Photocatalytic Oxidation of Diethyl Sulfide Vapor over TiO2-Based Composite Photocatalysts



Abstract
:
1. Introduction
2. Results and Discussion
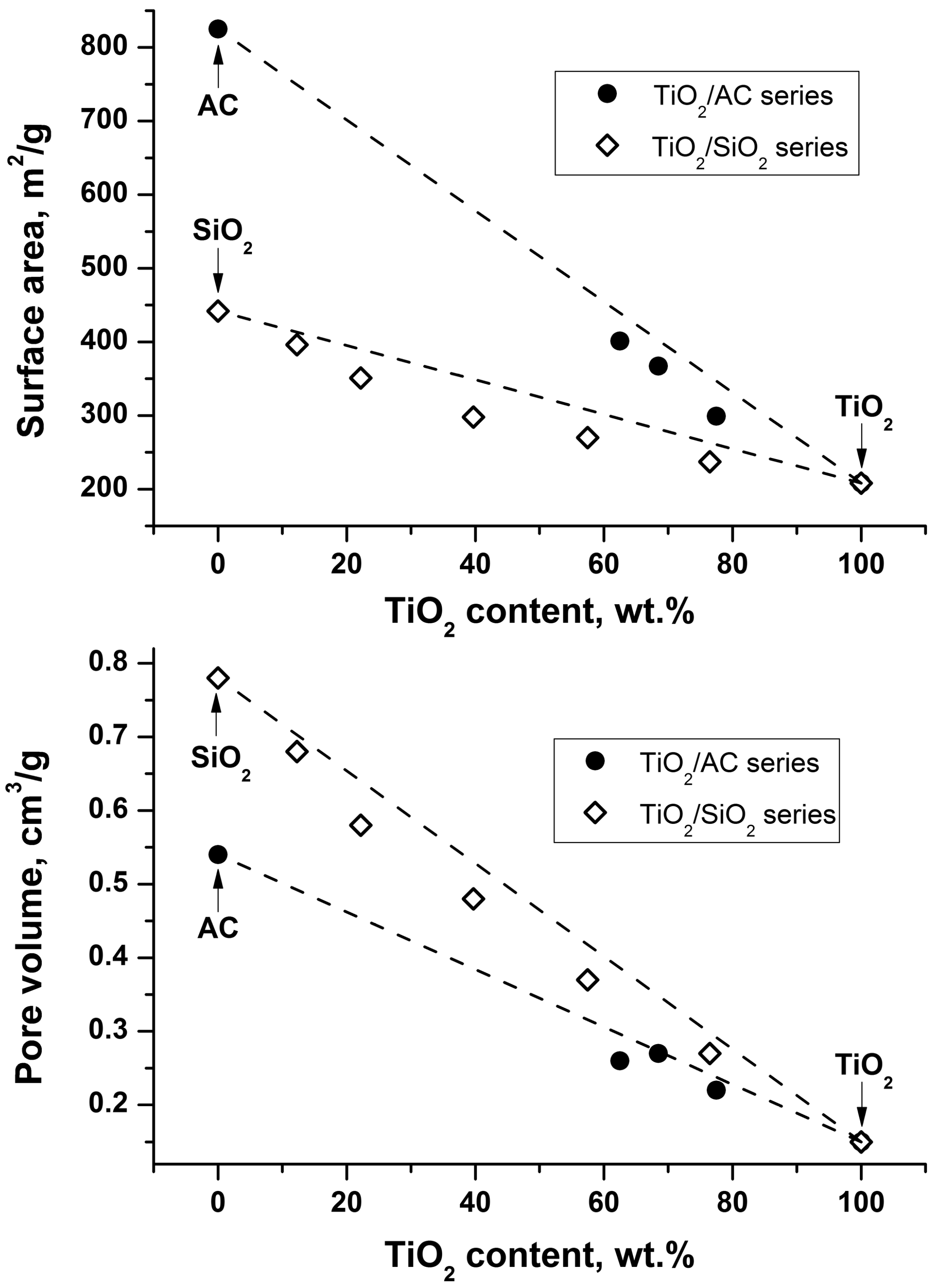
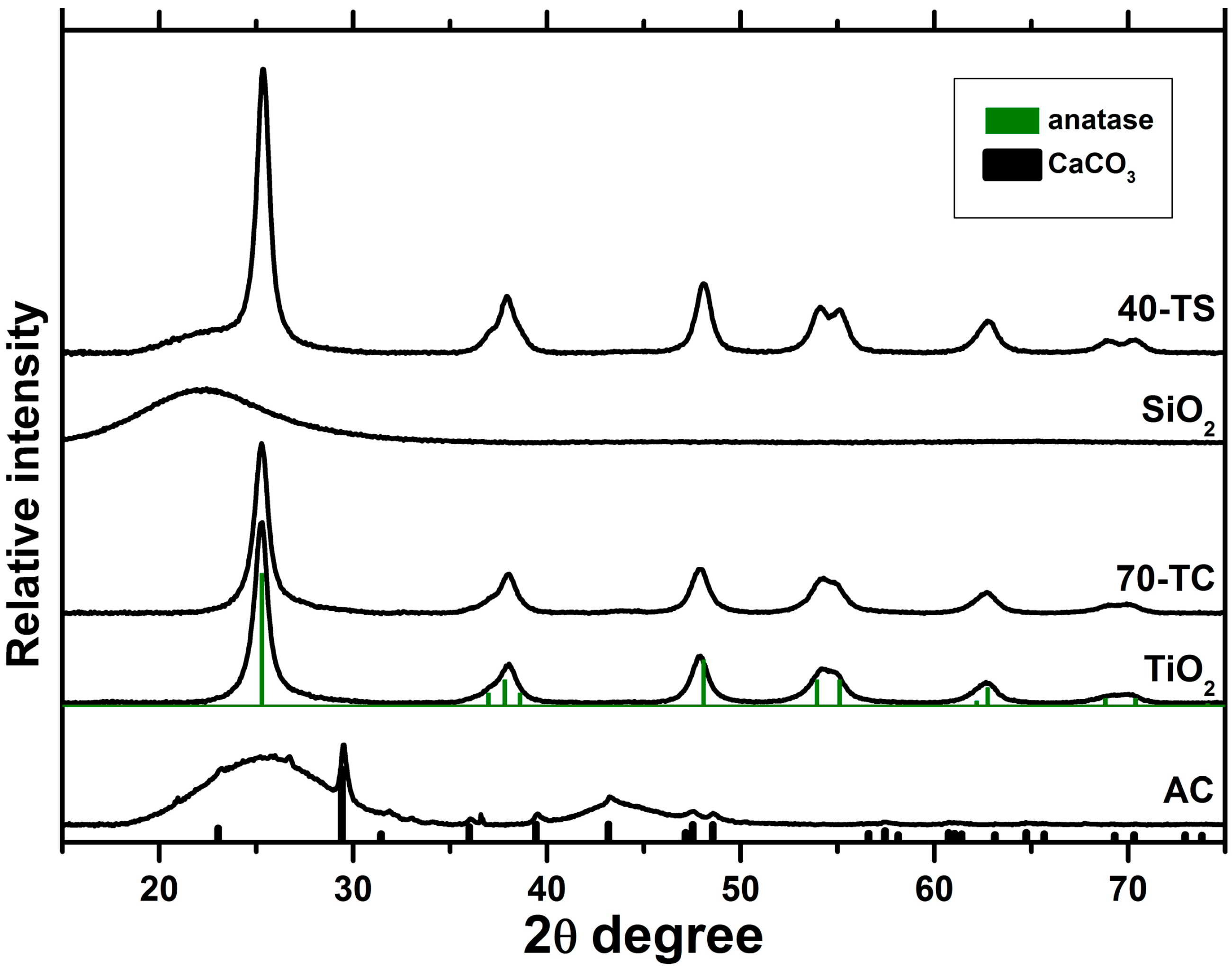
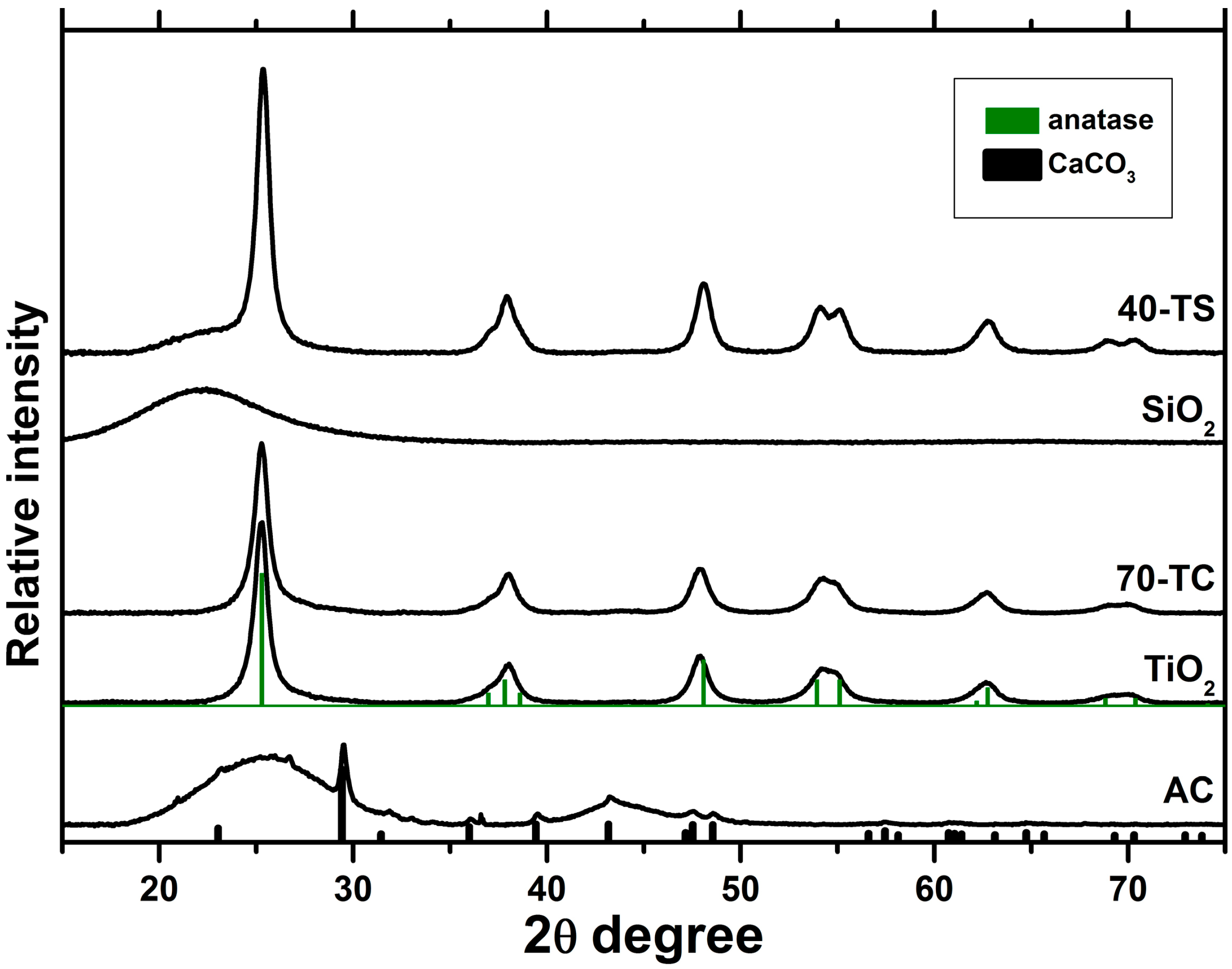
2.1. Characterization of the Synthesized Photocatalysts

{kind=link}
{kind=link}
{kind=link}
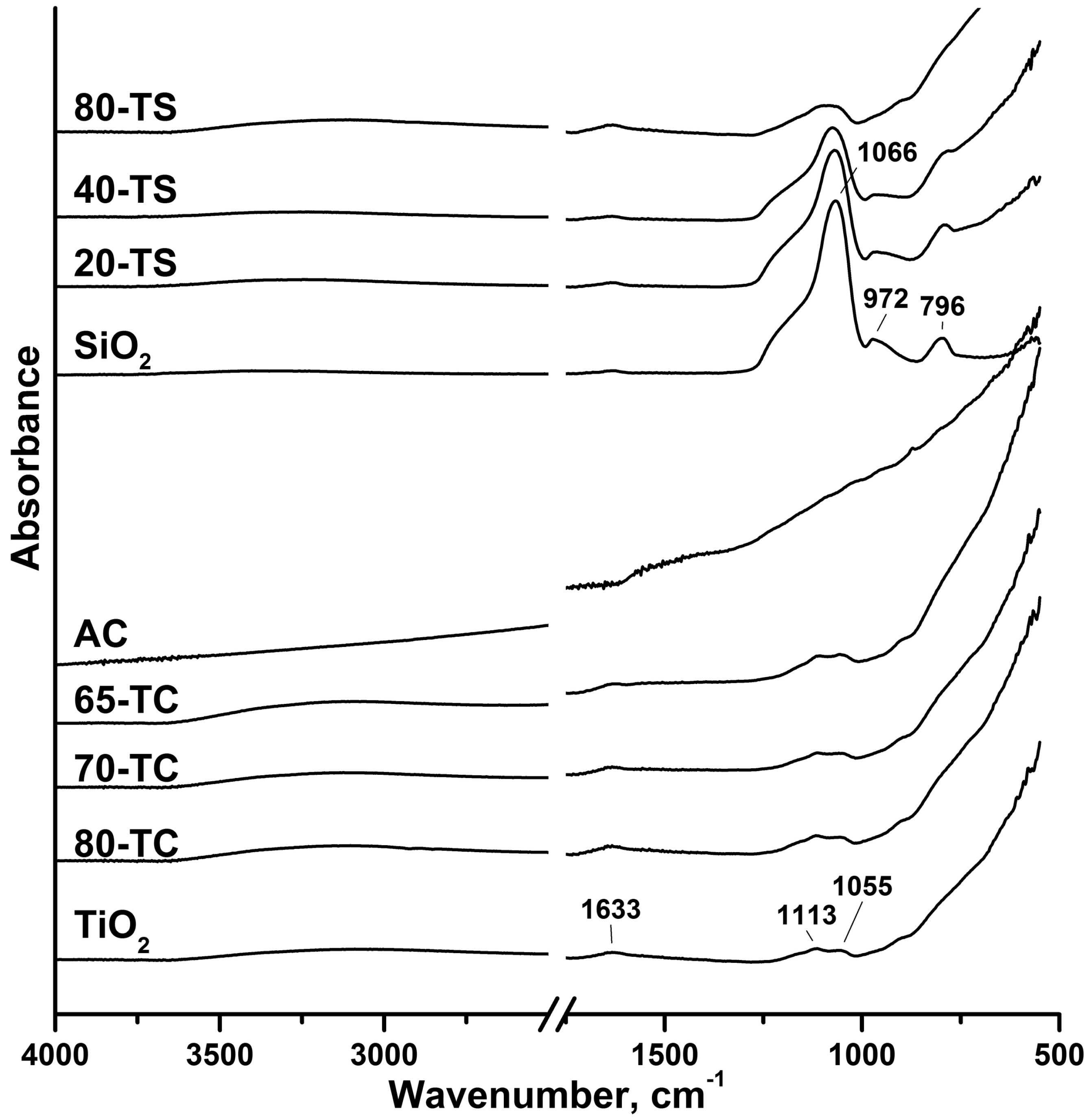{kind=link}
{kind=link}
{kind=link}
{kind=link}
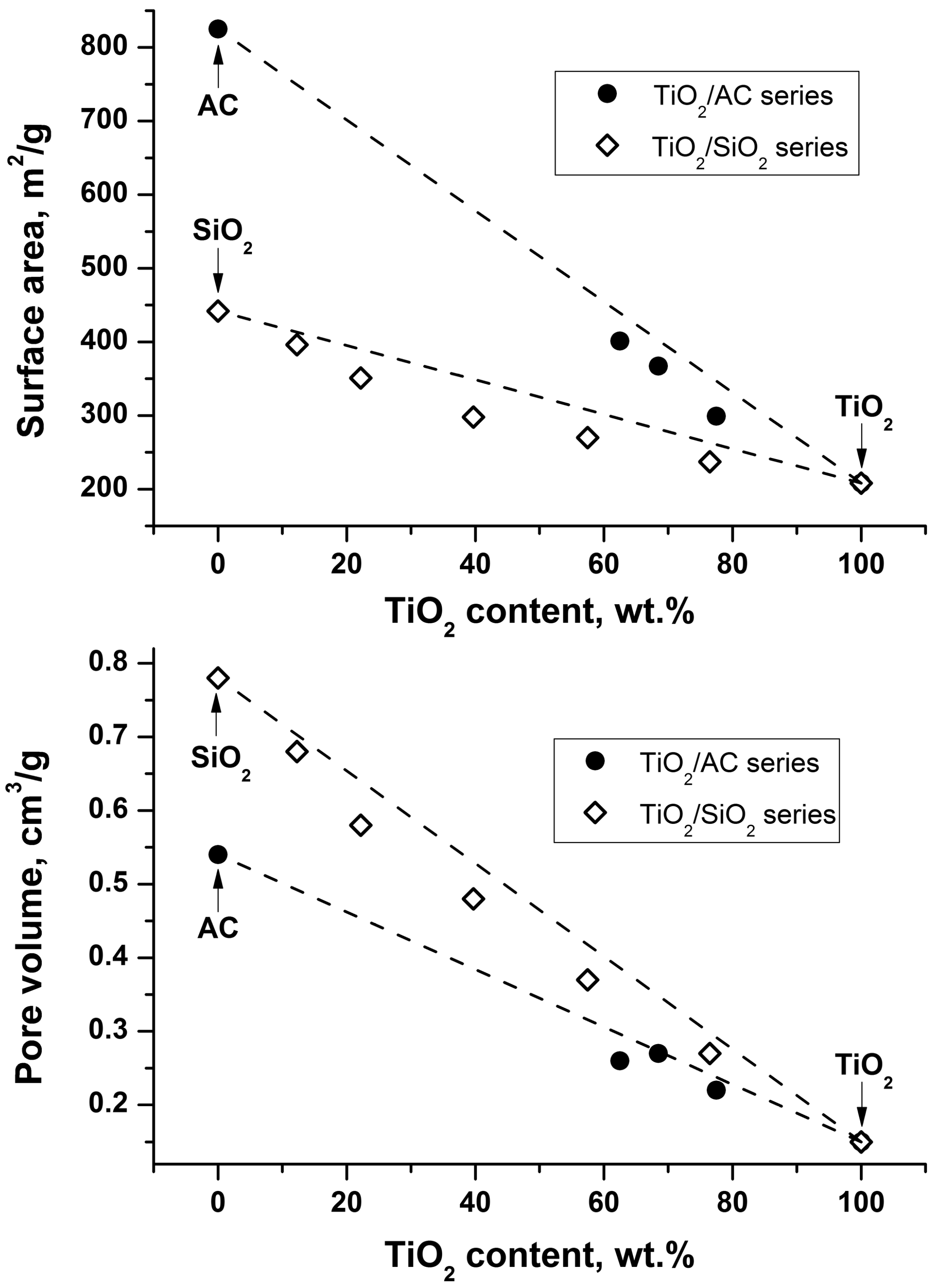
{kind=link}
{kind=link}
| Series | Sample * | TiO2 Content, wt. % | Surface Area, m2/g | Pore Volume, cm3/g |
|---|---|---|---|---|
| Supports | AC | -- | 825 | 0.54 |
| SiO2 | -- | 442 | 0.78 | |
| TiO2 | TiO2 | 100 | 208 | 0.15 |
| TiO2/AC | 80-TC | 77.5 | 299 | 0.22 |
| 70-TC | 68.5 | 367 | 0.27 | |
| 65-TC | 62.5 | 401 | 0.26 | |
| TiO2/SiO2 | 80-TS | 76.5 | 237 | 0.27 |
| 60-TS | 57.5 | 270 | 0.37 | |
| 40-TS | 39.7 | 298 | 0.48 | |
| 20-TS | 22.2 | 351 | 0.58 | |
| 10-TS | 12.3 | 396 | 0.68 |
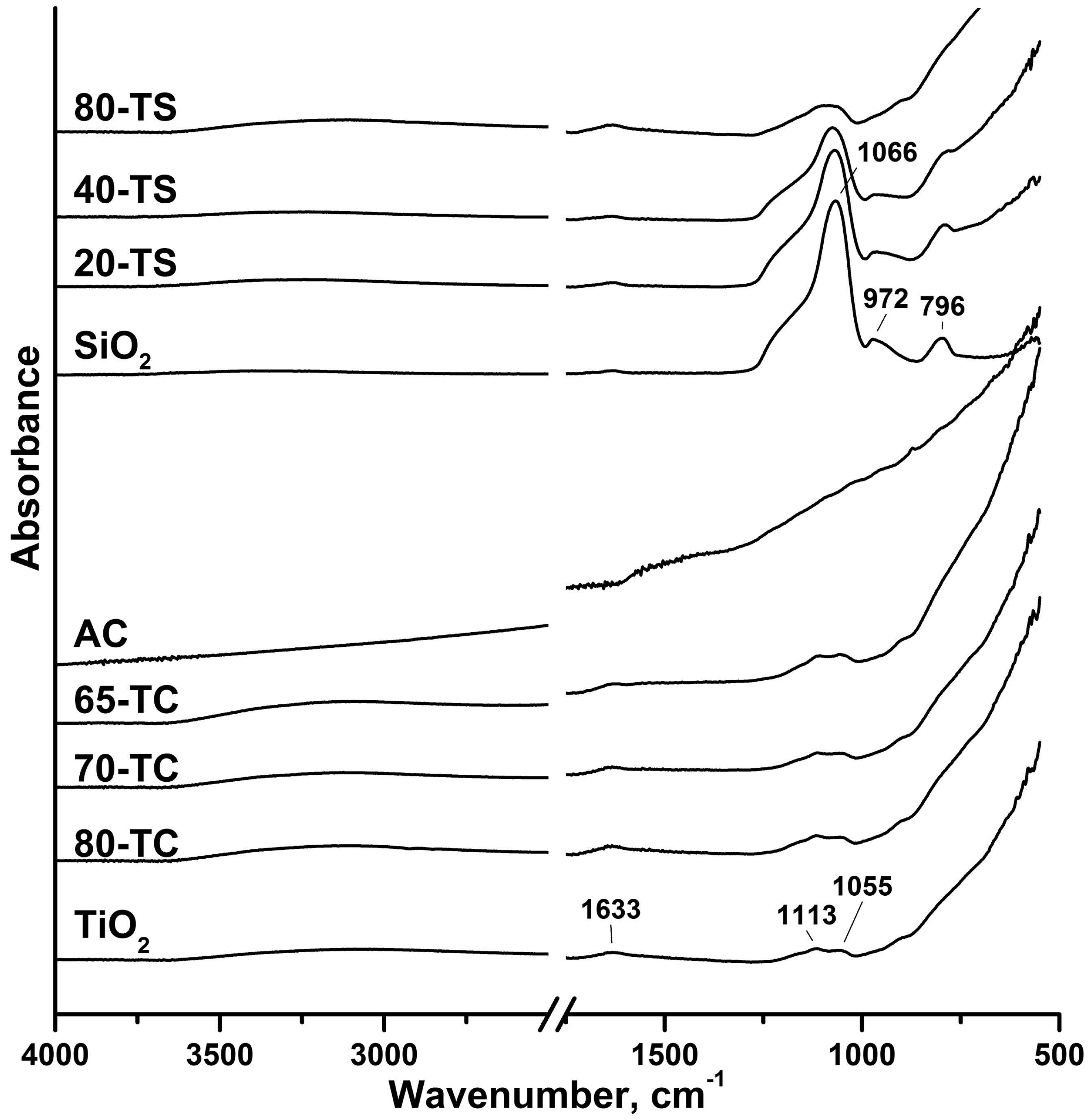
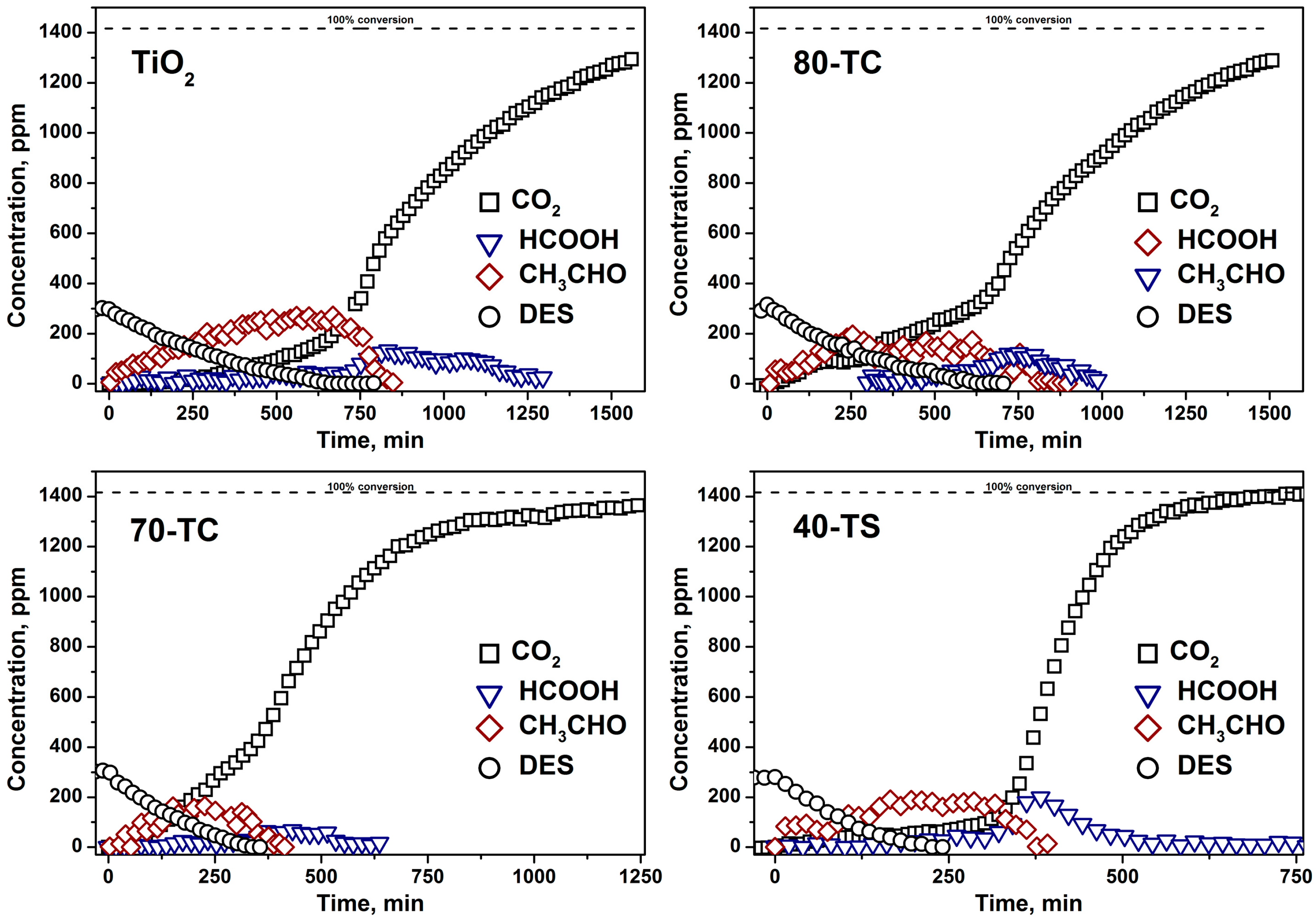
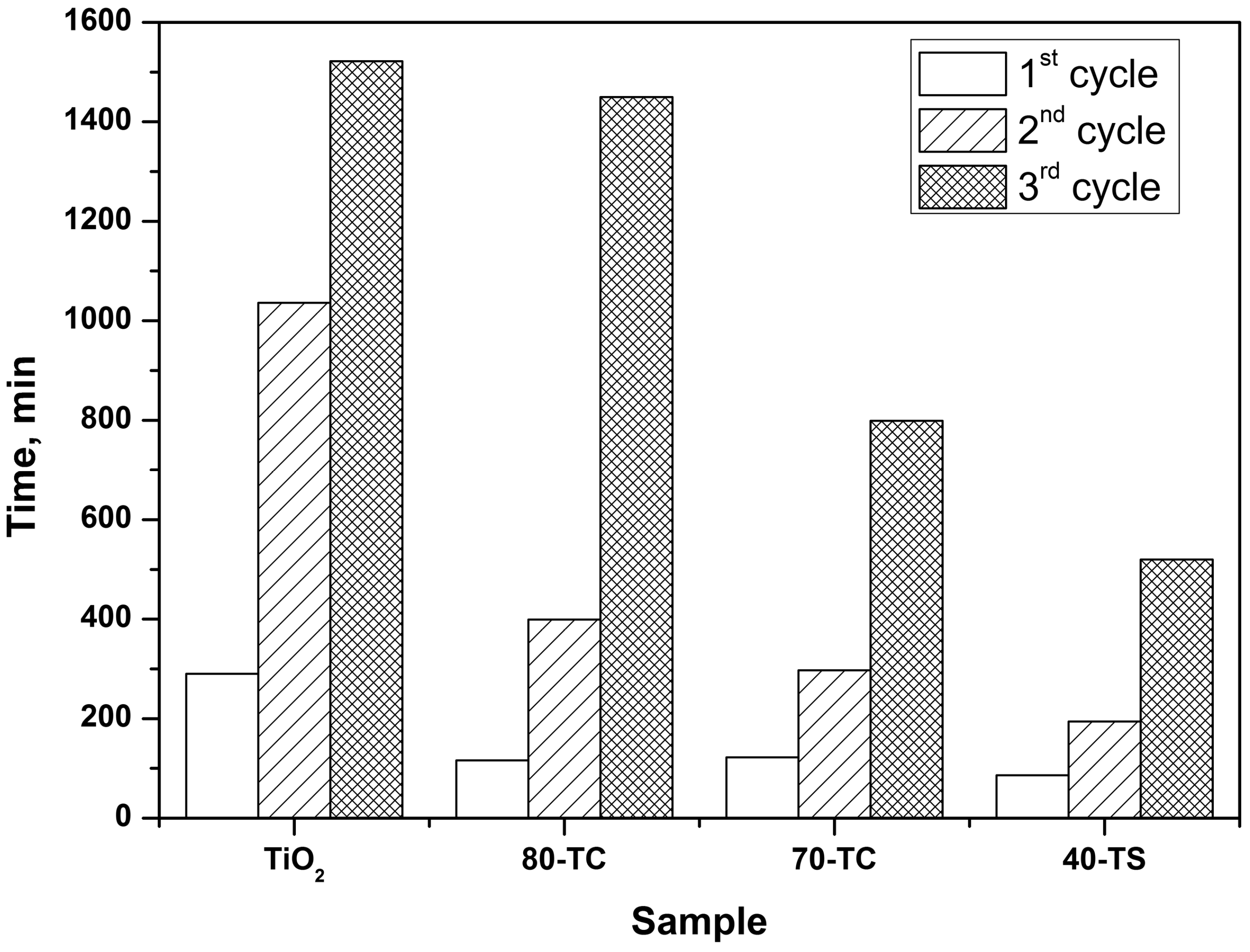
2.2. Kinetic Experiments
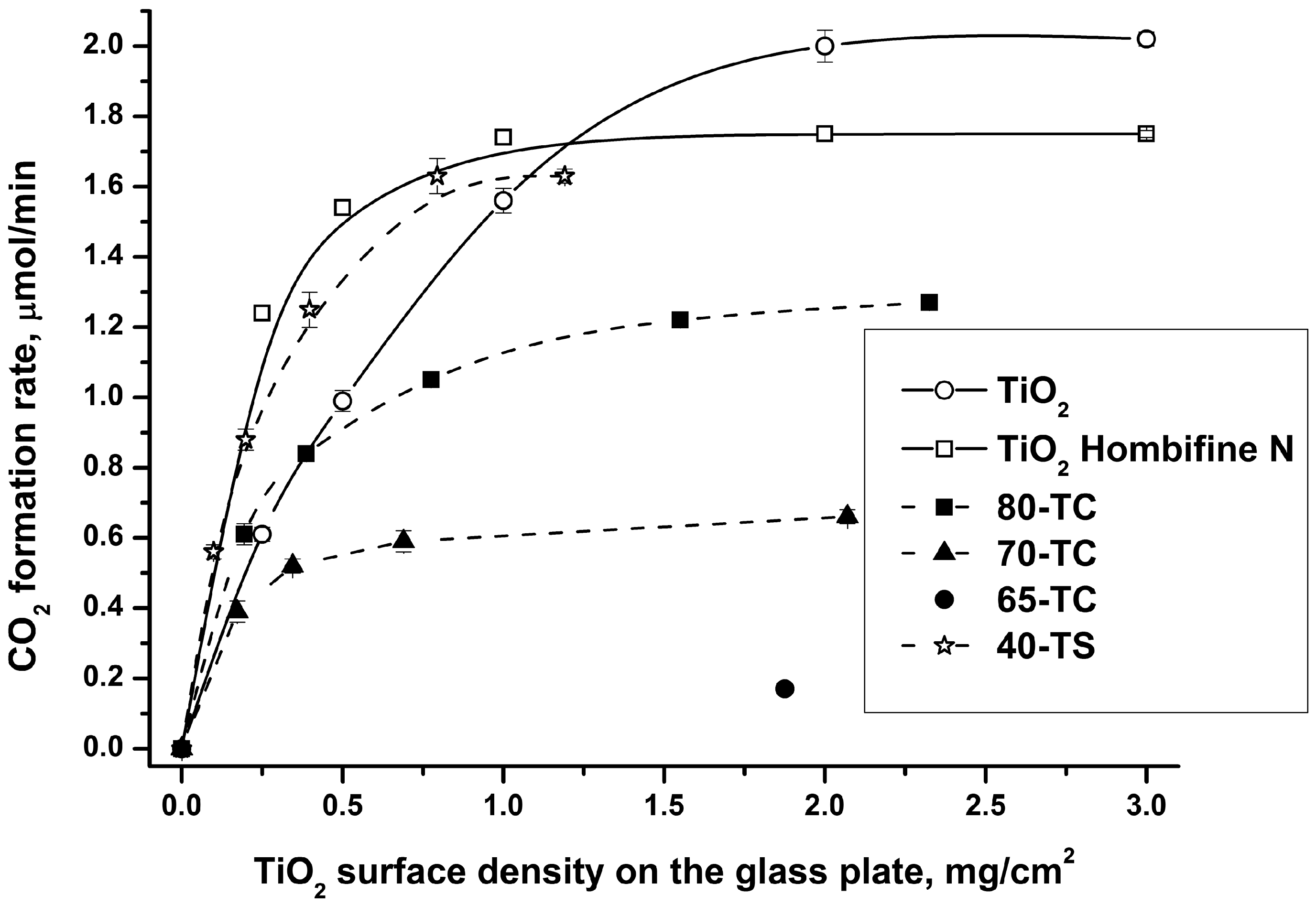
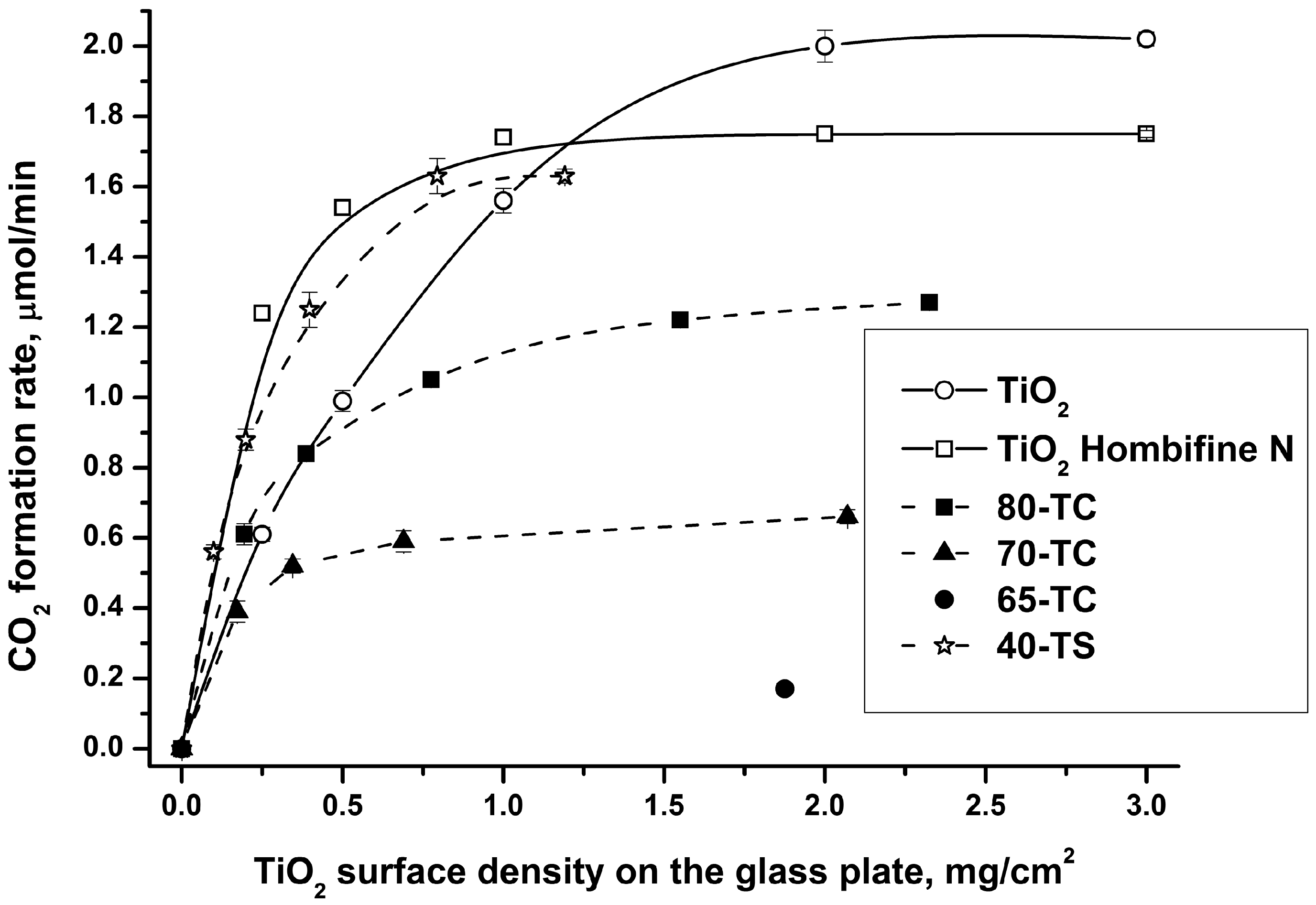
2.2.1. Effect of the Sample Quantity on the Photocatalytic Activity
- (1)
- Comparison of the photocatalysts’ activity should be done using high quantities when the thickness of the photocatalyst layer is sufficient for complete light absorption (e.g., 2–3 mg/cm2). We used this approach when choosing a photocatalyst with good adsorptivity and photocatalytic activity (see Section 2.2.2);
- (2)
- Studies of long-term photocatalyst use should be done using a relatively low TiO2 quantity (e.g., 0.5 mg/cm2) because in this case we can assume that the entire photocatalyst surface is irradiated and is involved in the reaction process. This is the reason why we investigated the diethyl sulfide oxidation with a 0.5 mg/cm2 quantity of TiO2.
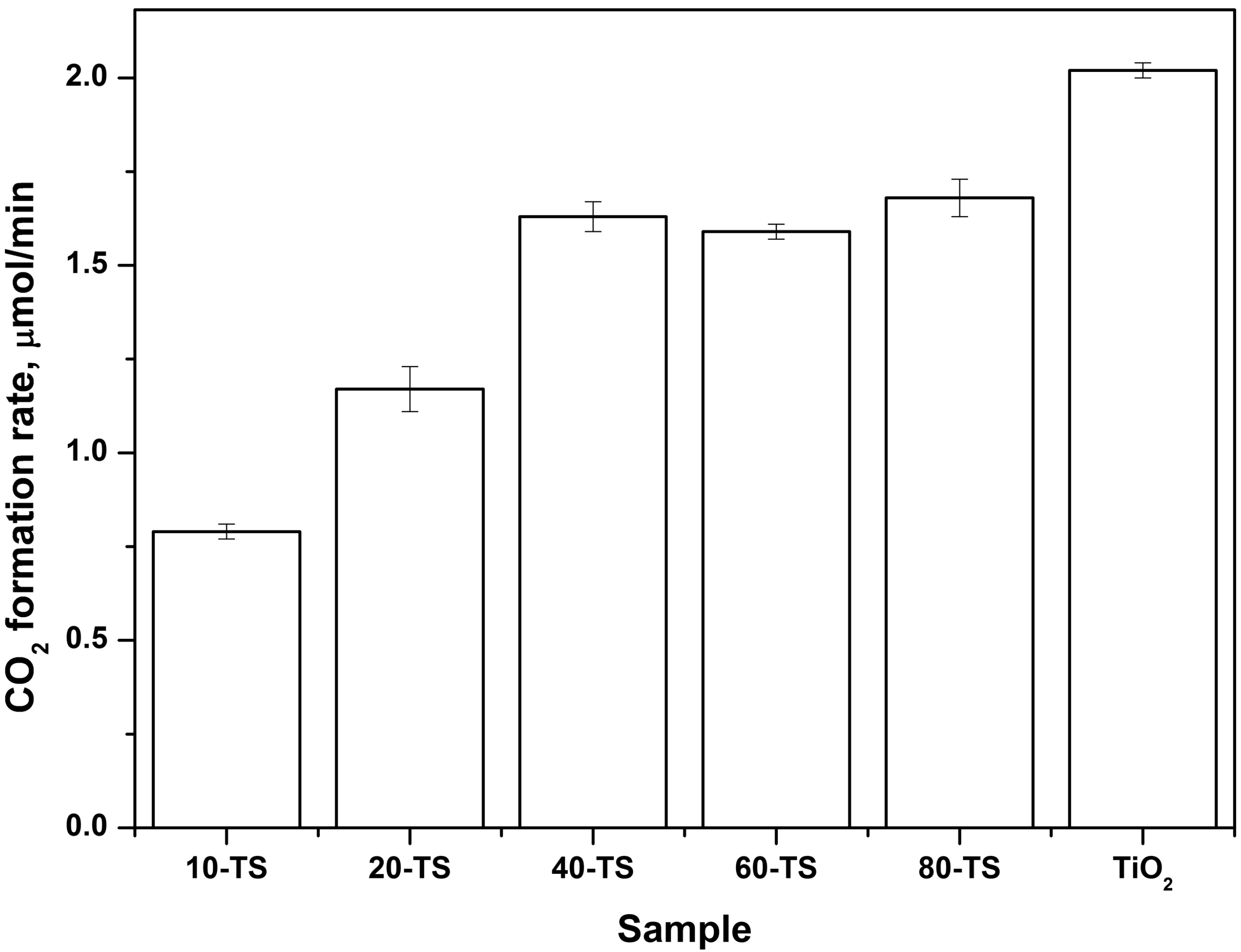
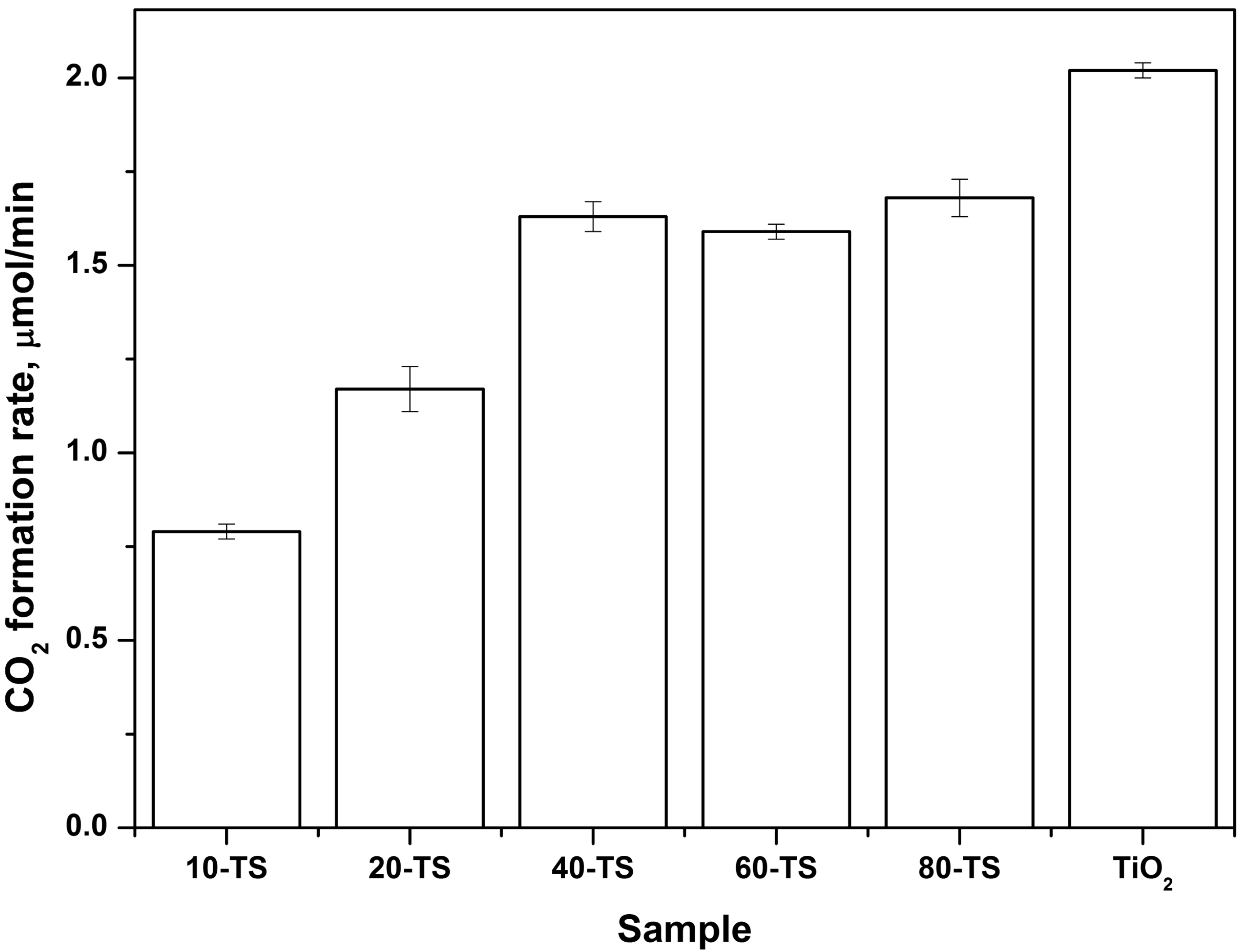
2.2.2. Effect of TiO2 Content on the Photocatalytic Activity of the TiO2/SiO2 Catalyst
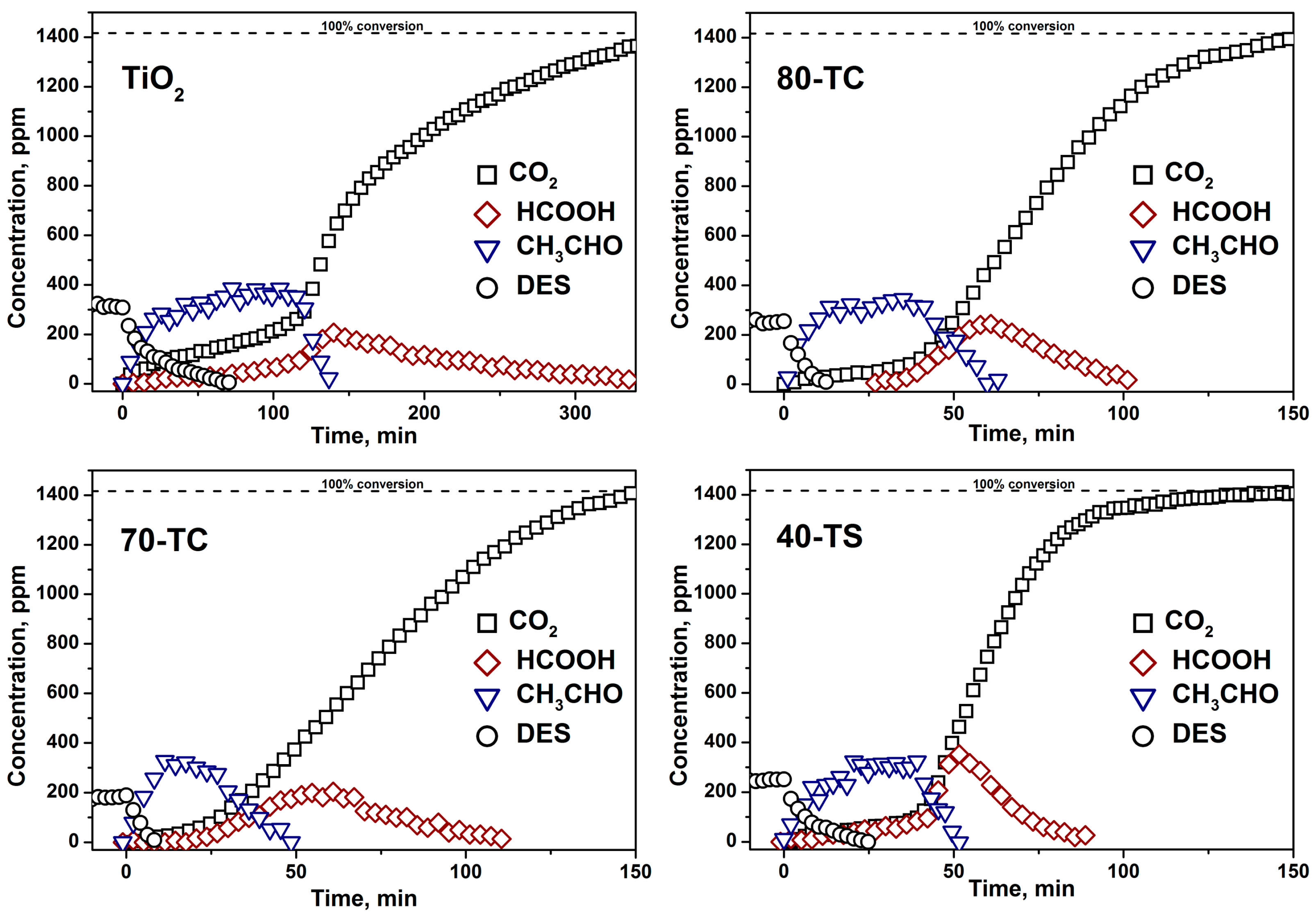
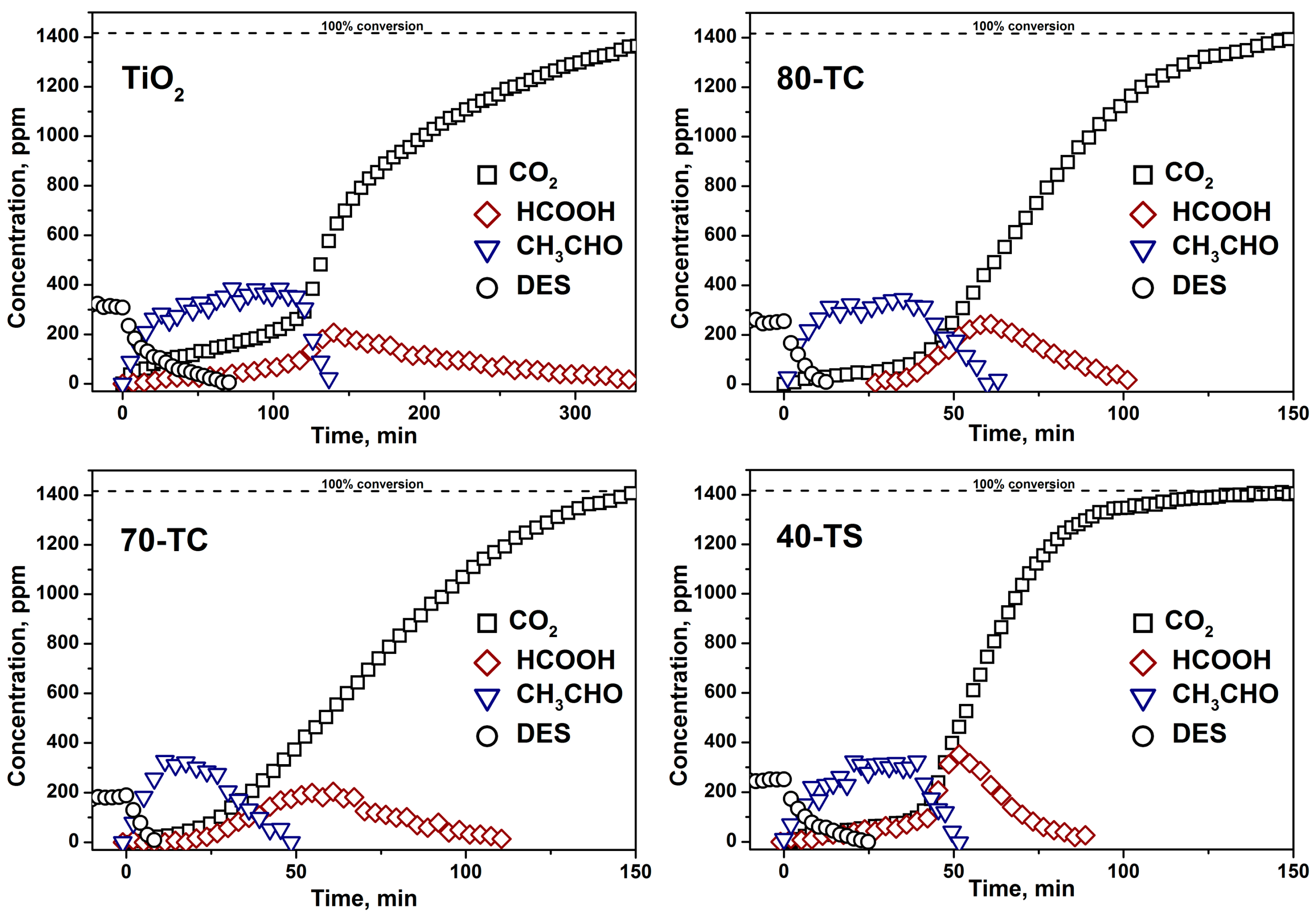
2.2.3. Kinetics of the DES PCO in a Static Reactor
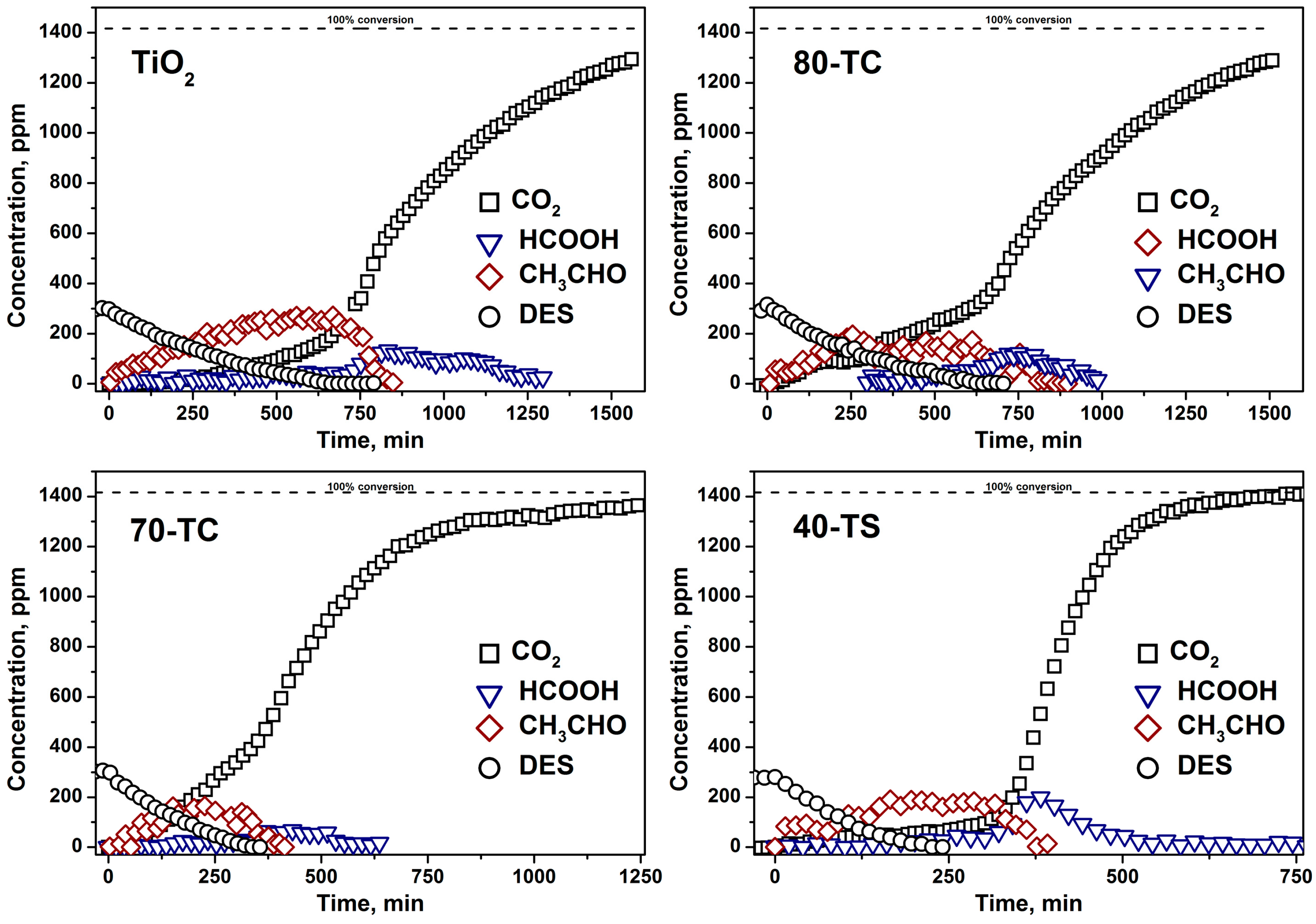
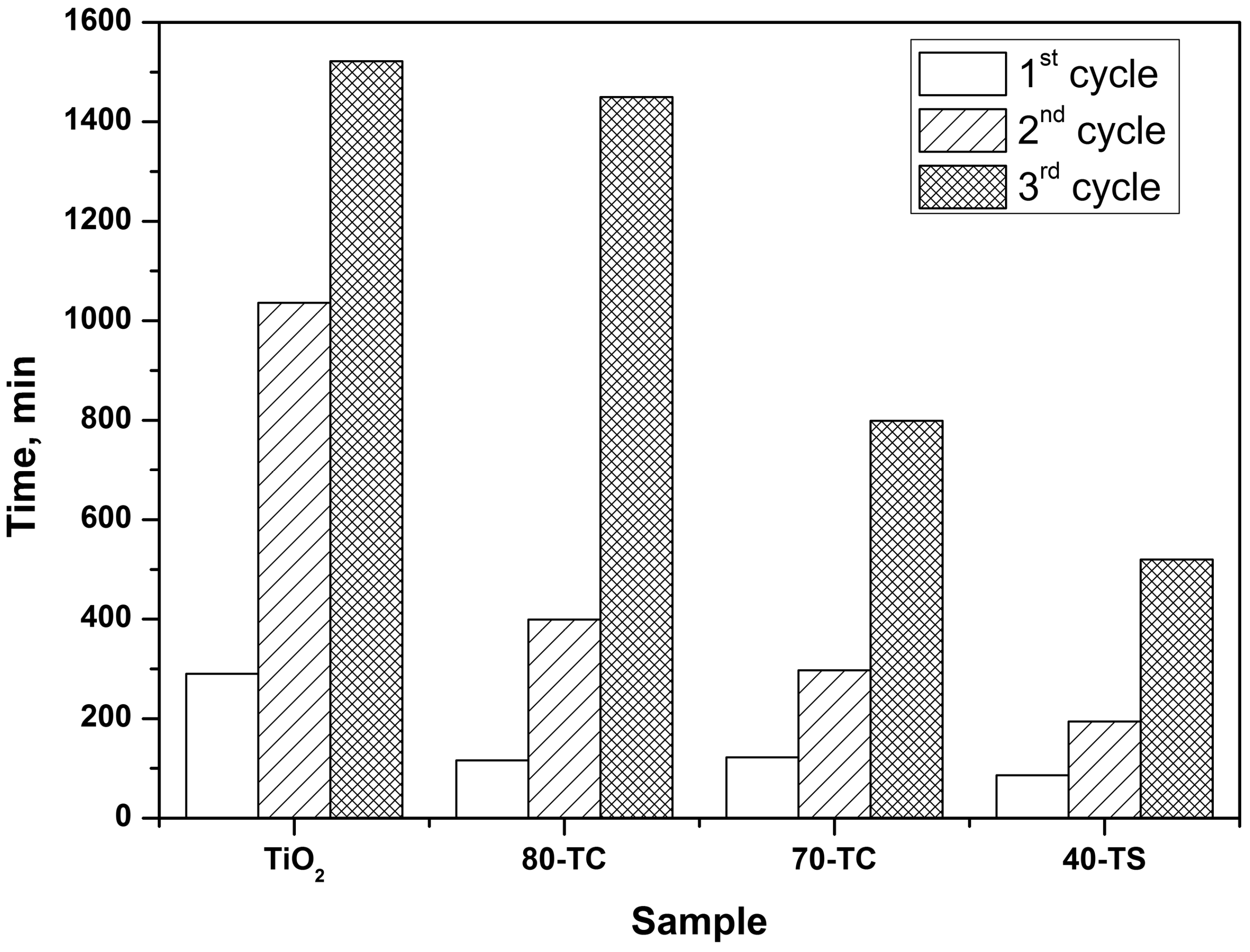
3. Experimental Section
3.1. Materials
3.2. Synthesis of the Composite Photocatalyst
3.3. Characterization Method
3.4. Kinetic Experiments
3.4.1. Acetone Oxidation
3.4.2. DES Oxidation
4. Conclusions
- (1)
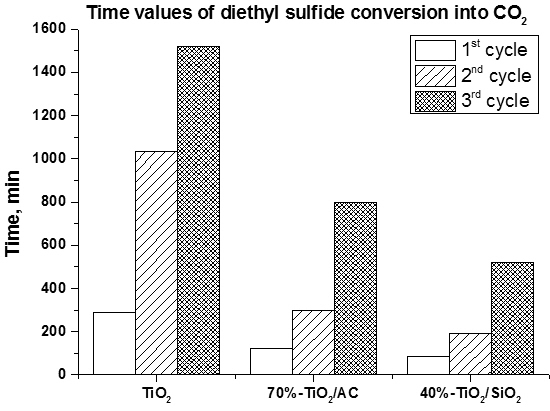
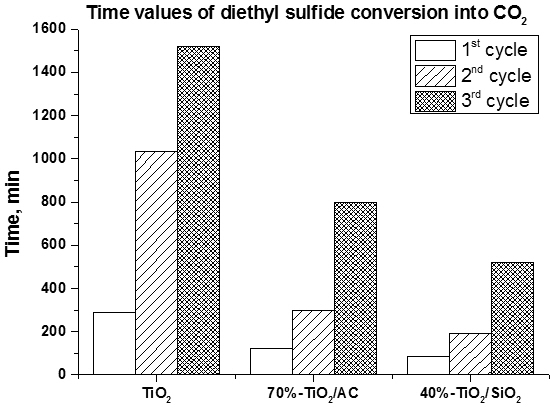
- The usage of composite photocatalyst results in up to an 8-fold decrease of DES removal time if compared with pure unmodified TiO2. This could be explained by an increase of the available surface area in the case of composite photocatalyst and reversible transfer of non-volatile intermediates from TiO2 surface to the support surface thus keeping the photocatalyst surface available for further interaction with substrate. Additionally the removal of intermediates—acetaldehyde and formic acid—occurs faster over composite photocatalyst;
- (2)
- The long-term oxidation of DES leads to a strong deactivation of the photocatalyst. The deactivation decreases in the following sequence: TiO2>TiO2/AC>TiO2/SiO2. The most active and stable catalyst is the TiO2/SiO2 one which contains 40 wt. % of TiO2. The calculated TOF number for this sample is 3.5 times higher than for pure TiO2.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watson, A.P.; Griffin, G.D. Toxicity of vesicant agents scheduled for destruction by the Chemical Stockpile Disposal Program. Environ. Health Perspect. 1992, 98, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Munro, N.B.; Watson, A.P.; Ambrose, K.R.; Griffin, G.D. Treating exposure to chemical warfare agents: Implications for health care providers and community emergency planning. Environ. Health Perspect. 1990, 89, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Munro, N. Toxicity of the organophosphate chemical warfare agents GA, GB, and VX: Implications for public protection. Environ. Health Perspect. 1994, 102, 18–38. [Google Scholar] [CrossRef] [PubMed]
- Perry Robinson, J.P. (Ed.) Public Health Response to Biological and Chemical Weapons: WHO Guidance, 2nd ed.; World Health Organization: Geneva, Switzerland, 2004; p. 340.
- Ivarsson, U.; Nilsson, H.; Santesson, J. A FOA Briefing book on Chemical Weapons: Threat, Effects, and Protection; National Defence Research Establishment: Umea, Sweden, 1992. [Google Scholar]
- Alexandrov, V.N.; Emel’yanov, V.I. Poisonous Compounds (in Russian); Voenizdat: Moscow, Russia, 1990; p. 271. [Google Scholar]
- Yang, Y.C.; Baker, J.A.; Ward, J.R. Decontamination of chemical warfare agents. Chem. Rev. 1992, 92, 1729–1743. [Google Scholar] [CrossRef]
- Zuo, G.M.; Cheng, Z.X.; Li, G.W.; Wang, L.Y.; Miao, T. Photoassisted Reaction of Sulfur Mustard under UV Light Irradiation. Environ. Sci. Technol. 2005, 39, 8742–8746. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.M.; Cheng, Z.X.; Li, G.W.; Shi, W.P.; Miao, T. Study on photolytic and photocatalytic decontamination of air polluted by chemical warfare agents (CWAs). Chem. Eng. J. 2007, 128, 135–140. [Google Scholar] [CrossRef]
- Bhatkhande, D.S.; Pangarkar, V.G.; Beenackers, A.A. Photocatalytic degradation for environmental applications—A review. J. Chem. Technol. Biotechnol. 2002, 77, 102–116. [Google Scholar] [CrossRef]
- Martyanov, I.N.; Klabunde, K.J. Photocatalytic Oxidation of Gaseous 2-Chloroethyl Ethyl Sulfide over TiO2. Environ. Sci. Technol. 2003, 37, 3448–3453. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.A.; Kim, Y.S.; Abdel-Wahab, A.A.; Dulay, M. Photocatalytic decontamination of sulfur-containing alkyl halides on irradiated semiconductor suspensions. Catal. Lett. 1990, 5, 369–376. [Google Scholar] [CrossRef]
- Vorontsov, A.V.; Panchenko, A.A.; Savinov, E.N.; Lion, C.; Smirniotis, P.G. Photocatalytic Degradation of 2-Phenethyl-2-chloroethyl Sulfide in Liquid and Gas Phases. Environ. Sci. Technol. 2002, 36, 5261–5269. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, D.; Vorontsov, A.; Smirniotis, P.; Savinov, E. Gas-phase photocatalytic oxidation of diethyl sulfide over TiO2: Kinetic investigations and catalyst deactivation. Appl. Catal. B Environ. 2003, 42, 77–87. [Google Scholar] [CrossRef]
- González-Garcı́a, N.; Ayllón, J.A.; Doménech, X.; Peral, J. TiO2 deactivation during the gas-phase photocatalytic oxidation of dimethyl sulfide. Appl. Catal. B Environ. 2004, 52, 69–77. [Google Scholar] [CrossRef]
- Vorontsov, A.V.; Savinov, E.V.; Davydov, L.; Smirniotis, P.G. Photocatalytic destruction of gaseous diethyl sulfide over TiO2. Appl. Catal. B Environ. 2001, 32, 11–24. [Google Scholar] [CrossRef]
- Vorontsov, A. TiO2 reactivation in photocatalytic destruction of gaseous diethyl sulfide in a coil reactor. Appl. Catal. B Environ. 2003, 44, 25–40. [Google Scholar] [CrossRef]
- Vorontsov, A.V. Photocatalytic transformations of organic sulfur compounds and H2S. Russ. Chem. Rev. 2008, 77, 909–926. [Google Scholar] [CrossRef]
- Vorontsov, A.V.; Besov, A.S.; Parmon, V.N. Fast purification of air from diethyl sulfide with nanosized TiO2 aerosol. Appl. Catal. B Environ. 2013, 129, 318–324. [Google Scholar] [CrossRef]
- Selishchev, D.; Kolinko, P.; Kozlov, D. Adsorbent as an essential participant in photocatalytic processes of water and air purification: Computer simulation study. Appl. Catal. A Gen. 2010, 377, 140–149. [Google Scholar] [CrossRef]
- Leary, R.; Westwood, A. Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis. Carbon 2011, 49, 741–772. [Google Scholar] [CrossRef]
- Selishchev, D.S.; Kolinko, P.A.; Kozlov, D.V. Influence of adsorption on the photocatalytic properties of TiO2/AC composite materials in the acetone and cyclohexane vapor photooxidation reactions. J. Photochem. Photobiol. A Chem. 2012, 229, 11–19. [Google Scholar] [CrossRef]
- Bouazza, N.; Lillo-Ródenas, M.A.; Linares-Solano, A. Photocatalytic activity of TiO2-based materials for the oxidation of propene and benzene at low concentration in presence of humidity. Appl. Catal. B Environ. 2008, 84, 691–698. [Google Scholar] [CrossRef]
- Pucher, P.; Benmami, M.; Azouani, R.; Krammer, G.; Chhor, K.; Bocquet, J.F.; Kanaev, A.V. Nano-TiO2 sols immobilized on porous silica as new efficient photocatalyst. Appl. Catal. A Gen. 2007, 332, 297–303. [Google Scholar] [CrossRef]
- Kitano, M.; Matsuoka, M.; Ueshima, M.; Anpo, M. Recent developments in titanium oxide-based photocatalysts. Appl. Catal. A Gen. 2007, 325, 1–14. [Google Scholar] [CrossRef]
- Xu, Y.; Langford, C.H. Photoactivity of titanium dioxide supported on MCM-41, zeolite X, and zeolite Y. J. Phys. Chem. B 1997, 101, 3115–3121. [Google Scholar] [CrossRef]
- Liu, S.X.; Chen, X.Y.; Chen, X. A TiO2/AC composite photocatalyst with high activity and easy separation prepared by a hydrothermal method. J. Hazard. Mater. 2007, 143, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Iwata, N.; Torimoto, T.; Yoneyama, H. Influence of carbon black as an adsorbent used in TiO2 photocatalyst films on photodegradation behaviors of propyzamide. J. Catal. 1998, 177, 240–246. [Google Scholar] [CrossRef]
- Anderson, C.; Bard, A.J. An Improved Photocatalyst of TiO2/SiO2 Prepared by a Sol-Gel Synthesis. J. Phys. Chem. 1995, 99, 9882–9885. [Google Scholar] [CrossRef]
- Kolinko, P.A.; Smirniotis, P.G.; Kozlov, D.V.; Vorontsov, A.V. Cr modified TiO2-loaded MCM-41 catalysts for UV-light driven photodegradation of diethyl sulfide and ethanol. J. Photochem. Photobiol. A Chem. 2012, 232, 1–7. [Google Scholar] [CrossRef]
- Panayotov, D.; Yates, J.T. Bifunctional Hydrogen Bonding of 2-Chloroethyl Ethyl Sulfide on TiO2-SiO2 Powders. J. Phys. Chem. B 2003, 107, 10560–10564. [Google Scholar] [CrossRef]
- Panayotov, D.A.; Paul, D.K.; Yates, J.T. Photocatalytic Oxidation of 2-Chloroethyl Ethyl Sulfide on TiO2-SiO2 Powders. J. Phys. Chem. B 2003, 107, 10571–10575. [Google Scholar] [CrossRef]
- Panayotov, D.; Kondratyuk, P.; Yates, J.T. Photooxidation of a Mustard Gas Simulant over TiO2-SiO2 Mixed-Oxide Photocatalyst: Site Poisoning by Oxidation Products and Reactivation. Langmuir 2004, 20, 3674–3678. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Inoue, S.; Kawada, H.; Hara, M.; Iwasaki, M.; Tada, H. Low-Temperature Synthesis of Nanometer-Sized Crystalline TiO2 Particles and Their Photoinduced Decomposition of Formic Acid. J. Colloid Interface Sci. 1999, 216, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Saur, O. The structure and stability of sulfated alumina and titania. J. Catal. 1986, 99, 104–110. [Google Scholar] [CrossRef]
- Carretero-Genevrier, A.; Boissiere, C.; Nicole, L.; Grosso, D. Distance dependence of the photocatalytic efficiency of TiO2 revealed by in situ ellipsometry. J. Am. Chem. Soc. 2012, 134, 10761–10764. [Google Scholar] [CrossRef] [PubMed]
- Childs, L.; Ollis, D. Is photocatalysis catalytic? J. Catal. 1980, 66, 383–390. [Google Scholar] [CrossRef]
- Korovin, E.; Selishchev, D.; Besov, A.; Kozlov, D. UV-LED TiO2 photocatalytic oxidation of acetone vapor: Effect of high frequency controlled periodic illumination. Appl. Catal. B Environ. 2015, 163, 143–149. [Google Scholar] [CrossRef]
- Kozlov, D.; Besov, A. Method of Spectral Subtraction of Gas-Phase Fourier Transform Infrared (FT-IR) Spectra by Minimizing the Spectrum Length. Appl. Spectrosc. 2011, 65, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are available from authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selishchev, D.; Kozlov, D. Photocatalytic Oxidation of Diethyl Sulfide Vapor over TiO2-Based Composite Photocatalysts. Molecules 2014, 19, 21424-21441. https://doi.org/10.3390/molecules191221424
Selishchev D, Kozlov D. Photocatalytic Oxidation of Diethyl Sulfide Vapor over TiO2-Based Composite Photocatalysts. Molecules. 2014; 19(12):21424-21441. https://doi.org/10.3390/molecules191221424
Chicago/Turabian StyleSelishchev, Dmitry, and Denis Kozlov. 2014. "Photocatalytic Oxidation of Diethyl Sulfide Vapor over TiO2-Based Composite Photocatalysts" Molecules 19, no. 12: 21424-21441. https://doi.org/10.3390/molecules191221424
APA StyleSelishchev, D., & Kozlov, D. (2014). Photocatalytic Oxidation of Diethyl Sulfide Vapor over TiO2-Based Composite Photocatalysts. Molecules, 19(12), 21424-21441. https://doi.org/10.3390/molecules191221424