the fact that
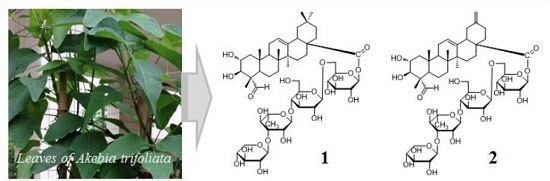
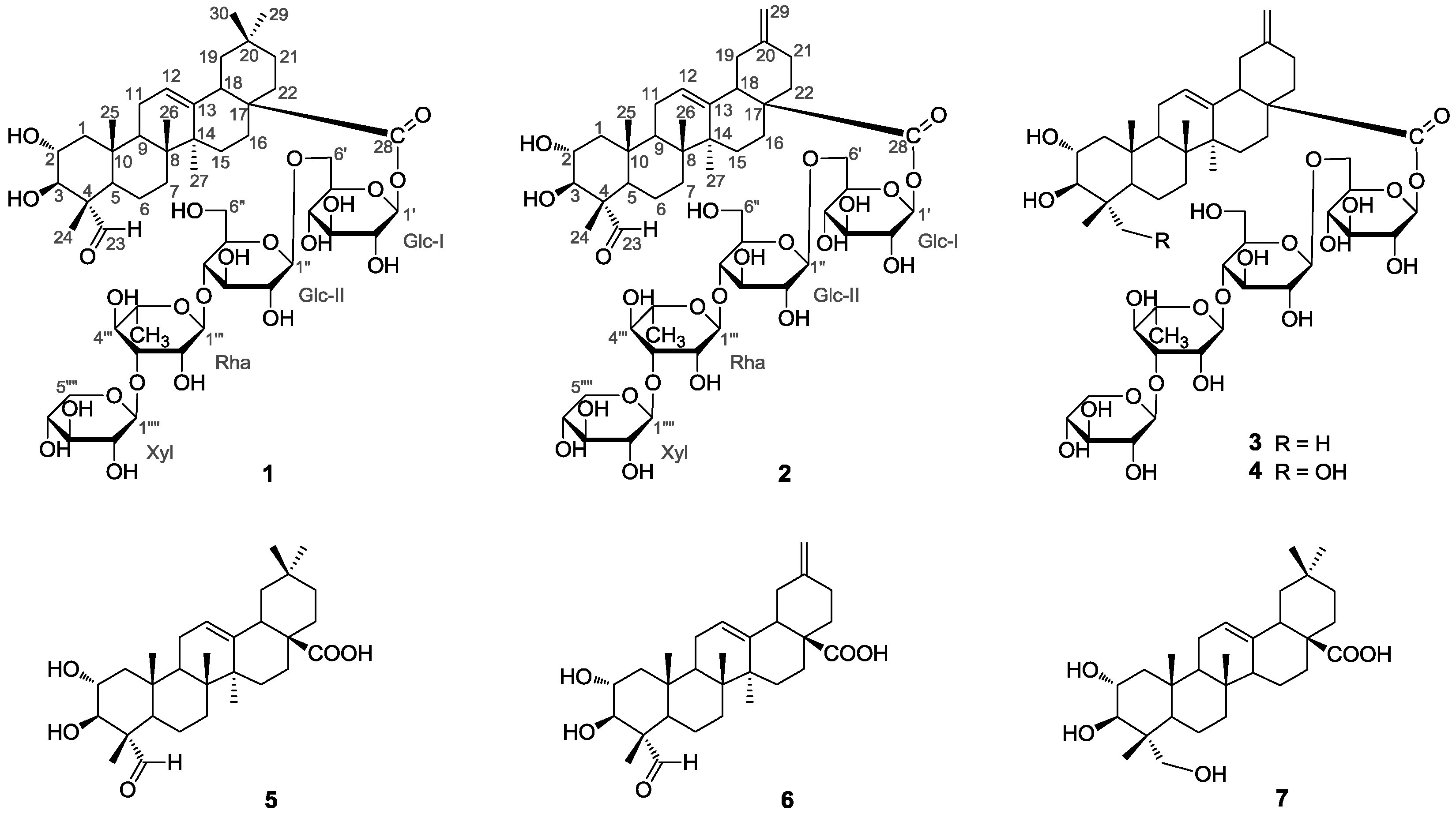
1, as shown in
Figure 1, was a monodesmoside saponin of
5 with an oligosaccharide chain linked at C-28 [
12]. This deduction was consistent with the molecular formula of
1, and further in complete accordance with the 2D NMR spectroscopic data. In the HMBC spectrum,
1H-
13C long-range correlation of H-1′ (δ
H 6.22) with C-28 (δ
C 176.3) was exhibited, which evidenced the glycoside linkage of Glc-I at C-1′ with the aglycone at C-28. The observation of HMBC correlations of H-1″ (δ
H 4.95) with C-6′ (δ
C 69.0), of H-1″′ (δ
H 5.87) with C-5″ (δ
C 77.0), and of H-1″″ (δ
H 5.24) with C-3″′ (δ
C 83.0) confirmed the sugar sequence as shown in
Figure 2. The β-anomeric configuration of the
d-glucose (Glc-I and Glc-II) and the xylose (Xyl) moieties were determined on the basis of their coupling constants of
3JH1′,H2′ (8.1 Hz),
3JH1″,H2″ (7.8 Hz) and
3JH1″″,H2″″ (7.5 Hz), respectively [
12,
13].
The α-configuration of the
l-rhamnose (Rha) unit was evidenced by the singlet signal δ
H 5.87 (br.s) observed for the anomeric proton H-1″′ [
12]. Therefore,
1 was identified as 2α,3β,23,29-tetrahydroxyolean-12-en-28-oic acid-
O-β-
d-xylpyranosyl-(1 → 3)-
O-α-
l-rhamnopyranosyl-(14)-
O-β-
d-glucopyranosyl-(1 → 6)-β-
d-glucopyranosyl ester (
1), trivially named akebiaoside K.
Compound
2 was obtained as a white amorphous powder with molecular formula C
52H
80O
23 as determined by HR-ESI-MS analysis ([M + Na]
+ m/
z 1095.4965, calcd. 1095.4983). Its spectral features and physicochemical properties suggested
2 was also a triterpenoid saponin. Careful comparison revealed that the
1H- and
13C-NMR spectral data of the aglycone part of
2 were quite close to those of 2α,3β-dihydroxy-23-oxo-30-norolean-12,20(29)-dien-28-oic acid [
14], a known noroleanane triterpene which was also obtained in this study as compound
6, suggesting that the aglycone unit of
2 was the same as
6. This assignment was supported by the acid hydrolysis of
2 which furnished the free aglycone identified as
6, and the monosaccharide compounents concomitantly obtained from the acid hydrolysis of
2 were identified as
d-glucose,
l-rhamnose and
d-xylose in the ratio of 2:1:1 based on GC-MS analysis of their chiral derivatives (see experimental section), supporting the presence of the same four sugar moieties as those in
1. On the basis of combined analysis of
1H-
1H COSY, HMQC and HMBC spectra, all proton and carbon signals were assigned (
Table 1 and
Table 2). The β-anomeric configuration of the
d-glucose units (Glc-I and Glc-II) and the xylose (Xyl) moiety were determined from their coupling constants of
3JH1′,H2′ (8.2 Hz),
3JH1″,H2″ (7.9 Hz) and
3JH1″″,H2″″ (7.5 Hz), respectively [
12]. And the α-anomeric configuration of the
l-rhamnose unit was judged by the singlet signal at δ
H 5.82 (br.s) for the anomeric proton H-1″′ [
12]. In the HMBC spectrum,
1H-
13C long-range correlation of H-1′ (δ
H 6.14) with C-28 (δ
C 175.6) was displayed, which evidenced the glycoside linkage of Glc-I at C-1′ with the aglycone at C-28. The exhibition of obvious HMBC correlations of H-1′′ (δ
H 4.91) with C-6′ (δ
C 69.1), of H-1″′ (δ
H 5.82) with C-5″ (δ
C 76.9), and of H-1″″ (δ
H 5.22) with C-3″′ (δ
C 83.0) confirmed the oligosaccharide sequence in
2 as shown in
Figure 2. Based upon the above information, the strcuture of compound
2 was elucidated as 2α,3β-dihydroxy-23-oxo-29-norolean-12,20(30)-dien-28-oic acid
O-β-
d-xylpyranosyl-(1 → 3)-
O-α-
l-rhamnopy-ranosyl-(1 → 4)-
O-β-
d-glucopyranosyl-(1 → 6)-β-
d-glucopyranosyl ester (
2), trivially named akebiaoside N.
The five known compounds were identified as akemisaponin I (
3) [
7], mutongsaponin B (
4) [
6], 2α,3β-dihydroxy-23-oxo-olean-12-en-28-oic acid (
5) [
11], 2α,3β-dihydroxy-23-oxo-30-norolean-12,20 (29)-dien-28-oic acid (
6) [
14] and arjunolic acid (
7) [
15], by comparison of their spectral data (
1H- and
13C-NMR and MS) to those reported in the literature. They were all isolated from the leaves of
A. trifoliata for the first time.
Compounds
1–
7 were evaluated for their in vitro cytotoxicity against human cancer cell lines A549, HeLa and HepG2, using a MTT method as described. The resulting IC
50 values are displayed in
Table 3, compared to adriamycin as positive control. Compounds
5 and
7 showed moderate cytotoxicity against all the three cancer cell lines, with IC
50 values ranging from 22.7 to 38.1 μM. Meanwhile, no obvious activity was detected for the four saponins
1–
4. This result reveals an obvious negative effect on the cytotoxicity when the triterpenes were linked with the linear tetrasaccharide chain at C-28. Comparison of the chemical structures and their cytotoxicity of
5 versus
6 indicated that the replacement of the structure fragment C-20(Me)
2 by the exocyclic double bond of C-20(29) also had a negative effect on the cytotoxicity.
These compounds were further tested for their α-glucosidase inhibitory activity with acarbose used as a reference compound. Compounds
5–
7 were revealed to show strong α-glucosidase inhibitory activity, with IC
50 values of 0.047, 0.220 and 0.040 mM, respectively, which were about two to ten-fold stronger than acarbose (IC
50 0.409 mM). These results suggested that triterpenes
5–
7 from the leaves of
A. trifoliata are effective α-glucosidase inhibitors with potential value for development as effective hypoglycemic agents for diabetes chemotherapy [
16]. Like in the cytotoxicity bioassay, no obvious α-glucosidase inhibitory activity was detected for the four triterpenoid saponins
1–
4, indicating that a negative effect of the tetrasaccharide chain linked at C-28 on the α-glucosidase inhibitory activity was also evident.
A. trifoliata is a medicinal plant naturally widely distributed in the eastern part of Asia. Its air-dried stems and fruits have long been used in China as an antiphlogistic, antineoplastic and diuretic agent. To date phytochemical investigations have revealed a series of triterpenes and triterpene saponins from this plant species. However, those studies were mainly focused on the stems and fruits, and seldom concentrated on the leaves. Our present study revealed that the leaves of A. trifoliata is also rich in triterpenes and triterpene saponins. At the same time, however, it is also demonstrated that much future work is needed to unravel the complexity of the chemical constituents in the leaves of A. trifoliata.
3. Materials and Methods
3.1. General Information
Optical rotation were measured on a Perkin-Elmer 341 polarimeter (Perkin-Elmer, Waltham, MA, USA) with MeOH as solvent at the wavelength of 589 nm and 20 °C to obtain their specific optical rotation [α] values after calculation. ESI-MS data were obtained using a MDS SCIEX API 2000 LC/MS/MS system (Applied Biosystems, Foster City, CA, USA) in both positive and negative ion modes in the range of m/z 50–1000 after the test solutions were directly injected into the ESI source by a syringe pump. HR-ESI-MS mass spectra were obtained on a Bruker maXis instrument (Bruker Daltonik GmbH, Bremen, Germany) in a positive ion mode after direct injection of the test solutions. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Advance 600 instrument (Bruker, Fällanden, Switzerland) or Bruker Ascend-500 spectrometer (Bruker BioSpin GmbH, Rheistetten, Germany). Preparative HPLC was carried out on a CXTH P3000 HPLC pump and a UV 3000 UV-Vis Detector with a Fuji-C18 column (10 μm–100 A, ChuangXinTongHeng Science and Technology Co., Ltd., Beijing, China); medium pressure liquid chromatography (MPLC) was performed using a CXTH P3000 HPLC pump, a UV 3000 UV-Vis Detector and a C18 column (400 × 25 mM i.d., 50 μM, YMC Co. Ltd., Kyoto, Japan).
Column chromatography (CC) was performed with silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Qingdao, China), YMC ODS-A (50 μm, YMC Co. Ltd.), and Sephadex LH-20 (Pharmacia Fine Chemical Co., Ltd., Uppsala, Sweden). Analytical grade petroleum ether (b.p. 60–90 °C), methanol, ethyl acetate, chloroform, n-butanol, acetone were purchased from Tianjin Fuyu Fine Chemical Industry Co. (Tianjin, China); HPLC grade methanol was purchased from J & K Chemical Ltd. (Beijing, China); Fractions were monitored by pre-coated HSGF254 TLC (Yantai Jiangyou Silica Gel Co., Ltd., Yantai, China), and spot detection was performed under fluorescent light (λ = 254 nm), and then spraying 10% H2SO4 in ethanol, followed by heating. Pyridine-d5, DMSO-d6, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and α-glucosidase were purchased from Sigma Chemical Co. (Sigma-Aldrich, St. Louis, MO, USA). RPMI-1640 medium and fetal calf serum were purchased from Gibco BRL (Gaithersburg, MD, USA). Adriamycin was obtained from Pfizer Italia SRL (Roma, Italy). p-Nitrophenyl-α-d-glucopyranoside (PNPG) and acarbose were obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan).
3.2. Plant Materials
The leaves of Akebia trifoliata were collected in August 2014, at Sangzhi, Hunan Province, China, were identified by Prof. Fu-Wu Xing at the South China Botanical Garden, the Chinese Academy of Sciences (CAS). A voucher specimen (No. 20140815) was deposited at the Laboratory of Bioorganic Chemistry of the South China Botanical Garden, Chinese Academy of Sciences.
3.3. Extraction and Isolation
Powdered air-dried leaves of A. trifoliate (dried weight 3.5 kg) were extracted three times with 95% EtOH (10 L × 3) at room temperature for 2 days each time. The extract was concentrated in vacuo to give a dark brown residue, which was suspended in 3 L of water and then sequentially extracted with petroleum ether (3 L × 3), ethyl acetate (EtOAc, 3 L × 3) and n-butanol (n-BuOH, 3 L × 3) to yield a petroleum ether-soluble fraction (31 g), an EtOAc-soluble fraction (168 g), and a n-BuOH-soluble fraction (245 g) after condensation to dryness in vacuo. The EtOAc-soluble fraction was subjected to silica gel CC (1000 mm × 105 mm i.d.) eluted with a gradient of CHCl3–MeOH (97:3–0:100, v/v) to give fractions F1–F10 after pooled according to their TLC profiles. Fraction F5 (7.1 g), obtained from the elution of CHCl3/MeOH of 85:15 (v/v), was subjected to a silica gel CC (800 × 50 mm i.d.) and successively eluted with CHCl3/MeOH (98:2–90:10, v/v) to yield sub-fractions F5-1–F5-6. Sub-fraction F5-3 (1.57 g) was separated by MPLC using MeOH/H2O (60:40–100:0, v/v) system at a flow rate of 10 mL/min, and further purified by passing through a Sephadex LH-20 column (1500 mm × 25 mm i.d.) eluted with MeOH to obtain compounds 5 (5.2 mg) and 6 (3.8 mg). Sub-fraction F5-5 (2.3 g) was separated by MPLC using a gradient of MeOH/H2O (30:70–80:20, v/v) at a flow rate of 10 mL/min to obtain six sub fractions (F5-5-1–F5-5-6), and sub fraction F5-5-5 was purified by preparative HPLC with a Fuji-C18 column (10 μm–100 A) using 73% methanol in water (v/v) as a mobile phase at flow rate of 8 mL/min to give compound 7 (15.6 mg).
The n-BuOH-soluble fraction (245 g) was dissolved in water and passed through a HP-20 column eluted with distilled water and 90% EtOH. Column chromatography of the 90% EtOH eluate portion (128 g) on silica gel (200–300 mesh) and elution with a gradient system of CHCl3/MeOH (85:15–0:100, v/v) to give six fractions (G1–G6) after pooled according to their TLC profiles. Fraction G3 (18 g), obtained from the elution of CHCl3/MeOH of 85:15 (v/v), was subjected to a silica gel CC (600 × 50 mm i.d.) and successively eluted with CHCl3/MeOH (85:15–40:60, v/v) to yield five sub-fractions (G3-1–F3-4). Sub-fraction G3-2 (2.7 g) was separated by MPLC using a gradient of MeOH/H2O (20:80–100:0, v/v) at a flow rate of 10 mL/min to obtain nine sub fractions (G3-2-1–G3-2-9), and G3-2-2 was purified by preparative HPLC with a Fuji-C18 column (10 μm–100 A) using a gradient of MeOH/H2O (50:50–70:30, v/v) as a mobile phase at flow rate of 10 mL/min to give compound 2 (6.4 mg). G3-2-6 was purified by preparative HPLC with a Fuji-C18 column (10 μm–100 A) using a gradient of methanol in water (45:55–63:37, v/v) as a mobile phase at flow rate of 10 mL/min to give compound 3 (10 mg). G3-2-7 was purified by a Sephadex LH-20 column (1500 mm × 25 mm i.d.) eluted with MeOH to afford compound 1 (12 mg). Fraction G5 was subjected to CC on silica gel (200–300 mesh) eluted with a gradient of CHCl3/MeOH (80:20–40:60) and Sephadex LH-20 with MeOH to furnish 4 (4.1 mg).
Compound
1. White amorphous powder.
: −7.3 (
c 0.33, MeOH). ESI-MS (+)
m/z: 1111 [M + Na]
+; ESI-MS (−)
m/
z: 1087 [M − H]
−. HR-ESI-MS (pos.)
m/
z: 1111.5297 (calcd for C
53H
84NaO
23, 1111.5296). For
1H-NMR (600 MHz, C
5D
5N) and
13C-NMR (150 MHz, C
5D
5N) data, see
Table 1 and
Table 2.
Compound
2. White amorphous powder.
: +5.03 (
c 0.34, MeOH). HR-ESI-MS (pos.)
m/
z: 1095.4965 (calcd for C
52H
80NaO
23, 1095.4983). For
1H-NMR (600 MHz, C
5D
5N) and
13C-NMR (150 MHz, C
5D
5N) data, see
Table 1 and
Table 2.
3.4. Acid Hydrolysis of 1 and 2
Each of compounds of 1 and 2 (3 mg) was heated in 2 M HCl (4 mL) at 90 °C for 2 h. The reaction mixture was extracted with EtOAc (3 × 4 mL). The EtOAc extract was purified by passing through a Sephadex LH-20 column (1500 mm × 25 mm i.d.) eluted with MeOH. By TLC comparison, the free aglycone of 1 was determined to be identical as 5, while that of 2 was identified as the same as 6, respectively. The aqueous layer was concentrated under reduced pressure to dryness to give a sugar-containing residue, which was reacted with l-cysteine methyl ester hydrochloride in pyridine at 60 °C for 2 h, then added with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) and stirred under reflux at 60 °C for 10 h. The supernatant was then analyzed by GC–MS using a GCMS-QP2010 PLUS instrument, HP-5ms capillary column (30 m, 0.25 mm ID), Helium at constant rate of 46.5 cm/s, 1 µL injection volume, injector temperature at 230 °C, temperature program as 2 °C/min to 180 °C, then 20 °C/min to 280 °C. Electron ionization mode was used at 70 eV. In the acid hydrolysate of 1 and 2, d-glucose, l-rhamnose and d-xylose were confirmed by comparison of their retention times of their derivatives with those of authentic d-glucose (tR 11.852 min), l-rhamnose (tR 5.292 min) and d-xylose (tR 6.842 min) derivatives prepared in the same way, respectively.
3.5. Cytotoxic Assay
Compounds
1–
7 were testeded for their cytotoxity against A549 (human lung adenocarcinoma), HeLa (human cervical carcinoma) and HepG2 (human liver hepatocellular carcinoma) cell lines. The three tumor cell lines were obtained from Kunming Institute of Zoology, Chinese Academy of Sciences. The cytotoxic activity of tested chemicals were assayed according to the MTT method using 96 well plates [
17]. Briefly, the cells were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO
2 at 37 °C. 100 μL adherent cells at the density of 5 × 10
4 cell/mL was seeded into each well of 96-well cell culture plates and incubated in 5% CO
2 at 37 °C for 24 h to form a monolayer on the flat bottoms. Then, the supernatant per well was removed and subsequently added with 100 μL fresh medium and 100 μL medium containing a test compound. The plate was then incubated in 5% CO
2 at 37 °C. After 72 h, 20 μL of 5 mg/mL MTT in DMSO was added into each well and incubated for 4 h. The supernatant per well was carefully removed and 150 μL DMSO was added. The plate was then vortex shaken for 15 min to dissolve blue formazan crystals. The optical density (OD) of each well was measured on a Genois microplate reader (Tecan GENios, Männedorf, Switzerland) at the wavelength of 570 nm. All experiments were performed in triplicate and adriamycin was used as a positive control. In each experiment, each of the tumor cell lines was exposed to the test compound at concentrations 50, 25, 12.5, 6.25, 3.125, 1.5625 µg/mL. The inhibitory rate of cell growth was calculated according to the following formula: Inhibition rate (%) = (OD
control − OD
treated)/OD
control × 100%. IC
50 values were calculated by SPSS 16.0 statistic software. The values were based on three individual experiments and expressed as means ± standard deviation (SD).
3.6. α-Glucosidase Inhibition Assay
The
a-glucosidase inhibitory activity of
1–
7 were determined spectrophotometrically in a 96-well microtiter plate based on
p-nitrophenyl-α-
d-glucopyranoside (PNPG) as a substrate following the method described in literature with slight modifications [
18,
19]. In brief, α
-glucosidase (20 μL, 0.8 U/mL) and various concentrations (500, 250, 125, 62.5, 31.25, 15.625 µg/mL) of tested compounds (120 μL) in 67 mM phosphate buffer (pH 6.8) were mixed at room temperature for 10 min. Reactions were initiated by addition of 5.0 mM PNPG (20 μL). The reaction mixture was incubated for 15 min at 37 °C in a final volume of 160 μL. Then, 0.2 M Na
2CO
3 (80 μL) was added to the incubation solution to stop the reaction. The activities were detected in a 96-well plate, and the absorbance was determined at 405 nm (for
p-nitrophenol). The negative blank was set by adding phosphate buffer instead of the sample via the same way as the test. Acarbose was utilized as positive control. The blank was set by adding phosphate buffer instead of the α
-glucosidase using the same method. Inhibition rate (%) = [(OD
negative control − OD
blank) − (OD
test − OD
test blank)]/(OD
negative blank − OD
blank) × 100%. IC
50 values of the samples were calculated using the Microsoft Office Excel soft.




{kind=link}
{kind=link}
{kind=link}