Results and Discussion
The synthesis of
7, a
cis-fused angularly substituted perhydrindane dione, in which one of the carbonyl groups is protected as cyclic ketal, involves two phases. First Meyers' methodology for the enantioselective synthesis of hydrindenones is applied in the preparation of
5 [
3]. In a second phase, the cyclohexanone carbonyl group of
5 is protected as an ethylene ketal and the
cis-fusion in
7 is obtained by stereoselective catalytic hydrogenation of
6.
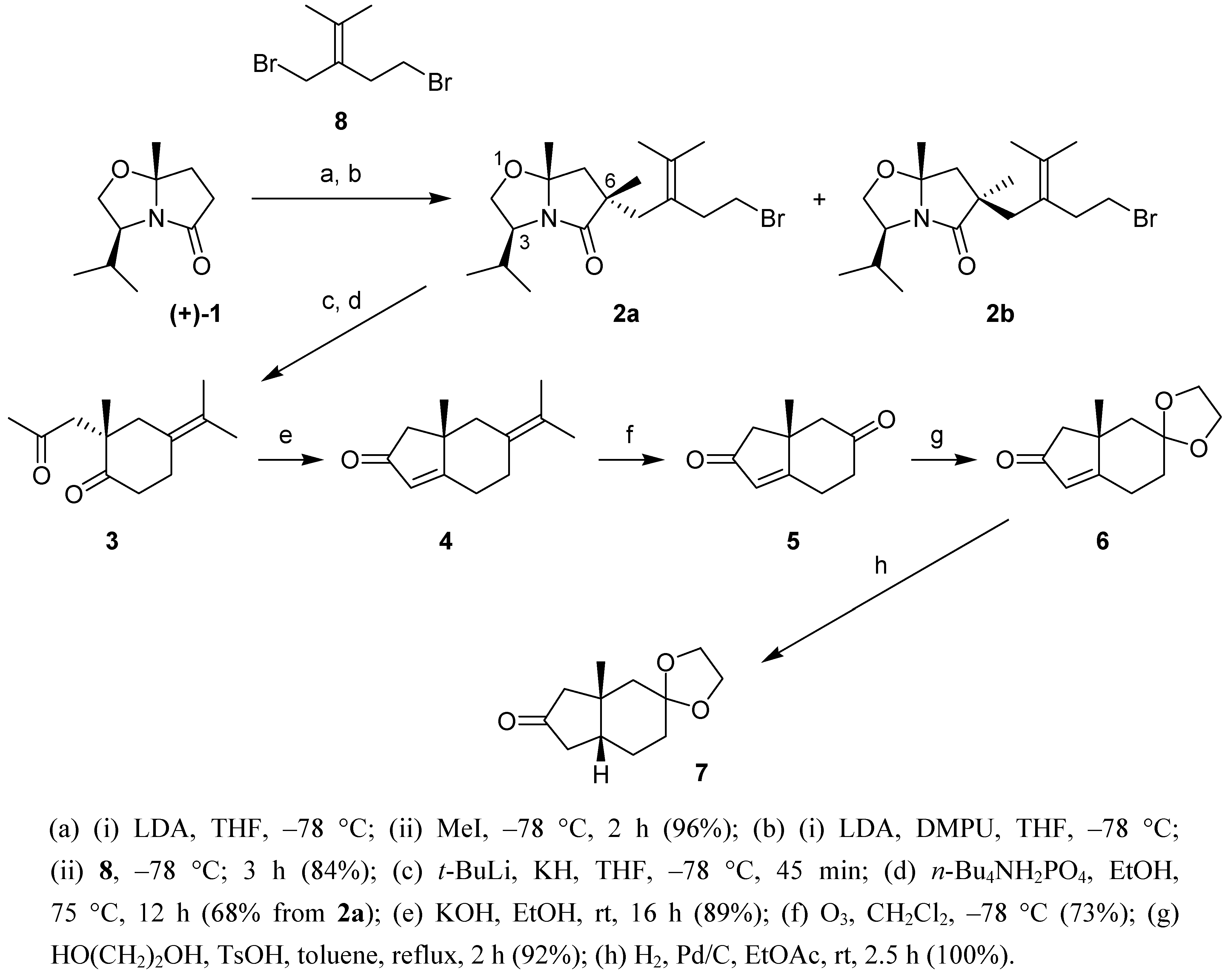
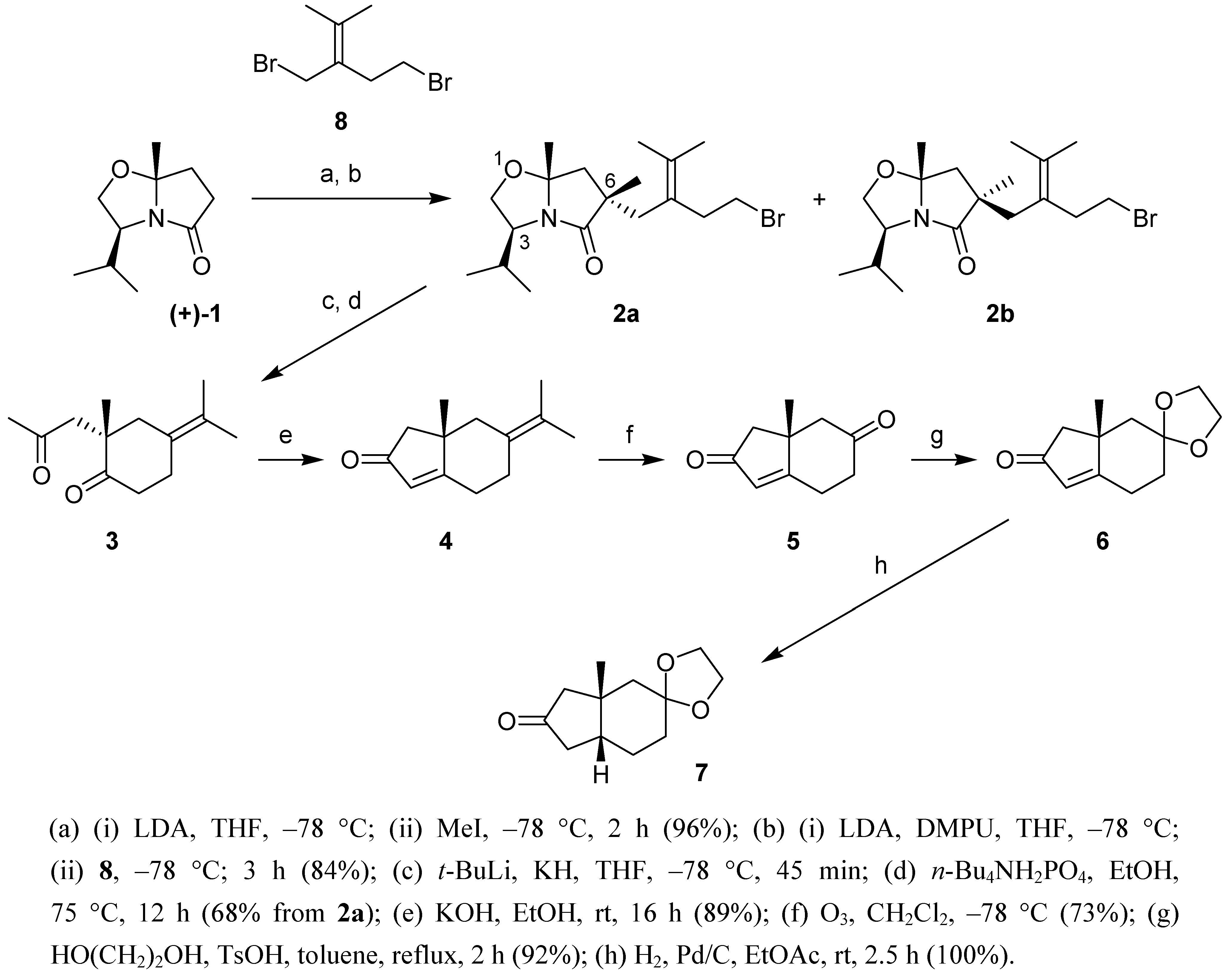
Meyers' approach for the asymmetric synthesis of angularly substituted hydrinden-2-ones proceeds in 3 stages: (i) the asymmetric introduction of the quaternary center using a chiral bicyclic lactam such as 1; (ii) the reductive intramolecular alkylation in which the 1,4-diketone is generated; (iii) the intramolecular aldolisation of the latter to the hydrinden-2-one. We chose to introduce the second carbonyl group in 5 via oxidative cleavage of the exocyclic double bond in 4. Following Meyers' methodology the latter is obtained from diastereomer 2a.
To stereoselectively obtain the correct configuration at the 6-position in
2a, the methyl group and the unsaturated side chain need to be introduced sequentially and in that precise order. Indeed, the prior introduction of the unsaturated side chain could eventually lead to the formation of spirocyclic derivatives [
4]. On the other hand, the preferred
endo-alkylation of Meyers' bicyclic lactam template has been well documented [
5]. Hence, enantiomerically pure
(+)-1 was required as starting material [
6].
Scheme 1.
Synthetic pathway to tetrahydro-1H-inden-2,6-dione 5 and derivatives.
Scheme 1.
Synthetic pathway to tetrahydro-1H-inden-2,6-dione 5 and derivatives.
(a) (i) LDA, THF, –78 °C; (ii) MeI, –78 °C, 2 h (96%); (b) (i) LDA, DMPU, THF, –78 °C; (ii) 8, –78 °C; 3 h (84%); (c) t-BuLi, KH, THF, –78 °C, 45 min; (d) n-Bu4NH2PO4, EtOH, 75 °C, 12 h (68% from 2a); (e) KOH, EtOH, rt, 16 h (89%); (f) O3, CH2Cl2, –78 °C (73%); (g) HO(CH2)2OH, TsOH, toluene, reflux, 2 h (92%); (h) H2, Pd/C, EtOAc, rt, 2.5 h (100%).
The methylation of
1 led to a 9:1 diastereomeric mixture, which was not separated. After further deprotonation with LDA (DMPU, THF) and alkylation with the known dibromide
8 [
7], a 7:3 mixture of
2a and
2b was obtained, which was separated by chromatography. The preferred formation of the
endo-isomer
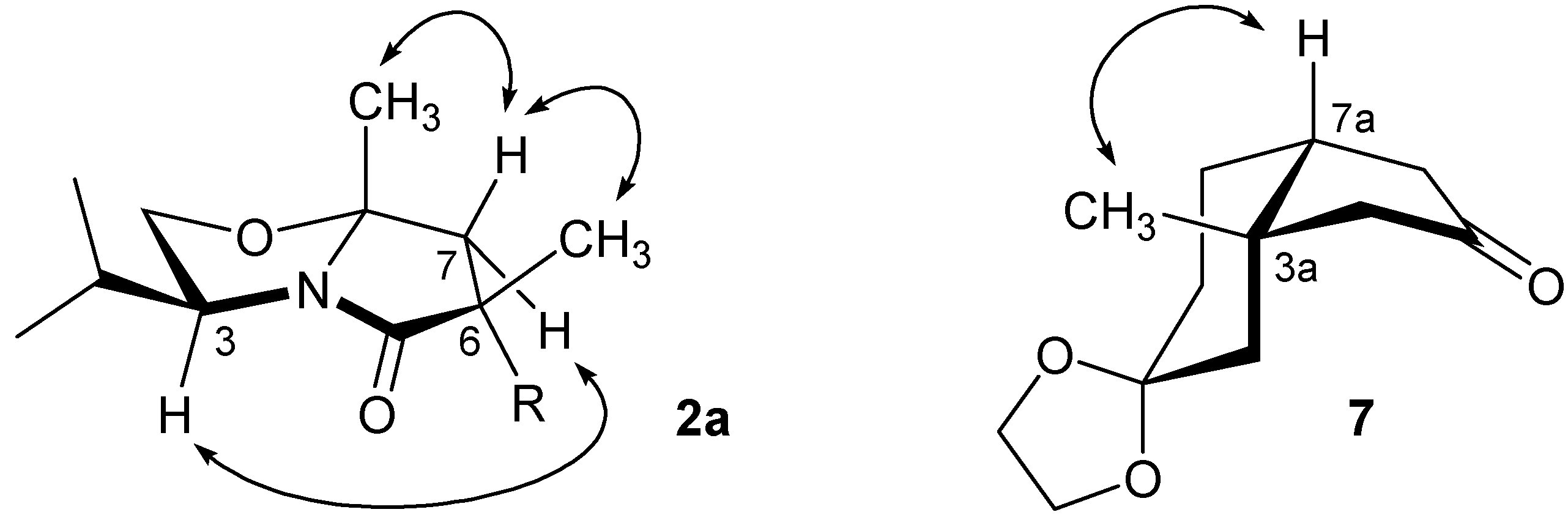
2a was proven by
1H-NMR nOe signal-enhancement studies establishing the relative positions of the Me groups and H atoms indicated in
Figure 1. The major isomer
2a was subjected to Meyers' protocol (KH,
t-BuLi) yielding diketone
3. Basic treatment (KOH, EtOH) led to hydrinden-2-one
4. Selective cleavage of the exocyclic double bond in
4 (ozone, Me
2S) gave diketone
5 in which the saturated carbonyl group was further protected to afford the ketal
6. Finally, catalytic hydrogenation of the latter occurred exclusively from the convex side of the bicyclic molecule leading to the
cis-fused perhydrindanone
7, the structure of which was confirmed by
1H-NMR structural analysis: upon irradiation of the angular Me group a clear nOe enhancement of the signal of the bridgehead hydrogen was observed, confirming the
cis-fusion of the hydrindane (
Figure 1).
Figure 1.
1H-NMR nOe signal-enhancement studies of 2a and 7.
Figure 1.
1H-NMR nOe signal-enhancement studies of 2a and 7.
Experimental
General
Tetrahydrofuran (THF) was distilled from benzophenone ketyl. Dichloromethane (DCM) was distilled from CaH2. Toluene was distilled from sodium. TLC were run on glass plates precoated with silica gel (Merck, 60F-254). Column chromatography was performed on silica gel (Merck, 230-400 mesh). IR spectra were recorded on a Perkin–Elmer series 1600 FT-IR spectrometer. 1H-NMR and 13C-NMR spectra were recorded on a Bruker AM-500 spectrometer. Hydrogen chemical shifts δ are reported in ppm relative to CDCl3 (7.26 ppm) as an internal reference. J values are given in Hz. Carbon chemical shifts δ are reported in ppm relative to CDCl3 (77.16 ppm) as an internal reference. Mass spectra (EI) were recorded on a Hewlett–Packard 5898A spectrometer at 70 eV.
(3S,6S,7aR)-6-[2-(2-Bromoethyl)-3-methylbut-2-en-1-yl]-3-isopropyl-6,7a-dimethyltetrahydro-pyrrolo[2,1-b][1,3]oxazol-5(6H)-one ((+)-2a) [8]
To i-Pr2NLi (LDA, 2 M solution in THF; 110 mL, 0.22 mol) was added dry THF (1100 mL) and the solution was cooled to –78 °C. (3S,7aR)-3-Isopropyl-7a-methyltetrahydropyrrolo[2,1-b][1,3]-oxazol-5(6H)-one ((+)-1; 20 g, 0.11 mol) was added dropwise and the mixture was stirred for 30 min. MeI (46.74 g, 20.5 mL, 0.33 mol) was then added dropwise, the reaction mixture was stirred for 2 h and then allowed to warm to rt. An aqueous saturated NH4Cl solution (50 mL) was added and the mixture was stirred for 1 h. H2O was added, the aqueous layer was extracted with Et2O (3×), the combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (isooctane/EtOAc, 7:3) to afford a 9:1 mixture of the (6R/6S)-diastereomers of the methylated bicyclic lactam (20.8 g, 96%).
To LDA (2 M solution in THF; 7.5 mL, 20 mmol) was added dry THF (100 mL) and the solution was cooled to –78 °C. 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU; 11 mL) was added and the mixture was stirred for 10 min. The above mixture of methylated bicyclic lactams (2.0 g, 10.1 mmol) in dry THF (10 mL) was added dropwise and the reaction mixture was stirred for 2 h. 5-Bromo-3-(bromomethyl)-2-methylpent-2-ene (8; 5.12 g, 20 mmol) in dry THF (10 mL) was then added dropwise and stirring was continued for 1 h. An aqueous saturated NH4Cl solution (20 mL) was added and the mixture was allowed to warm to rt. H2O was added, the aqueous layer was extracted with Et2O, the combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The obtained 7:3 mixture of isomers of the dialkylated bicyclic lactam (3.1 g, 84%) was separated by column chromatography on silica gel (isooctane/ EtOAc, 7:3) to give the major (6S)-isomer (+)-2a as a solid: mp 45 °C; Rf (isooctane/EtOAc, 95:5) 0.22; [α]Drt +106.5 (c 1.15, CHCl3); IR (KBr pellet) ν 2963, 2871, 1708, 1463, 1376, 1381, 1298, 1252, 1218, 1176, 1130, 1037, 1006), 967, 903, 842, 808, 771, 684, 650 cm–1; 1H-NMR (500 MHz, CDCl3) δ 4.18 (1 H, dd, J = 8.8, 8.8 Hz), 3.77 (1 H, dd, J = 7.9, 7.9 Hz), 3.59–3.54 (1 H, m), 3.39–3.34 (1 H, m), 3.28–3.22 (1 H, m), 2.39 (2 H, s), 2.68–2.62 (1 H, m), 2.29–2.23 (1 H, m), 2.24 (1 H, d, J = 13.5 Hz), 1.88 (1 H, d, J = 13.3 Hz), 1.71 (3 H, s), 1.66–1.63 (1 H, m), 1.63 (3 H, s), 1.50 (3 H, s), 1.29 (3 H, s), 1.04 (3 H, d, J = 6.6 Hz), 0.88 (3 H, d, J = 6.6 Hz) ppm; 13C-NMR/DEPT (50 MHz, CDCl3) δ 183.7 (C), 132.9 (C ), 126.8 (C), 96.8 (C), 70.6 (CH2), 61.6 (CH), 48.3 (C), 45.5 (CH2), 38.2 (CH2), 35.6 (CH2), 34.2 (CH), 30.9 (CH2), 27.5 (CH3), 25.8 (CH3), 21.4 (CH3), 20.7 (CH3), 20.6 (CH3), 18.9 (CH3) ppm; MS m/z (%) 372 (M+, 17), 359 (17), 358 (88), 357 (20), 356 (88), 341 (14), 331 (11), 330 (60), 328 (100), 326 (46), 310 (7), 300 (21). Anal. Calcd for C18H30BrNO2: C, 58.10; H, 8.06; N, 3.76. Found: C, 57.95; H, 8.25, N, 3.70.
(2S)-2-Methyl-4-(1-methylethylidene)-2-(2-oxopropyl)cyclohexanone ((+)-3)
To a solution of dialkylated bicyclic lactam (+)-2a (1.0 g, 2.7 mmol) in dry THF (54 mL) were added t-BuLi (1.7 M solution in pentane; 3.32 mL, 5.6 mmol) and KH (0.324 g, 8.1 mmol) at –78 °C and the reaction mixture was stirred for 45 min. H2O (20 mL) was added and the mixture was allowed to warm to rt under stirring for 20 min. The solution was concentrated under reduced pressure to 1/3 of its volume and EtOH (20 mL) was added to obtain a homogeneous solution. A 1 M n-Bu4NH2PO4 solution (26.5 mL, 0.081 mmol) was added and the mixture was refluxed for 12 h. After cooling, H2O was added, the aqueous layer was extracted with Et2O (2×), the combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (isooctane/EtOAc, 8:2) to give diketone (+)-3 (0.388 g, 68%): Rf (isooctane/EtOAc, 8:2) 0.33; [α]Drt +20.5 (c 1.10, CHCl3); IR (KBr film) ν 2963, 2921, 1710, 1456, 1360, 1167, 1104 cm–1; 1H-NMR (500 MHz, CDCl3) δ 2.98 (1 H, AB, J = 17.7 Hz), 2.69–2.63 (2 H, m), 2.56–2.47 (2 H, m), 2.49 (1 H, AB, J = 17.7 Hz), 2.44–2.38 (1 H, m), 2.34 (1 H, d, J = 14.1 Hz), 2.10 (3 H, s), 1.71 (3 H, s), 1.70 (3 H, s), 1.03 (3 H, s) ppm; 13C-NMR/APT (125 MHz, CDCl3) δ 206.9 (C), 206.9 (C), 126.1 (C), 125.3 (C), 52.2 (CH2), 47.0 (C), 38.6 (CH2), 38.6 (CH2), 30.7 (CH3), 27.2 (CH2), 24.1 (CH3), 20.4 (CH3), 20.3 (CH3); MS m/z (%) 208 (M+, 1), 150 (49), 135 (34), 107 (43), 93 (21), 79 (26), 67 (21), 43 (100).
(7aS)-7a-Methyl-6-(1-methylethylidene)-1,4,5,6,7,7a-hexahydro-2H-inden-2-one ((+)-4)
To a solution of diketone (+)-3 (2.5 g, 12 mmol) in EtOH (12 mL) was added KOH (0.64 g, 12 mmol) and the reaction mixture was stirred at rt for 16 h. H2O was added and the aqueous layer was extracted with Et2O (3×). The combined organic layers were washed with a saturated NaCl solution, dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (isooctane/EtOAc, 9:1) to give enone (+)-4 (2.0 g, 89%) as yellow crystals: mp 84 °C; Rf (isooctane/EtOAc, 8:2) 0.37; [α]Drt +53.15 (c 0.75, CHCl3); IR (KBr film) ν 2956, 2916, 2860, 2358, 1709, 1620, 1437, 1409, 1372, 1274, 1220, 1171, 840, 750, 670 cm–1; 1H-NMR (500 MHz, CDCl3) δ 5.79 (1 H, d, J = 1.6 Hz), 2.96 (1 H, m), 2.89 (1 H, dd, J = 13.0, 2.3 Hz), 2.72 (1 H, ddd, J = 13.6, 4.6, 2.3 Hz), 2.36 (1 H, ddd, J = 13.5, 13.5, 6.3 Hz), 2.33 (1 H, AB, J = 18.4 Hz), 2.28 (1 H, AB, J = 18.4 Hz), 1.82–1.75 (2 H, m), 1.77 (3 H, s), 1.72 (3 H, s), 1.13 (3H, s) ppm; 13C-NMR/APT (125 MHz, CDCl3) δ 208.2 (C), 188.0 (C), 126.8 (C), 126.2 (C), 126.0 (CH), 51.1 (CH2), 45.2 (C), 43.9 (CH2), 30.6 (CH2), 28.9 (CH2), 24.2 (CH3), 20.5 (CH3), 20.4 (CH3) ppm; MS m/z (%) 190 (M+, 100), 175 (32), 162 (10), 147 (76), 133 (15), 119 (57), 105 (44), 91 (47), 79 (38), 55 (28), 41 (45).
(7aS)-7a-Methyl-4,5,7,7a-tetrahydro-1H-indene-2,6-dione ((–)-5)
O3 was bubbled through a solution of alkene (+)-4 (0.48 g, 2.5 mmol) in DCM (126 mL) at –78 °C for 8 min. Me2S (0.3 mL, 3.4 mmol) was added, the reaction mixture was allowed to warm to 0 °C, stirred for 1 h and then allowed to warm to rt. The solvent was evaporated and the residue was purified by column chromatography on silica gel (Et2O/isooctane, 9:1) to give ketone (–)-5 (0.298 g, 73%): Rf (Et2O/isooctane, 9:1) 0.23; [α]Drt –62.0 (c 1.10, CHCl3); IR (KBr film) ν 3070, 2953, 1710, 1678, 1622, 1432, 1337, 1296, 1198, 1090, 1070, 902 cm–1; 1H-NMR (500 MHz, CDCl3) δ 6.02 (1 H, d, J = 1.7 Hz), 3.06 (1 H, ddd, J = 14.6, 7.5, 1.4 Hz), 2.83 (1 H, dddd, J = 14.6, 13.1, 6.7, 1.7 Hz), 2.67–2.61 (2 H, m), 2.52–2.44 (2 H, m), 2.43 (1 H, AB, J = 18.6 Hz), 2.39 (1 H, AB, J = 18.6 Hz), 1.25 (3 H, s) ppm; 13C-NMR/APT (125 MHz, CDCl3) δ 207.2 (C), 206.4 (C), 181.2 (C), 128.3 (C), 54.2 CH2), 51.2 (CH2), 45.4 CH), 40.0 (CH2), 26.09 (CH2), 25.5 (CH3) ppm; MS m/z (%) 164 (M+, 86), 149 (18), 136 (14), 121 (39), 107 (44), 93 (56), 79 (100), 55 (25), 53 (30).
(3a'S)-3a'-Methyl-3a',4',6',7'-tetrahydrospiro[1,3-dioxolane-2,5'-inden]-2'(3'H)-one ((+)-6)
To a solution of ketone (–)-5 (0.77 g, 4.7 mmol) in dry toluene (47 mL) were added HOCH2CH2OH (0.3 g, 0.27 mL, 4.7 mmol) and TsOH (90 mg, 0.47 mmol). The reaction mixture was refluxed for 2 h with azeotropic removal of the generated H2O (Dean–Stark). The solution was allowed to cool, concentrated under reduced pressure and an aqueous saturated NaHCO3 solution (10 mL) was added. The aqueous layer was extracted with Et2O (3 × 20 mL), the combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (Et2O/isooctane, 9:1) to give ketal (+)-6 (0.9 g, 92%) as white-yellow crystals: mp 72 °C; Rf (Et2O/isooctane, 9:1) 0.46; [α]Drt +6.28 (c 1.03, CHCl3); IR (KBr pellet) ν 2989, 2965, 2925, 1707, 1621, 1448, 1363, 1290, 1262, 1181, 1105, 1069, 1010, 952, 934, 907), 866, 844, 716 cm–1; 1H-NMR (500 MHz, CDCl3) δ 5.83 (1 H, s), 4.05–4.00 (2 H, m), 3.92 (2 H, t, J = 6.09 Hz), 2.69 (2 H, dd, J = 8.8, 3.2 Hz), 2.31 (1 H, AB, J = 18.3 Hz), 2.24 (1 H, AB, J = 18.3 Hz), 2.07–2.00 (2 H, m), 1.70–1.62 (2 H, m), 1.37 (3 H, s) ppm; 13C-NMR/APT (125 MHz, CDCl3) δ 185.97 (C), 126.8 (CH), 107.9 (C), 107.9 (C), 64.9 (CH2), 64.0 (CH2), 53.3 (CH2), 47.3 (CH2), 43.5 (C), 35.7 (CH2), 26.3 (CH3), 25.2 (CH2) ppm; MS m/z (%) 208 (M+, 26), 193 (34), 152 (13), 121 (13), 107 (26), 86 (100), 79 (35), 55 (18), 53 (21). Anal. Calcd for C12H16O3: C, 69.21; H, 7.74. Found: C, 68.86; H, 7.95.
(3a'S,7a'R)-3a'-Methylhexahydrospiro[1,3-dioxolane-2,5'-inden]-2'(3'H)-one ((+)-7)
To a solution of enone (+)-6 (1.0 g, 4.8 mmol) in EtOAc (96 mL) was added Pd/C (10%; 0.5 g, 0.48 mmol) and the reaction mixture was shaken under 4 bar H2 pressure (Parr apparatus) at rt for 2.5 h. The mixture was filtered through Celite® and the solvent was evaporated under reduced pressure to afford saturated ketone (+)-7 (1.0 g, 100%): Rf (isooctane/EtOAc, 9:1) 0.09; [α]Drt +43.4 (c 1.5, CHCl3); IR (KBr film) ν 2931, 2884, 2250, 1736, 1453, 1407, 1364, 1256, 1211, 1145, 1098, 1068, 1018, 994, 911, 732, 648, 608 cm–1; 1H NMR (500 MHz, CDCl3) δ 3.93–3.90 (4 H, m), 2.73 (1 H, AB, J = 18.4 Hz), 2.47 (1 H, dd, J = 18.8, 7.7 Hz), 2.04 (1 H, dd, J = 18.8, 4.2 Hz), 1.95–1.93 (1 H, m), 1.87 (1 H, AB, J = 18.4 Hz), 1.85–1.80 (1 H, m), 1.71–1.66 (2 H, m), 1.59–1.48 (3 H, m), 1.12 (3 H, s) ppm; 13C NMR/APT (125 MHz, CDCl3) δ 219.4 (C), 108.4 (C), 64.2 (CH2), 63.9 (CH2), 49.9 (CH2), 43.0 (CH2), 42.0 (CH2), 40.8 (CH), 39.7 (CH2), 32.0 (C), 28.8 (CH3), 26.0 (CH2) ppm; MS m/z (%) 210 (M+, 3), 153 (9), 126 (28), 99 (100), 86 (30), 55 (18), 42 (15).

{kind=link}
{kind=link}