First Principles Calculation of the Entropy of Liquid Aluminum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. General Theory
2.1. One-Body Term
2.2. Two- and Three-Body Terms
3. Results
3.1. One-Body Term
3.2. Two-Body Term
3.3. Electronic Entropy
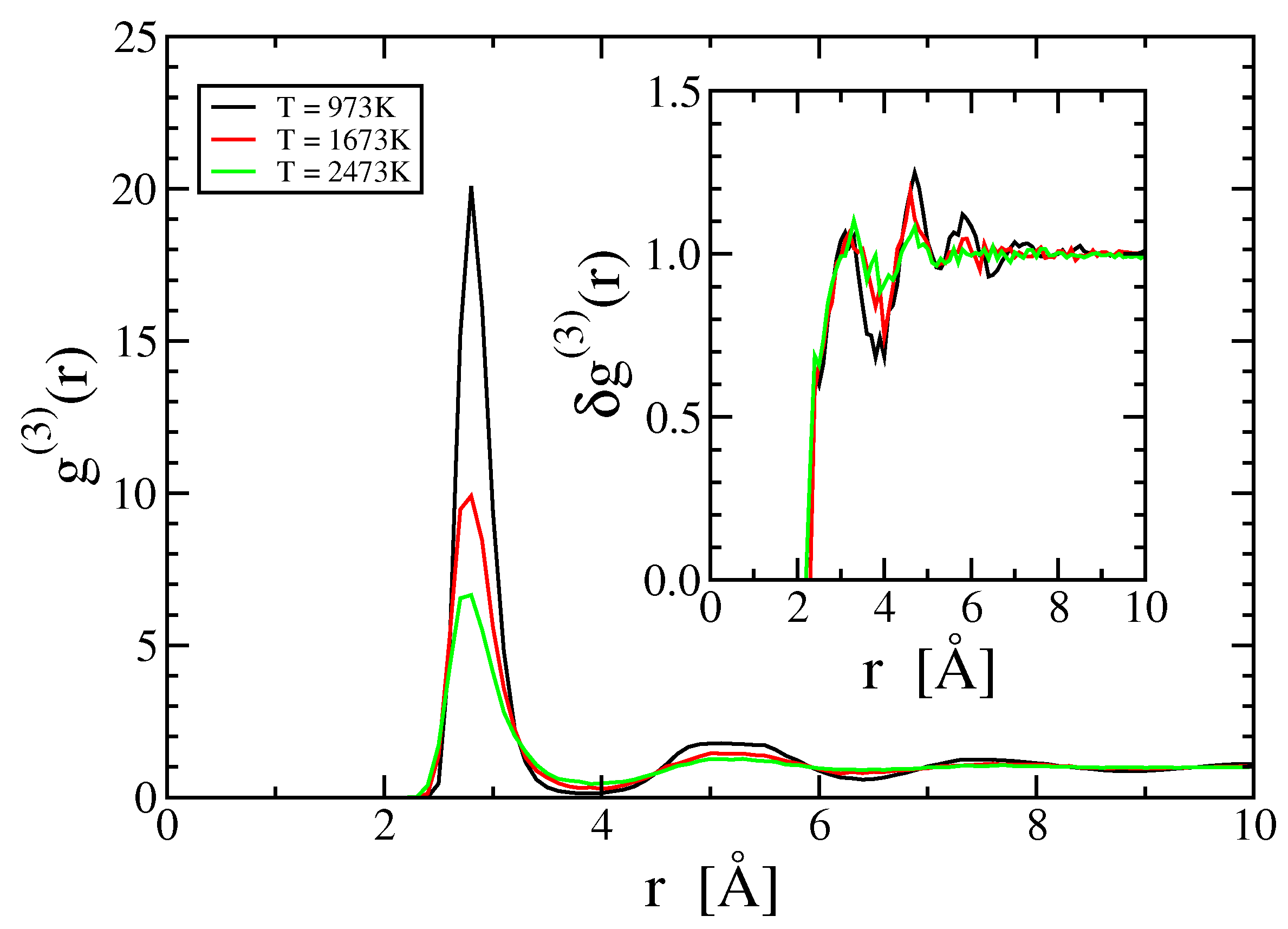
3.4. Three- and Higher-Body Terms
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Gibbs, J.W. Elementary Principles in Statistical Mechanics; Dover: New York, NY, USA, 1960. [Google Scholar]
- von Neumann, J. Thermodynamik quantenmechanischer Gesamtheiten. Nachr. Ges. Wiss. Gott. Math. Phys. Kl. 1927, 1927, 273–291. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Jaynes, E.T. Information Theory and Statistical Mechanics. Phys. Rev. 1957, 106, 620–630. [Google Scholar] [CrossRef]
- Gao, M.; Widom, M. Information entropy of liquid metals. J. Phys. Chem. B 2018, 122, 3550–3555. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, R.R. Selected Values of the Thermodynamic Properties of the Elements; American Society for Metals: Metals Park, OH, USA, 1973. [Google Scholar]
- Chase, M.; Davies, C.; Downey, J.; Frurip, D.; McDonald, R.; Syverud, A. NIST-JANAF Thermochemical Tables. 1985. Available online: http://kinetics.nist.gov/janaf/ (accessed on 5 July 2017).
- Green, H.S. The Molecular Theory of Fluids; North-Holland: Amsterdam, The Netherlands, 1952. [Google Scholar]
- Yvon, J. Correlations and Entropy in Classical Statistical Mechanics; Pergamon: Oxford, UK, 1969. [Google Scholar]
- Wallace, D.C. On the role of density fluctuations in the entropy of a fluid. J. Chem. Phys. 1987, 87, 2282–2284. [Google Scholar] [CrossRef]
- Baranyai, A.; Evans, D.J. Direct entropy calculation from computer simulation of liquids. Phys. Rev. A 1989, 40, 3817–3822. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1973; Chapter 5-3, 10-7 and 13-2. [Google Scholar]
- Rowlinson, J.S.; Widom, B. Molecular Theory of Capilarity; Clarendon Press: Oxford, UK, 1982. [Google Scholar]
- Ben-Naim, A. An informational theoretical approach to the entropy of liquids and solutions. Entropy 2018, 20, 514. [Google Scholar] [CrossRef]
- Nettleton, R.E.; Green, M.S. Expression in Terms of Molecular Distribution Functions for the Entropy Density in an Infinite System. J. Chem. Phys. 1958, 29, 1365–1370. [Google Scholar] [CrossRef]
- Raveche, H.J. Entropy and Molecular Correlation Functions in Open Systems. I. Derivation. J. Chem. Phys. 1971, 55, 2242–2250. [Google Scholar] [CrossRef]
- Mountain, R.D.; Raveche, H.J. Entropy and Molecular Correlation Functions in Open Systems. II Two- and Three-Body Correlations. J. Chem. Phys. 1971, 55, 2250–2255. [Google Scholar] [CrossRef]
- Laird, B.B.; Haymet, A.D.J. Calculation of the entropy of binary hard sphere mixtures from pair correlation functions. J. Chem. Phys. 1992, 97, 2153–2155. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Widom, M. Modeling the structure and thermodynamics of high-entropy alloys. J. Mater. Res. 2018, 33, 2881–2898. [Google Scholar] [CrossRef]
- Baranyai, A.; Evans, D.J. Three-particle contribution to the configurational entropy of simple fluids. Phys. Rev. A 1990, 42, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Laird, B.B.; Haymet, A.D.J. Calculation of the entropy from multiparticle correlation functions. Phys. Rev. A 1992, 45, 5680–5689. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, J.A.; Widom, M. First Principles Interatomic Potentials for Transition Metal Aluminides. Theory and Trends Across the 3d Series. Phys. Rev. B 1997, 56, 7905–7917. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widom, M.; Gao, M. First Principles Calculation of the Entropy of Liquid Aluminum. Entropy 2019, 21, 131. https://doi.org/10.3390/e21020131
Widom M, Gao M. First Principles Calculation of the Entropy of Liquid Aluminum. Entropy. 2019; 21(2):131. https://doi.org/10.3390/e21020131
Chicago/Turabian StyleWidom, Michael, and Michael Gao. 2019. "First Principles Calculation of the Entropy of Liquid Aluminum" Entropy 21, no. 2: 131. https://doi.org/10.3390/e21020131
APA StyleWidom, M., & Gao, M. (2019). First Principles Calculation of the Entropy of Liquid Aluminum. Entropy, 21(2), 131. https://doi.org/10.3390/e21020131