
The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation

Abstract
:1. Introduction
2. Chemistry and Pharmacokinetics
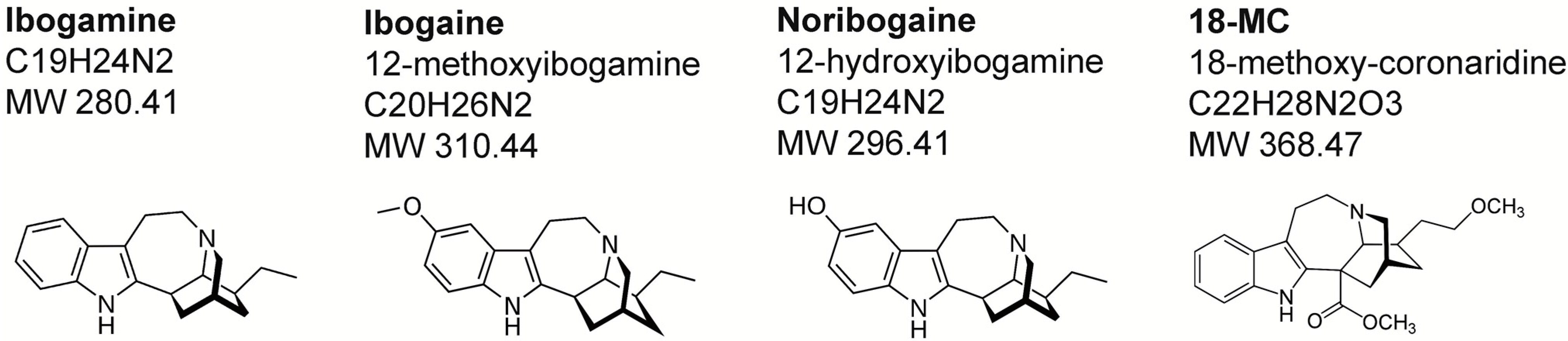

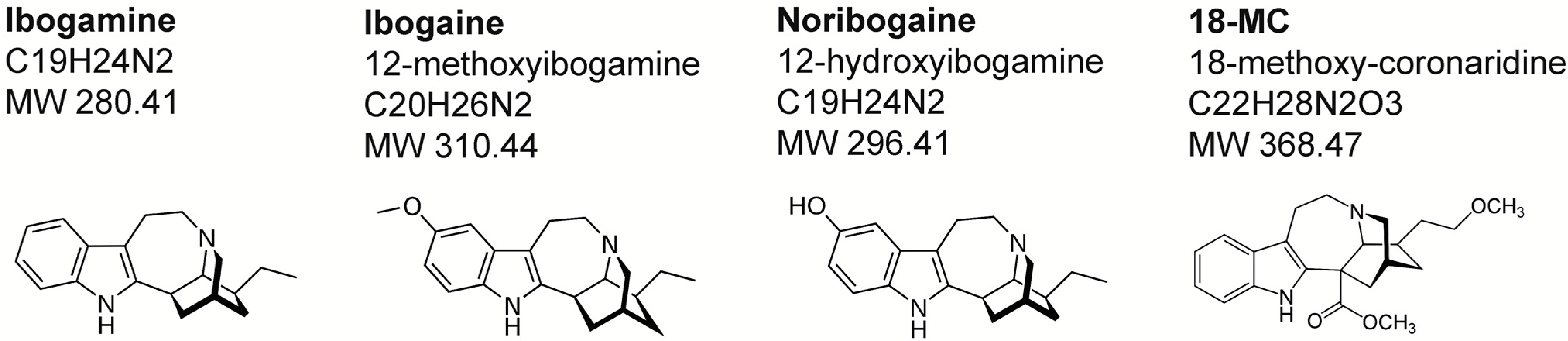{kind=link}
{kind=link}
| Age (years) | Sex | Year of Publication | Time after Drug Intake | Diagnosis | Dosage (mg/kg) | Ibogaine (blood, mg/L) $ | Electrolyte Levels | QTc (ms) | History/Pathology other Intoxication | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 24–54 | m+f | 1990–2007 | 1.5–76 h | sudden death | 4.5–29.0 | 0.24–9.3 | na | na | cardiovascular diseases, hepatitis, liver cirrhosis, opiates, cocaine, alcohol, diazepam | [1] |
| 52 | m | 2013 | 12–24 h | sudden death | na | 2 | low K, Mg # | na | alcoholism, atherosclerosis, liver cirrhosis na | [24] |
| 27 | m | 2013 | 12 h | sudden death | 1.5–2.2 | 0.65–1.27 | na | na | multiple substance addiction, no cardiac pathology no methadone >48 h, diazepam | [25] |
| 25 | m | 2013 | 48 h | sudden death | 6.25 * | na | na | na | heroin addiction, supraventricular tachycardia na | [26] |
| 31 | f | 2009 | long QT, VT | 8.75 * | na | low K, Mg | 616 | alcohol addiction no alcohol, no other drugs | [21] | |
| 49 | m | 2012 | long QT, VT, TdP | na | na | low K | >700 | heroin addiction, hyperthyroidism traces of opioids | [27] | |
| 31 | f | 2012 | long QT, TdP | 8.75 * | na | low K, Mg | 616 | alcohol addiction no other medication | [27] | |
| 43 | f | 2012 | long QT | na | 0.37 | low K | 480 | heroin/benzodiazepin addiction, on methadone methadone | [27] | |
| 33 | m | 2012 | long QT, VF | 10 * | 0.68 | na | 593 | na no cocaine, heroin, methadone > 48 h | [28] | |
| 63 | m | 2012 | long QT, VT, TdP | 10.5 § | na | low K | 498 | heroin addiction short-acting opioids # | [29] | |
| young | m | 2013 | long QT, VT, TdP | 17.5 | na | na | 600 | na no alcohol; no heroin, methadone for >72 h | [30] | |
| 26 | m | 2014 | long QT, VT, VF | 35 | 0.95 | low K, Mg | 663 | healthy no other drugs | [31] |
3. Effects on the Cardiovascular System
3.1. Cardiac Electrophysiology
3.1.1. Ibogaine Effects on the Heart Rate
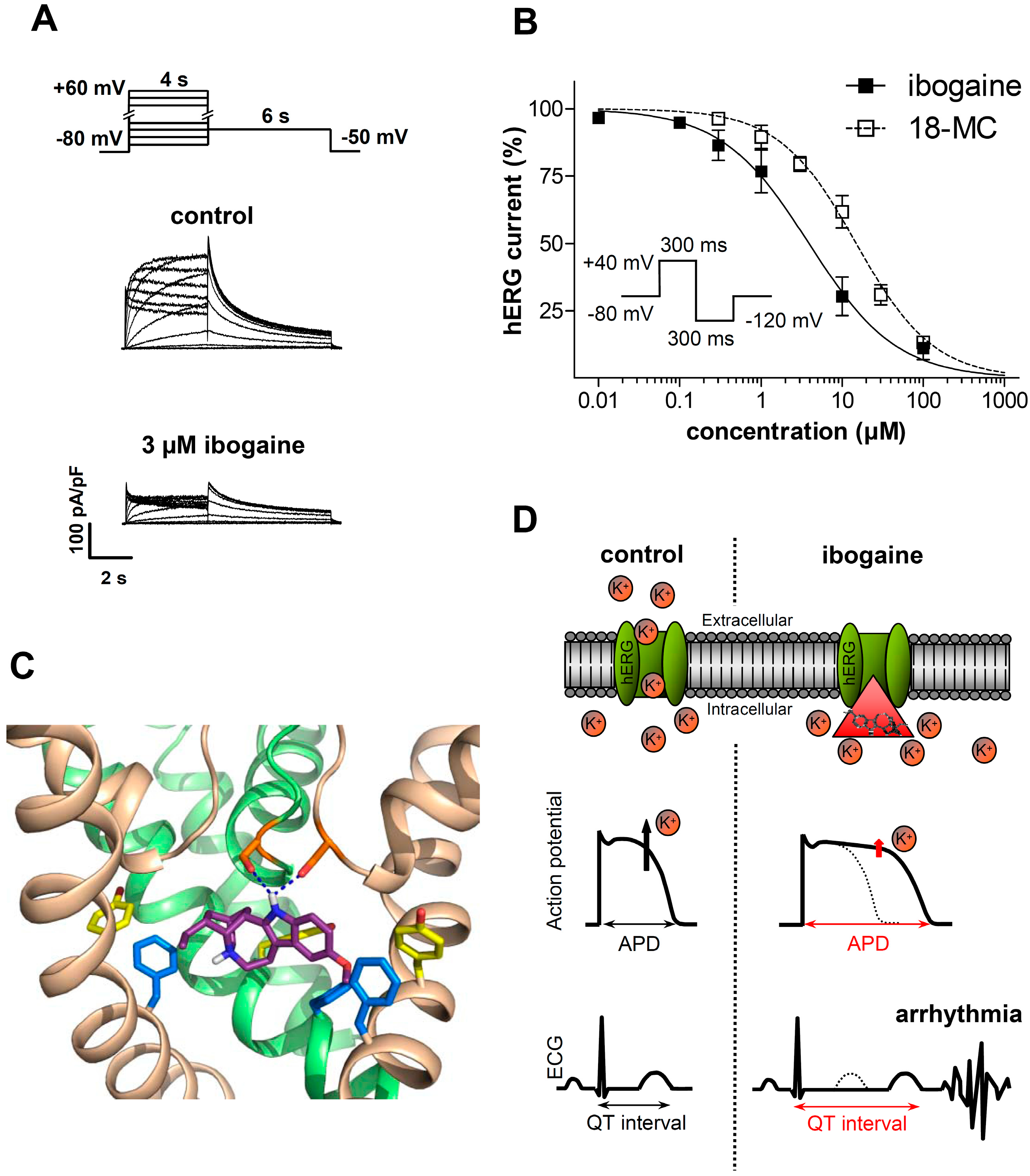
3.1.2. Ibogaine Effects on Cardiac Ion Channels
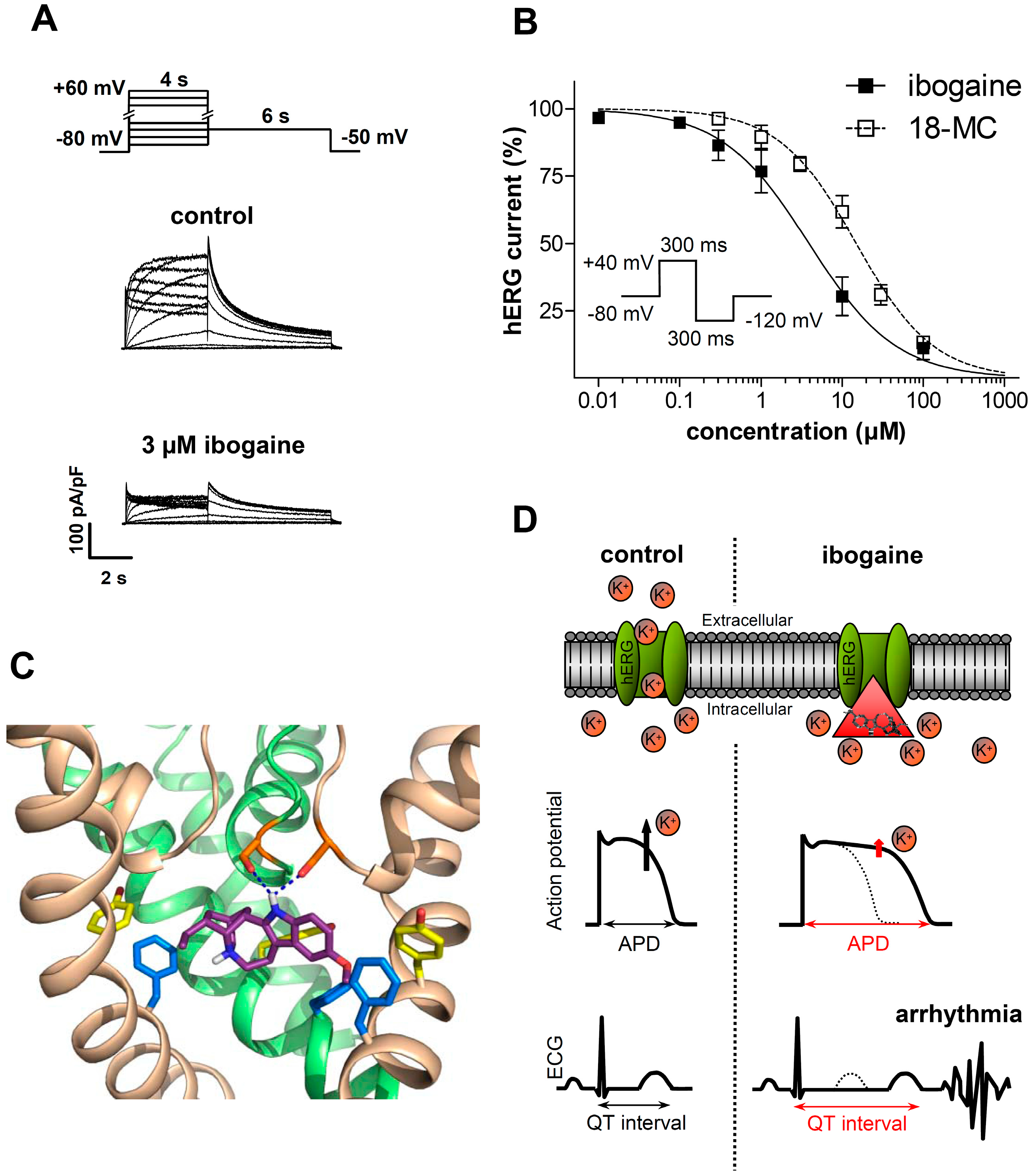
3.2. Clinical Evidence of Cardiotoxicity
3.2.1. Clinical Reports
3.2.2. Risk Factors
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alper, K.R.; Stajic, M.; Gill, J.R. Fatalities Temporally Associated with the Ingestion of Ibogaine. J. Forensic. Sci. 2012, 57, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Alper, K.R. Ibogaine: A review. Alkaloids Chem. Biol. 2001, 56, 1–38. [Google Scholar] [PubMed]
- Glick, S.D.; Maisonneuve, I.S. Mechanisms of antiaddictive actions of ibogaine. Ann. N. Y. Acad. Sci. 1998, 844, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Glick, S.D.; Maisonneuve, I.M.; Szumlinski, K.K. 18-Methoxycoronaridine (18-MC) and ibogaine: Comparison of antiaddictive efficacy, toxicity, and mechanisms of action. Ann. N. Y. Acad. Sci. 2000, 914, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Maciulaitis, R.; Kontrimaviciute, V.; Bressolle, F.M.; Briedis, V. Ibogaine, an anti-addictive drug: Pharmacology and time to go further in development. A narrative review. Hum. Exp. Toxicol. 2008, 27, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.K. Ibogaine in the treatment of substance dependence. Curr. Drug Abuse Rev. 2013, 6, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Pablo, J.P.; Ali, S.F.; Rothman, R.B.; Mash, D.C. Noribogaine (12-hydroxyibogamine): A biologically active metabolite of the antiaddictive drug ibogaine. Ann. N. Y. Acad. Sci. 2000, 914, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Rothman, R.B.; Pablo, J.P.; Mash, D.C. In vivo neurobiological effects of ibogaine and its O-desmethyl metabolite, 12-hydroxyibogamine (noribogaine), in rats. J. Pharmacol. Exp. Ther. 2001, 297, 531–539. [Google Scholar] [PubMed]
- Mash, D.C.; Kovera, C.A.; Pablo, J.; Tyndale, R.F.; Ervin, F.D.; Williams, I.C.; Singleton, E.G.; Mayor, M. Ibogaine: Complex pharmacokinetics, concerns for safety, and preliminary efficacy measures. Ann. N. Y. Acad. Sci. 2000, 914, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Rinehart, R.K. Analysis of the cardiovascular action of ibogaine hydrochlorid. Arch. Int. Pharmacodyn. Ther. 1957, 110, 92–102. [Google Scholar] [PubMed]
- Vastag, B. Addiction research. Ibogaine therapy: A “vast, uncontrolled experiment”. Science 2005, 308, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Antonio, T.; Childers, S.R.; Rothman, R.B.; Dersch, C.M.; King, C.; Kuehne, M.; Bornmann, W.G.; Eshleman, A.J.; Janowsky, A.; Simon, E.R.; et al. Effect of Iboga alkaloids on micro-opioid receptor-coupled G protein activation. PLoS One 2013, 8, e77262. [Google Scholar] [CrossRef]
- National Institutes of Health. (2013) ND-Enabling Studies and GMP Scale-Up of 18-Methoxycoronaridine Hydrochloride (18 MC) Project Number: 1U01DA034986-01. Available online: http://projectreporter.nih.gov/project_info_description.cfm?aid=8448461&icde=16047111&ddparam=&ddvalue=&ddsub=&cr=41&csb=default&cs=ASC (accessed on 28 September 2013).
- Glick, S.D.; Kuehne, M.E.; Maisonneuve, I.M.; Bandarage, U.K.; Molinari, H.H. 18-Methoxycoronaridine, a non-toxic iboga alkaloid congener: Effects on morphine and cocaine self-administration and on mesolimbic dopamine release in rats. Brain Res. 1996, 719, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Glick, S.D.; Maisonneuve, I.M.; Szumlinski, K.K. Mechanisms of action of ibogaine: Relevance to putative therapeutic effects and development of a safer iboga alkaloid congener. Alkaloids Chem. Biol. 2001, 56, 39–53. [Google Scholar] [PubMed]
- Glick, S.D.; Sell, E.M.; McCallum, S.E.; Maisonneuve, I.M. Brain regions mediating alpha3beta4 nicotinic antagonist effects of 18-MC on nicotine self-administration. Eur. J. Pharmacol. 2011, 669, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Kovar, M.; Rubi, L.; Mike, A.K.; Lukacs, P.; Gawali, V.S.; Todt, H.; Hilber, K.; Sandtner, W. Anti-addiction drug ibogaine inhibits voltage-gated ionic currents: A study to assess the drug’s cardiac ion channel profile. Toxicol. Appl. Pharmacol. 2013, 273, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Molinari, H.H.; Maisonneuve, I.M.; Glick, S.D. Ibogaine neurotoxicity: A re-evaluation. Brain Res. 1996, 737, 255–262. [Google Scholar] [CrossRef] [PubMed]
- O’Hearn, E.; Molliver, M.E. Degeneration of Purkinje cells in parasagittal zones of the cerebellar vermis after treatment with ibogaine or harmaline. Neuroscience 1993, 55, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chang, L.W.; Slikker, W.; Ali, S.F.; Rountree, R.L.; Scallet, A.C. A dose-response study of ibogaine-induced neuropathology in the rat cerebellum. Toxicol. Sci. 2000, 57, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Hoelen, D.W.; Spiering, W.; Valk, G.D. Long-QT syndrome induced by the antiaddiction drug ibogaine. N. Engl. J. Med. 2009, 360, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; Lynch, W.G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.; Strang, I.; Sullivan, A.T.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Papadodima, S.A.; Dona, A.; Evaggelakos, C.I.; Goutas, N.; Athanaselis, S.A. Ibogaine related sudden death: A case report. J. Forensic Leg. Med. 2013, 20, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Mazoyer, C.; Carlier, J.; Boucher, A.; Peoc’h, M.; Lemeur, C.; Gaillard, Y. Fatal case of a 27-year-old male after taking iboga in withdrawal treatment: GC-MS/MS determination of ibogaine and ibogamine in iboga roots and postmortem biological material. J. Forensic Sci. 2013, 58, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Jalal, S.; Daher, E.; Hilu, R. A case of death due to ibogaine use for heroin addiction: Case report. Am. J. Addict. 2013, 22, 302. [Google Scholar] [CrossRef] [PubMed]
- Paling, F.P.; Andrews, L.M.; Valk, G.D.; Blom, H.J. Life-threatening complications of ibogaine: Three case reports. Neth. J. Med. 2012, 70, 422–424. [Google Scholar] [PubMed]
- Pleskovic, A.; Gorjup, V.; Brvar, M.; Kozelj, G. Ibogaine-associated ventricular tachyarrhythmias. Clin. Toxicol. 2012, 50, 157. [Google Scholar] [CrossRef]
- Shawn, L.K.; Alper, K.; Desai, S.P.; Stephenson, K.; Olgun, A.M.; Nelson, L.S.; Hoffman, R.S. Pause-dependent ventricular tachycardia and torsades de pointes after ibogaine ingestion. Clin. Toxicol. 2012, 50, 654. [Google Scholar]
- Asua, I. Growing menace of ibogaine toxicity. Br. J. Anaesth. 2013, 111, 1029–1030. [Google Scholar] [CrossRef] [PubMed]
- Vlaanderen, L.; Martial, L.C.; Franssen, E.J.; van der Voort, P.H.; Oosterwerff, E.; Somsen, G.A. Cardiac arrest after ibogaine ingestion. Clin. Toxicol. 2014, 52, 642–643. [Google Scholar] [CrossRef]
- Jenks, C.W. Extraction studies of Tabernanthe iboga and Voacanga africana. Nat. Prod. Lett. 2002, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Büchi, G.; Coffen, D.L.; Kocsis, K.; Sonnet, P.E.; Ziegler, F.E. The total synthesis of Iboga alkaloids. J. Am. Chem. Soc. 1966, 88, 3099–3109. [Google Scholar] [CrossRef]
- Jana, G.K.; Sinha, S. Total synthesis of ibogaine, epiibogaine and their analogues. Tetrahedron 2012, 68, 7155–7165. [Google Scholar] [CrossRef]
- Mash, D.C.; Kovera, C.A.; Pablo, J.; Tyndale, R.; Ervin, F.R.; Kamlet, J.D.; Hearn, W.L. Ibogaine in the treatment of heroin withdrawal. Alkaloids Chem. Biol. 2001, 56, 155–171. [Google Scholar] [PubMed]
- Obach, R.S.; Pablo, J.; Mash, D.C. Cytochrome P4502D6 catalyzes the O-demethylation of the psychoactive alkaloid ibogaine to 12-hydroxyibogamine. Drug Metab. Dispos. 1998, 26, 764–768. [Google Scholar] [PubMed]
- Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin. Pharmacokinet. 2009, 48, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Hough, L.B.; Pearl, S.M.; Glick, S.D. Tissue distribution of ibogaine after intraperitoneal and subcutaneous administration. Life Sci. 1996, 58, L119–L122. [Google Scholar] [CrossRef]
- Mash, D.C.; Kovera, C.A.; Buck, B.E.; Norenberg, M.D.; Shapshak, P.; Hearn, W.L.; Sanchez-Ramos, J. Medication development of ibogaine as a pharmacotherapy for drug dependence. Ann. N. Y. Acad. Sci. 1998, 844, 274–292. [Google Scholar] [CrossRef] [PubMed]
- Glue, P.; Lockhart, M.; Lam, F.; Hung, N.; Hung, C.T.; Friedhoff, L. Ascending-dose study of noribogaine in healthy volunteers: Pharmacokinetics, pharmacodynamics, safety, and tolerability. J. Clin. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Kontrimaviciute, V.; Breton, H.; Mathieu, O.; Mathieu-Daude, J.C.; Bressolle, F.M. Liquid chromatography-electrospray mass spectrometry determination of ibogaine and noribogaine in human plasma and whole blood. Application to a poisoning involving Tabernanthe iboga root. J. Chromatogr. B 2006, 843, 131–141. [Google Scholar] [CrossRef]
- Zhang, W.; Ramamoorthy, Y.; Tyndale, R.F.; Glick, S.D.; Maisonneuve, I.M.; Kuehne, M.E.; Sellers, E.M. Metabolism of 18-methoxycoronaridine, an ibogaine analog, to 18-hydroxycoronaridine by genetically variable CYP2C19. Drug Metab. Dispos. 2002, 30, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Hajo, N.; Dupont, C.; Wepierre, J. Effects of tabernanthine on various cardiovascular parameters in the rat and dog (author's transl). J. Pharmacol. 1981, 12, 441–453. [Google Scholar] [PubMed]
- Hamon, G.; Castillon, A.; Gaignault, J.C.; Worcel, M. Peripheral cardiovascular effects of tabernanthine tartrate in anaesthetized rats. Arch. Int. Pharmacodyn. Ther. 1985, 276, 60–72. [Google Scholar] [PubMed]
- Hajo-Tello, N.; Dupont, C.; Wepierre, J.; Cohen, Y.; Miller, R.; Godfraind, T. Effects of tabernanthine on calcium and catecholamine stimulated contractions of isolated vascular and cardiac muscle. Arch. Int. Pharmacodyn. Ther. 1985, 276, 35–43. [Google Scholar] [PubMed]
- Binienda, Z.; Beaudoin, M.A.; Thorn, B.T.; Prapurna, D.R.; Johnson, J.R.; Fogle, C.M.; Slikker, W.; Ali, S.F. Alteration of electroencephalogram and monoamine concentrations in rat brain following ibogaine treatment. Ann. N. Y. Acad. Sci. 1998, 844, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Mash, D.C.; Allen-Ferdinand, K.; Mayor, M.; Kovera, C.A.; Ayafor, J.F.; Williams, I.C.; Ervin, F.R. Ibogaine: Clinical observations of safety after single dose administrations. In Problems of Drug Dependence, Proceedings of the 60th Annual Scientific Meeting, The College on Problems of Drug Dependence Inc., Scottsdale, Arizona, 12–17 June 1998; Harris, L.S., Ed.; National Institute on Drug Abuse: Bethesda, MD, USA, 1998. [Google Scholar]
- Zhang, H.; Cuevas, J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J. Neurophysiol. 2002, 87, 2867–2879. [Google Scholar] [PubMed]
- Glick, S.D.; Maisonneuve, I.M. Development of novel medications for drug addiction. The legacy of an African shrub. Ann. N. Y. Acad. Sci. 2000, 909, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Alper, K.; Reith, M.E.; Sershen, H. Ibogaine and the inhibition of acetylcholinesterase. J. Ethnopharmacol. 2012, 139, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Sweetnam, P.M.; Lancaster, J.; Snowman, A.; Collins, J.L.; Perschke, S.; Bauer, C.; Ferkany, J. Receptor binding profile suggests multiple mechanisms of action are responsible for ibogaine’s putative anti-addictive activity. Psychopharmacology (Berl) 1995, 118, 369–376. [Google Scholar] [CrossRef]
- Deecher, D.C.; Teitler, M.; Soderlund, D.M.; Bornmann, W.G.; Kuehne, M.E.; Glick, S.D. Mechanisms of action of ibogaine and harmaline congeners based on radioligand binding studies. Brain Res. 1992, 571, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Letienne, R.; Vie, B.; Le, G.B. Pharmacological characterisation of sodium channels in sinoatrial node pacemaking in the rat heart. Eur. J. Pharmacol. 2006, 530, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Kolecki, P.F.; Curry, S.C. Poisoning by sodium channel blocking agents. Crit. Care Clin. 1997, 13, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Haufe, V.; Chamberland, C.; Dumaine, R. The promiscuous nature of the cardiac sodium current. J. Mol. Cell. Cardiol. 2007, 42, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.N.; de la Rosa, A.; Navarro, F.; Franco, D.; Aranega, A.E. Tissue distribution and subcellular localization of the cardiac sodium channel during mouse heart development. Cardiovasc. Res. 2008, 78, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.G.; Westenbroek, R.E.; Maass, A.H.; Lange, V.; Renner, A.; Wischmeyer, E.; Bonz, A.; Muck, J.; Ertl, G.; Catterall, W.A.; et al. Distribution and function of sodium channel subtypes in human atrial myocardium. J. Mol. Cell. Cardiol. 2013, 61, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Marger, L.; Toyoda, F.; Rizzetto, R.; Audoubert, M.; Dubel, S.; Torrente, A.G.; Difrancesco, M.L.; Muller, J.C.; Leoni, A.L.; et al. The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 2013, 142, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Kovar, M.; Boehm, S.; Sandtner, W.; Hilber, K. Anti-addiction drug ibogaine inhibits hERG channels: a cardiac arrhythmia risk. Addict. Biol. 2014, 19, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Thurner, P.; Stary-Weinzinger, A.; Gafar, H.; Gawali, V.S.; Kudlacek, O.; Zezula, J.; Hilber, K.; Boehm, S.; Sandtner, W.; Koenig, X. Mechanism of hERG channel block by the psychoactive indole alkaloid ibogaine. J. Pharmacol. Exp. Ther. 2014, 348, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Michel, D.; Wegener, J.W.; Nawrath, H. Effects of quinine and quinidine on the transient outward and on the L-type Ca2+ current in rat ventricular cardiomyocytes. Pharmacology 2002, 65, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Po, S.S.; Wang, D.W.; Yang, I.C.; Johnson, J.P.; Nie, L.; Bennet, P.B. Modulation of HERG potassium channels by extracellular magnesium and quinidine. J. Cardiovasc. Pharmacol. 1999, 33, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Bai, R.; Liu, N.; Fowler, S.J.; Priori, S.G.; Wang, L.; Lin, L.; Yu, R.H.; Ma, C.S.; Alper, K. Herg blockade by iboga alkaloids. In Proceedings of the European Society of Cardiology Congress 2014, Barcelona, Spain, 30 August–3 September 2014.
- Wible, B.A.; Hawryluk, P.; Ficker, E.; Kuryshev, Y.A.; Kirsch, G.; Brown, A.M. HERG-Lite: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods 2005, 52, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Hondeghem, L.M.; Carlsson, L.; Duker, G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 2001, 103, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.M.; Kors, J.A.; de Bruin, M.L.; van der Hooft, C.S.; Hofman, A.; Heeringa, J.; Deckers, J.W.; Kingma, J.H.; Sturkenboom, M.C.; Stricker, B.H.; et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J. Am. Coll. Cardiol. 2006, 47, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Fermini, B.; Fossa, A.A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Drug-induced prolongation of the QT interval: Regulatory dilemmas and implications for approval and labelling of a new chemical entity. Fundam. Clin. Pharmacol. 2002, 16, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc. Res. 2001, 50, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Roden, D.M. Extracellular potassium modulation of drug block of IKr implications for torsade de pointes and reverse use-dependence. Circulation 1996, 93, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.; Roden, D.M.; Darbar, D. Drug-induced long QT syndrome. Pharmacol. Rev. 2010, 62, 760–781. [Google Scholar] [CrossRef] [PubMed]
- Osadchii, O.E. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam. Clin. Pharmacol. 2010, 24, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Massaeli, H.; Xu, J.; Jia, Z.; Wigle, J.T.; Mesaeli, N.; Zhang, S. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J. Clin. Investig. 2009, 119, 2745–2757. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Snyders, D.J.; Roden, D.M. Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ. Res. 1997, 80, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Rossinen, J.; Sinisalo, J.; Partanen, J.; Nieminen, M.S.; Viitasalo, M. Effects of acute alcohol infusion on duration and dispersion of QT interval in male patients with coronary artery disease and in healthy controls. Clin. Cardiol. 1999, 22, 591–594. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.E. Inhibition of human ether-a-go-go potassium channels by cocaine. Mol. Pharmacol. 2001, 59, 269–277. [Google Scholar] [PubMed]
- Kuryshev, Y.A.; Bruening-Wright, A.; Brown, A.M.; Kirsch, G.E. Increased cardiac risk in concomitant methadone and diazepam treatment: Pharmacodynamic interactions in cardiac ion channels. J. Cardiovasc. Pharmacol. 2010, 56, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Romero, J.; Taub, C.C. Methadone, QTc prolongation and torsades de pointes: Current concepts, management and a hidden twist in the tale? J. Cardiovasc. Dis. Res. 2013, 4, 229–235. [Google Scholar] [PubMed]
- Brown, R.; Kraus, C.; Fleming, M.; Reddy, S. Methadone: Applied pharmacology and use as adjunctive treatment in chronic pain. Postgrad. Med. J. 2004, 80, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Coller, J.K.; Michalakas, J.R.; James, H.M.; Farquharson, A.L.; Colvill, J.; White, J.M.; Somogyi, A.A. Inhibition of CYP2D6-mediated tramadol O-demethylation in methadone but not buprenorphine maintenance patients. Br. J. Clin. Pharmacol. 2012, 74, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.; Zhao, X.; Shin, J.G.; Flockhart, D.A. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.J.; Ackerman, M.J.; Funk, M.; Gibler, W.B.; Kligfield, P.; Menon, V.; Philippides, G.J.; Roden, D.M.; Zareba, W. Prevention of Torsade de Pointes in Hospital Settings: A scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation 2010, 121, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koenig, X.; Hilber, K. The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation. Molecules 2015, 20, 2208-2228. https://doi.org/10.3390/molecules20022208
Koenig X, Hilber K. The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation. Molecules. 2015; 20(2):2208-2228. https://doi.org/10.3390/molecules20022208
Chicago/Turabian StyleKoenig, Xaver, and Karlheinz Hilber. 2015. "The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation" Molecules 20, no. 2: 2208-2228. https://doi.org/10.3390/molecules20022208
APA StyleKoenig, X., & Hilber, K. (2015). The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation. Molecules, 20(2), 2208-2228. https://doi.org/10.3390/molecules20022208