
Food Phenotyping: Recording and Processing of Non-Targeted Liquid Chromatography Mass Spectrometry Data for Verifying Food Authenticity

Abstract

1. Introduction
2. Data Acquisition of Non-Targeted LC-MS Data Sets
3. From Non-Targeted Data Sets to Marker Compounds
3.1. Data Preprocessing
3.1.1. Peak Detection/Peak Picking
3.1.2. Retention Time Alignment
3.1.3. Calculation of a Feature Matrix
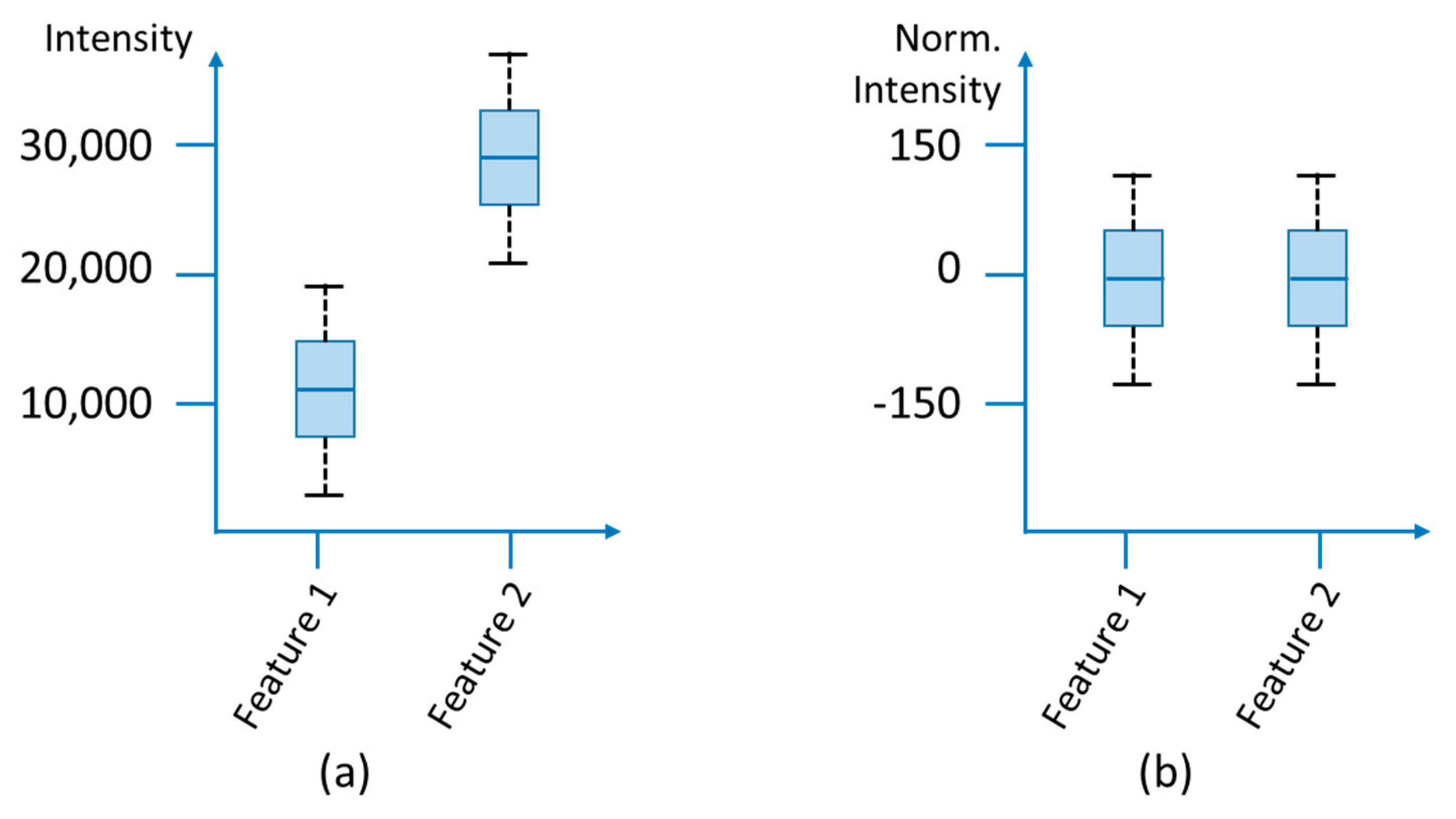
3.1.4. Normalization
3.1.5. Data Transformation
3.1.6. Scaling
- (1)
- Auto scaling (unit variance scaling): This approach is one of the most used scaling methods and has the consequence that the standard deviation becomes 1 for all features, so that all variables are equally important. This enables a comparison of the features based on their correlations and not on the basis of the covariances.
- (2)
- Pareto scaling: Compared to auto scaling, pareto scaling does not use the standard deviation, but the square root of the standard deviation as scaling factor. In this way, the data structure is better preserved because large measurement errors are reduced more than small ones. However, this method is comparatively sensitive to large fluctuations in concentration. Nevertheless, pareto scaling offers a good starting point for uncertainties about which scaling method is the most suitable.
- (3)
- Range scaling: In range scaling, the biological range is used as a scaling factor, which is calculated from the difference between the largest and the smallest value for each variable. However, this procedure is very sensitive to outliers, since the biological range is determined by only two measured values.
- (4)
- Vast scaling (variable stability scaling): The features are divided by the ratio of standard deviation and mean. As a result, the influence of variables with a small standard deviation is increased. Vast Scaling is relatively robust, but not suitable if larger variances occur.
- (5)
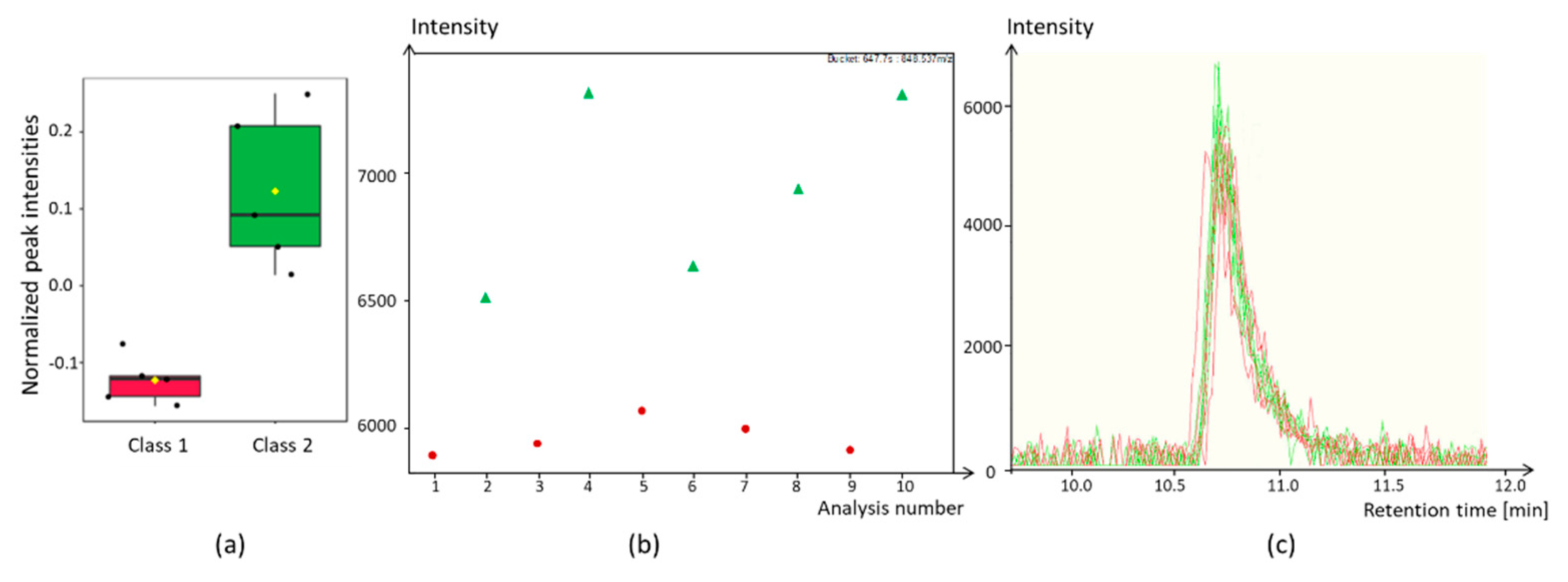
3.1.7. Dealing with Batch Effects
- (1)
- (2)
- Absolute quantitation strategy: Another option is the absolute quantitation of the analytes, if the corresponding reference substances are available, which is often not the case. In addition, the non-targeted approach is lost, since quantitation is usually not possible for all features of a non-targeted experiment [14].
- (3)
3.2. Data Processing–Application of Multivariate Analysis Methods
3.2.1. Principal Component Analysis
3.2.2. Partial Least Square Discriminant Analysis
3.2.3. Random Forests
3.2.4. Support Vector Machines
4. Evaluation of Marker Compounds
5. Identification of Marker Compounds
6. Pathway Analysis
7. Conclusions
Funding
Conflicts of Interest
References
- Ulberth, F. Tools to combat food fraud—A gap analysis. Food Chem. 2020, 330, 127044. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Fischer, M. Omics approaches for food authentication. Electrophoresis 2018, 39, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Fischer, M. Blockchain and more—Algorithm driven food traceability. Food Control 2019, 105, 45–51. [Google Scholar] [CrossRef]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Ernst, M.; Silva, D.B.; Silva, R.R.; Vêncio, R.Z.; Lopes, N.P. Mass spectrometry in plant metabolomics strategies: From analytical platforms to data acquisition and processing. Nat. Prod. Rep. 2014, 31, 784–806. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Hollywood, K.; Brison, D.R.; Goodacre, R. Metabolomics: Current technologies and future trends. Proteomics 2006, 6, 4716–4723. [Google Scholar] [CrossRef]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar] [CrossRef]
- Lv, H. Mass spectrometry-based metabolomics towards understanding of gene functions with a diversity of biological contexts. Mass Spectrom. Rev. 2013, 32, 118–128. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2001, 2, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Piñero, M.-Y.; Amo-González, M.; Ballesteros, R.D.; Pérez, L.R.; De la Mora, G.F.; Arce, L. Chemical fingerprinting of olive oils by electrospray ionization-differential mobility analysis-mass spectrometry: A new alternative to food authenticity testing. J. Am. Soc. Mass Spectrom. 2020, 31, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Hudzik, D.; Rurik, M.; Kohlbacher, O.; Fischer, M. Food authentication: Small-molecule profiling as a tool for the geographic discrimination of German white asparagus. J. Agric. Food Chem. 2018, 66, 13328–13339. [Google Scholar] [CrossRef] [PubMed]
- Klockmann, S.; Reiner, E.; Cain, N.; Fischer, M. Food Targeting: Geographical origin determination of hazelnuts (Corylus avellana) by LC-QqQ-MS/MS-based targeted metabolomics application. J. Agric. Food Chem. 2017, 65, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- England, P.; Tang, W.; Kostrzewa, M.; Shahrezaei, V.; Larrouy-Maumus, G. Discrimination of bovine milk from non-dairy milk by lipids fingerprinting using routine matrix-assisted laser desorption ionization mass spectrometry. Sci. Rep. 2020, 10, 5160. [Google Scholar] [CrossRef]
- Cao, M.; Han, Q.A.; Zhang, J.; Zhang, R.; Wang, J.; Gu, W.; Kang, W.; Lian, K.; Ai, L. An untargeted and pseudotargeted metabolomic combination approach to identify differential markers to distinguish live from dead pork meat by liquid chromatography–mass spectrometry. J. Chromatogr. A 2020, 1610, 460553. [Google Scholar] [CrossRef]
- Castro-Puyana, M.; Pérez-Míguez, R.; Montero, L.; Herrero, M. Application of mass spectrometry-based metabolomics approaches for food safety, quality and traceability. TrAC Trends Anal. Chem. 2017, 93, 102–118. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Thomas, F.; Donarski, J.; Ingallina, C.; Circi, S.; Cesare Marincola, F.; Capitani, D.; Mannina, L. Use of NMR applications to tackle future food fraud issues. Trends Food Sci. Technol. 2019, 91, 347–353. [Google Scholar] [CrossRef]
- Eisenreich, W.; Bacher, A. Advances of high-resolution NMR techniques in the structural and metabolic analysis of plant biochemistry. Phytochemistry 2007, 68, 2799–2815. [Google Scholar] [CrossRef]
- Stringer, K.A.; McKay, R.T.; Karnovsky, A.; Quémerais, B.; Lacy, P. Metabolomics and its application to acute lung diseases. Front. Immunol. 2016, 7, 44. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Duraipandian, S.; Petersen, J.; Lassen, M. Authenticity and concentration analysis of extra virgin olive oil using spontaneous raman spectroscopy and multivariate data analysis. Appl. Sci. 2019, 9, 2433. [Google Scholar] [CrossRef]
- Achten, E.; Schütz, D.; Fischer, M.; Fauhl-Hassek, C.; Riedl, J.; Horn, B. Classification of grain maize (Zea mays L.) from different geographical origins with FTIR spectroscopy—A suitable analytical tool for feed authentication? Food Anal. Methods 2019, 12, 2172–2184. [Google Scholar] [CrossRef]
- Richter, B.; Rurik, M.; Gurk, S.; Kohlbacher, O.; Fischer, M. Food monitoring: Screening of the geographical origin of white asparagus using FT-NIR and machine learning. Food Control 2019, 104, 318–325. [Google Scholar] [CrossRef]
- Arndt, M.; Rurik, M.; Drees, A.; Bigdowski, K.; Kohlbacher, O.; Fischer, M. Comparison of different sample preparation techniques for NIR screening and their influence on the geographical origin determination of almonds (Prunus dulcis MILL.). Food Control 2020, 115, 107302. [Google Scholar] [CrossRef]
- Segelke, T.; Schelm, S.; Ahlers, C.; Fischer, M. Food authentication: Truffle (Tuber spp.) species differentiation by FT-NIR and chemometrics. Foods 2020, 9, 922. [Google Scholar] [CrossRef]
- Gika, H.; Virgiliou, C.; Theodoridis, G.; Plumb, R.S.; Wilson, I.D. Untargeted LC/MS-based metabolic phenotyping (metabonomics/metabolomics): The state of the art. J. Chromatogr. B 2019, 1117, 136–147. [Google Scholar] [CrossRef]
- Jandera, P.; Janás, P. Recent advances in stationary phases and understanding of retention in hydrophilic interaction chromatography. A review. Anal. Chim. Acta 2017, 967, 12–32. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T. Our favorite materials: Silica hydride stationary phases. J. Sep. Sci. 2009, 32, 3999–4011. [Google Scholar] [CrossRef]
- Rojo, D.; Barbas, C.; Rupérez, F.J. LC-MS metabolomics of polar compounds. Bioanalysis 2012, 4, 1235–1243. [Google Scholar] [CrossRef]
- Aydoğan, C. Nanoscale separations based on LC and CE for food analysis: A. review. TrAC Trends Anal. Chem. 2019, 121, 115693. [Google Scholar] [CrossRef]
- Aszyk, J.; Byliński, H.; Namieśnik, J.; Kot-Wasik, A. Main strategies, analytical trends and challenges in LC-MS and ambient mass spectrometry–based metabolomics. TrAC Trends Anal. Chem. 2018, 108, 278–295. [Google Scholar] [CrossRef]
- Holčapek, M.; Jirásko, R.; Lísa, M. Recent developments in liquid chromatography–mass spectrometry and related techniques. J. Chromatogr. A 2012, 1259, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Knolhoff, A.M.; Kneapler, C.N.; Croley, T.R. Optimized chemical coverage and data quality for non-targeted screening applications using liquid chromatography/high-resolution mass spectrometry. Anal. Chim. Acta 2019, 1066, 93–101. [Google Scholar] [CrossRef]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef]
- Dodds, J.N.; Baker, E.S. Ion mobility spectrometry: Fundamental concepts, instrumentation, applications, and the road ahead. J. Am. Soc. Mass Spectrom. 2019, 30, 2185–2195. [Google Scholar] [CrossRef]
- Cajka, T.; Hajslova, J.; Mastovska, K. Mass spectrometry and hyphenated instruments in food analysis. In Handbook of Food Analysis Instruments; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2008; pp. 197–228. [Google Scholar]
- Pól, J.; Strohalm, M.; Havlíček, V.; Volný, M. Molecular mass spectrometry imaging in biomedical and life science research. Histochem. Cell Biol. 2010, 134, 423–443. [Google Scholar] [CrossRef]
- Soares, R.; Franco, C.; Pires, E.; Ventosa, M.; Palhinhas, R.; Koci, K.; Almeida, A.; Coelho, A. Mass spectrometry and animal science: Protein identification strategies and particularities of farm animal species. J. Proteomics 2012, 75, 4190–4206. [Google Scholar] [CrossRef]
- Shao, B.; Li, H.; Shen, J.; Wu, Y. Nontargeted detection methods for food safety and integrity. Annu. Rev. Food Sci. Technol. 2019, 10, 429–455. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Holzgrabe, U.; Diehl, B.W.K. Current role and future perspectives of multivariate (chemometric) methods in NMR spectroscopic analysis of pharmaceutical products. J. Pharm. Biomed. Anal. 2018, 147, 580–589. [Google Scholar] [CrossRef]
- McGrath, T.F.; Haughey, S.A.; Patterson, J.; Fauhl-Hassek, C.; Donarski, J.; Alewijn, M.; Van Ruth, S.; Elliott, C.T. What are the scientific challenges in moving from targeted to non-targeted methods for food fraud testing and how can they be addressed?—Spectroscopy case study. Trends Food Sci. Technol. 2018, 76, 38–55. [Google Scholar] [CrossRef]
- Nyamundanda, G.; Gormley, I.; Fan, Y.; Gallagher, W.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Van Iterson, M.; Hoen, P.A.T.; Pedotti, P.; Hooiveld, G.J.; Den Dunnen, J.T.; Van Ommen, G.J.; Boer, J.M.; Menezes, R.X. Relative power and sample size analysis on gene expression profiling data. BMC Genomics 2009, 10, 439. [Google Scholar] [CrossRef] [PubMed]
- Billoir, E.; Navratil, V.; Blaise, B.J. Sample size calculation in metabolic phenotyping studies. Briefings Bioinform. 2015, 16, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Blaise, B.; Correia, G.; Tin, A.; Young, J.; Vergnaud, A.-C.; Lewis, M.; Pearce, J.; Elliott, P.; Nicholson, J.; Holmes, E.; et al. A novel method for power analysis and sample size determination in metabolic phenotyping. Anal. Chem. 2016, 88, 5179–5188. [Google Scholar] [CrossRef]
- Ćwiek-Kupczyńska, H.; Altmann, T.; Arend, D.; Arnaud, E.; Chen, D.; Cornut, G.; Fiorani, F.; Frohmberg, W.; Junker, A.; Klukas, C.; et al. Measures for interoperability of phenotypic data: Minimum information requirements and formatting. Plant Methods 2016, 12, 44. [Google Scholar] [CrossRef]
- Taylor, C.F.; Field, D.; Sansone, S.A.; Aerts, J.; Apweiler, R.; Ashburner, M.; Ball, C.A.; Binz, P.A.; Bogue, M.; Booth, T.; et al. Promoting coherent minimum reporting guidelines for biological and biomedical investigations: The MIBBI project. Nat. Biotechnol. 2008, 26, 889–896. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Fiehn, O.; Sumner, L.W.; Rhee, S.Y.; Ward, J.; Dickerson, J.; Lange, B.M.; Lane, G.; Roessner, U.; Last, R.; Nikolau, B. Minimum reporting standards for plant biology context information in metabolomic studies. Metabolomics 2007, 3, 195–201. [Google Scholar] [CrossRef]
- Creydt, M.; Fischer, M. Metabolic imaging: Analysis of different sections of white Asparagus officinalis shoots using high-resolution mass spectrometry. J. Plant Physiol. 2020, 250, 153179. [Google Scholar] [CrossRef] [PubMed]
- Speiser, B. Leitfaden für die Probenahme und Rückstandsanalyse von Biolebensmitteln. 2013. Available online: https://orgprints.org/34117/1/speiser-2013-Leitfaden_Probenahme-Mai-2013.pdf (accessed on 1 August 2020).
- Paetz, A.; Wilke, B.-M. Soil sampling and storage. In Monitoring and Assessing Soil Bioremediation; Margesin, R., Schinner, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–45. [Google Scholar]
- European Union Law, Commission Regulation (EC) No 401/2006 of 23 February 2006 Laying Down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02006R0401-20140701 (accessed on 1 August 2020).
- European Union Law, Commission Regulation (EU) No 691/2013 of 19 July 2013 Amending Regulation (EC) No 152/2009 as Regards Methods of Sampling and Analysis. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1596292531098&uri=CELEX:32013R0691 (accessed on 1 August 2020).
- Ueda, S.; Iwamoto, E.; Kato, Y.; Shinohara, M.; Shirai, Y.; Yamanoue, M. Comparative metabolomics of Japanese Black cattle beef and other meats using gas chromatography-mass spectrometry. Biosci. Biotechnol. Biochem. 2019, 83, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Fischer, M. Food authentication in real life: How to link nontargeted approaches with routine analytics? Electrophoresis 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Klockmann, S.; Reiner, E.; Bachmann, R.; Hackl, T.; Fischer, M. Food Fingerprinting: Metabolomic approaches for geographical origin discrimination of hazelnuts (Corylus avellana) by UPLC-QTOF-MS. J. Agric. Food Chem. 2016, 64, 9253–9262. [Google Scholar] [CrossRef]
- Liu, K.H.; Nellis, M.; Uppal, K.; Ma, C.; Tran, V.; Liang, Y.; Walker, D.I.; Jones, D.P. Reference standardization for quantification and harmonization of large-scale metabolomics. Anal. Chem. 2020, 92, 8836–8844. [Google Scholar] [CrossRef]
- Beger, R.D.; Dunn, W.B.; Bandukwala, A.; Bethan, B.; Broadhurst, D.; Clish, C.B.; Dasari, S.; Derr, L.; Evans, A.; Fischer, S.; et al. Towards quality assurance and quality control in untargeted metabolomics studies. Metabolomics 2019, 15, 4. [Google Scholar] [CrossRef]
- Stoll, D.R. Contaminants everywhere! Tips and tricks for reducing background signals when using LC–MS. LC GC N. Am. 2018, 36, 498–504. [Google Scholar]
- Pyke, J.S.; Callahan, D.L.; Kanojia, K.; Bowne, J.; Sahani, S.; Tull, D.; Bacic, A.; McConville, M.J.; Roessner, U. A tandem liquid chromatography–mass spectrometry (LC–MS) method for profiling small molecules in complex samples. Metabolomics 2015, 11, 1552–1562. [Google Scholar] [CrossRef]
- Flanagan, J.M. Mass Spectrometry Calibration Using Homogeneously Substituted Fluorinated Tiazatriphosphorines. U.S. Patent 5872357A, 16 February 1999. [Google Scholar]
- Juo, C.-G.; Chen, C.-L.; Lin, S.-T.; Fu, S.-H.; Chen, Y.-T.; Chang, Y.-S.; Yu, J.-S. Mass accuracy improvement of reversed-phase liquid chromatography/electrospray ionization mass spectrometry based urinary metabolomic analysis by post-run calibration using sodium formate cluster ions. Rapid Commun. Mass Spectrom. 2014, 28, 1813–1820. [Google Scholar] [CrossRef]
- Zhou, F.; Shui, W.; Lu, Y.; Yang, P.; Guo, Y. High accuracy mass measurement of peptides with internal calibration using a dual electrospray ionization sprayer system for protein identification. Rapid Commun. Mass Spectrom. 2002, 16, 505–511. [Google Scholar] [CrossRef]
- Hannis, J.C.; Muddiman, D.C. A dual electrospray ionization source combined with hexapole accumulation to achieve high mass accuracy of biopolymers in fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 2000, 11, 876–883. [Google Scholar] [CrossRef]
- Martínez-Sena, T.; Luongo, G.; Sanjuan-Herráez, D.; Castell, J.V.; Vento, M.; Quintás, G.; Kuligowski, J. Monitoring of system conditioning after blank injections in untargeted UPLC-MS metabolomic analysis. Sci. Rep. 2019, 9, 9822. [Google Scholar] [CrossRef] [PubMed]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O’Hagan, S.; Knowles, J.D.; Halsall, A.; Wilson, I.D.; et al. Development of a robust and repeatable UPLC−MS method for the long-term metabolomic study of human serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Begou, O.; Gika, H.G.; Theodoridis, G.A.; Wilson, I.D. Quality Control and Validation Issues in LC-MS Metabolomics. Methods Mol. Biol. 2018, 1738, 15–26. [Google Scholar]
- United States Pharmacopeia (USP). Appendix XVIII: USP Guidance on Developing and Validating Non-Targeted Methods for Adulteration Detection. 2018. Available online: https://members.aoac.org/AOAC_Docs/StandardsDevelopment/Food_Auth/2019USPC-Appendix%20XVIII_Guidance_on_Developing_and_Validating_Non-Targeted_Methods_for_Adulteration_Detection-FCC_Forum_December_2018.pdf (accessed on 25 August 2020).
- Bijlsma, S.; Bobeldijk, I.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; Van Ommen, B.; Smilde, A.K. Large-Scale Human Metabolomics Studies: A Strategy for Data (Pre-) Processing and Validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar] [CrossRef]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef]
- Winkler, R. Chapter 1 Introduction. In Processing Metabolomics and Proteomics Data with Open Software: A Practical Guide; The Royal Society of Chemistry: Croydon, UK, 2020; pp. 1–25. [Google Scholar]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- ThermoFisher Scientific. Compound Discoverer Software. Available online: https://www.thermofisher.com/de/de/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/compound-discoverer-software.html (accessed on 25 August 2020).
- Bruker Daltonics. MetaboScape. Available online: https://www.bruker.com/products/mass-spectrometry-and-separations/ms-software/metaboscape.html (accessed on 25 August 2020).
- Agilent Technologies. Mass Profiler Professional. Available online: https://www.agilent.com/en/products/software-informatics/mass-spectrometry-software/data-analysis/mass-profiler-professional-software (accessed on 25 August 2020).
- Waters Corporation. Progenesis QI. Available online: https://www.waters.com/waters/en_US/Progenesis-QI/nav.htm?cid=134790652&lset=1&locale=en_US&changedCountry=Y (accessed on 25 August 2020).
- Davidson, R.L.; Weber, R.J.M.; Liu, H.; Sharma-Oates, A.; Viant, M.R. Galaxy-M: A Galaxy workflow for processing and analyzing direct infusion and liquid chromatography mass spectrometry-based metabolomics data. GigaScience 2016, 5, 10. [Google Scholar] [CrossRef]
- Liggi, S.; Hinz, C.; Hall, Z.; Santoru, M.L.; Poddighe, S.; Fjeldsted, J.; Atzori, L.; Griffin, J.L. KniMet: A pipeline for the processing of chromatography-mass spectrometry metabolomics data. Metabolomics 2018, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 37, 14. [Google Scholar]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Pfeuffer, J.; Sachsenberg, T.; Alka, O.; Walzer, M.; Fillbrunn, A.; Nilse, L.; Schilling, O.; Knut, R.; Kohlbacher, O. OpenMS—A platform for reproducible analysis of mass spectrometry data. J. Biotechnol. 2017, 261. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2014, 31, 1493–1495. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Kachlicki, P.; Stobiecki, M. Analytical methods for detection of plant metabolomes changes in response to biotic and abiotic stresses. Int. J. Mol. Sci. 2019, 20, 379. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.; Salek, R.M.; Moreno, P.; Cañueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef]
- Tian, H.; Li, B.; Shui, G. Untargeted LC–MS data preprocessing in metabolomics. J. Anal. Test. 2017, 1, 187–192. [Google Scholar] [CrossRef]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of metabolomic data: Tools, current strategies and future challenges for omics data integration. Briefings Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef]
- Misra, B.B.; Mohapatra, S. Tools and resources for metabolomics research community: A 2017–2018 update. Electrophoresis 2019, 40, 227–246. [Google Scholar] [CrossRef]
- Chernick, M.R.; González-Manteiga, W.; Crujeiras, R.M.; Barrios, E.B. Bootstrap methods. In International Encyclopedia of Statistical Science; Lovric, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 169–174. [Google Scholar]
- Ramaker, H.-J.; Van Sprang, E.N.M.; Westerhuis, J.A.; Smilde, A.K. Dynamic time warping of spectroscopic BATCH data. Anal. Chim. Acta 2003, 498, 133–153. [Google Scholar] [CrossRef]
- Skov, T.; Van den Berg, F.; Tomasi, G.; Bro, R. Automated alignment of chromatographic data. J. Chemom. 2006, 20, 484–497. [Google Scholar] [CrossRef]
- Eilers, P.H. Parametric time warping. Anal. Chem. 2004, 76, 404–411. [Google Scholar] [CrossRef]
- Lee, L.C.; Liong, C.-Y.; Jemain, A.A. Partial least squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: A review of contemporary practice strategies and knowledge gaps. Analyst 2018, 143, 3526–3539. [Google Scholar] [CrossRef]
- Xu, Y.; Goodacre, R. On splitting training and validation set: A comparative study of cross-validation, bootstrap and systematic sampling for estimating the generalization performance of supervised learning. J. Anal. Test. 2018, 2, 249–262. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; Van Velzen, E.J.J.; Van Duijnhoven, J.P.M.; Van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; Van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef]
- De Livera, A.M.; Dias, D.A.; De Souza, D.; Rupasinghe, T.; Pyke, J.; Tull, D.; Roessner, U.; McConville, M.; Speed, T.P. Normalizing and integrating metabolomics data. Anal. Chem. 2012, 84, 10768–10776. [Google Scholar] [CrossRef]
- Jauhiainen, A.; Madhu, B.; Narita, M.; Narita, M.; Griffiths, J.; Tavaré, S. Normalization of metabolomics data with applications to correlation maps. Bioinformatics 2014, 30, 2155–2161. [Google Scholar] [CrossRef]
- Cuevas-Delgado, P.; Dudzik, D.; Miguel, V.; Lamas, S.; Barbas, C. Data-dependent normalization strategies for untargeted metabolomics—A case study. Anal. Bioanal. Chem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.; Lim, M.S.; Seong, S.J.; Seo, J.J.; Park, S.M.; Lee, H.W.; Yoon, Y.R. Quantile normalization approach for liquid chromatography-mass spectrometry-based metabolomic data from healthy human volunteers. Anal. Sci. 2012, 28, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Rao, R.S.P.; Zhang, Y.; Zhong, C.X.; Thelen, J.J. A modified data normalization method for GC-MS-based metabolomics to minimize batch variation. SpringerPlus 2014, 3, 439. [Google Scholar] [CrossRef] [PubMed]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Creydt, M.; Fischer, M. Mass-spectrometry-based food metabolomics in routine applications: A basic standardization approach using housekeeping metabolites for the authentication of asparagus. J. Agric. Food Chem. 2020, in press. [Google Scholar] [CrossRef]
- Karnovsky, A.; Li, S. Pathway analysis for targeted and untargeted metabolomics. Methods Mol. Biol. 2020, 2104, 387–400. [Google Scholar]
- Khatri, P.; Drăghici, S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics 2005, 21, 3587–3595. [Google Scholar] [CrossRef]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]
- Rosato, A.; Tenori, L.; Cascante, M.; De Atauri Carulla, P.R.; Martins dos Santos, V.A.P.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef]
- Granato, D.; Santos, J.S.; Escher, G.B.; Ferreira, B.L.; Maggio, R.M. Use of principal component analysis (PCA) and hierarchical cluster analysis (HCA) for multivariate association between bioactive compounds and functional properties in foods: A critical perspective. Trends Food Sci. Technol. 2018, 72, 83–90. [Google Scholar] [CrossRef]
- Kemsley, E.K.; Defernez, M.; Marini, F. Multivariate statistics: Considerations and confidences in food authenticity problems. Food Control 2019, 105, 102–112. [Google Scholar] [CrossRef]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef]
- Triba, M.N.; Le Moyec, L.; Amathieu, R.; Goossens, C.; Bouchemal, N.; Nahon, P.; Rutledge, D.N.; Savarin, P. PLS/OPLS models in metabolomics: The impact of permutation of dataset rows on the K-fold cross-validation quality parameters. Mol. BioSyst. 2015, 11, 13–19. [Google Scholar] [CrossRef]
- Stolt, R.; Torgrip, R.J.O.; Lindberg, J.; Csenki, L.; Kolmert, J.; Schuppe-Koistinen, I.; Jacobsson, S.P. Second-order peak detection for multicomponent high-resolution LC/MS data. Anal. Chem. 2006, 78, 975–983. [Google Scholar] [CrossRef]
- Bedia, C.; Tauler, R.; Jaumot, J. Compression strategies for the chemometric analysis of mass spectrometry imaging data. J. Chemom. 2016, 30, 575–588. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chawla, N.; Bowyer, K.; Hall, L.; Kegelmeyer, W. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Sanchez-Pinto, L.N.; Venable, L.R.; Fahrenbach, J.; Churpek, M.M. Comparison of variable selection methods for clinical predictive modeling. Int. J. Med. Inform. 2018, 116, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, A.; Morlet, J. Decomposition of Hardy Functions into Square Integrable Wavelets of Constant Shape. SIAM J. Math. Anal. 1984, 15, 723–736. [Google Scholar] [CrossRef]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. TrAC, Trends Anal. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jones, D.P. Improving peak detection in high-resolution LC/MS metabolomics data using preexisting knowledge and machine learning approach. Bioinformatics 2014, 30, 2941–2948. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.D.; Tsentalovich, Y.P.; Yanshole, V.V. Deep learning for the precise peak detection in high-resolution LC–MS Data. Anal. Chem. 2020, 92, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gonzalez, E.; Hestilow, T.; Haskins, W.; Huang, Y. Review of peak detection algorithms in liquid-chromatography-mass spectrometry. Curr. Genomics 2009, 10, 388–401. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef]
- Treviño, V.; Yañez-Garza, I.-L.; Rodriguez-López, C.E.; Urrea-López, R.; Garza-Rodriguez, M.-L.; Barrera-Saldaña, H.-A.; Tamez-Peña, J.G.; Winkler, R.; Díaz de-la-Garza, R.-I. GridMass: A fast two-dimensional feature detection method for LC/MS. J. Mass Spectrom. 2015, 50, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.-Y.; Guo, J.-W.; Yu, Y.-J.; Li, H.-D.; Cui, H.-P.; Liu, P.-P.; Wang, B.; Wang, S.; Lu, P. A simple multi-scale Gaussian smoothing-based strategy for automatic chromatographic peak extraction. J. Chromatogr. A 2016, 1452, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, N.G.; Patti, G.J. Systems-level annotation of a metabolomics data set reduces 25,000 features to fewer than 1000 unique metabolites. Anal. Chem. 2017, 89, 10397–10406. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.L.; Luu, G.T.; Pierce, E.C.; Dutton, R.J.; Sanchez, L.M. BLANKA: An algorithm for blank subtraction in mass spectrometry of complex biological samples. J. Am. Soc. Mass Spectrom. 2019, 30, 1426–1434. [Google Scholar] [CrossRef]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 data-preprocessing to enhance molecular networking reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef]
- Nielsen, N.-P.V.; Carstensen, J.M.; Smedsgaard, J. Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping. J. Chromatogr. A 1998, 805, 17–35. [Google Scholar] [CrossRef]
- Tomasi, G.; Van den Berg, F.; Andersson, C. Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data. J. Chemom. 2004, 18, 231–241. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. bioeng. biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef]
- Campos, M.P.; Reis, M.S. Data preprocessing for multiblock modelling – A systematization with new methods. Chemom. Intell. Lab. Syst. 2020, 199, 103959. [Google Scholar] [CrossRef]
- Smith, R.; Ventura, D.; Prince, J.T. LC-MS alignment in theory and practice: A comprehensive algorithmic review. Briefings Bioinform. 2015, 16, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Podwojski, K.; Fritsch, A.; Chamrad, D.C.; Paul, W.; Sitek, B.; Stühler, K.; Mutzel, P.; Stephan, C.; Meyer, H.E.; Urfer, W.; et al. Retention time alignment algorithms for LC/MS data must consider non-linear shifts. Bioinformatics 2009, 25, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xie, P.; Chau, F. Chromatographic fingerprinting and related chemometric techniques for quality control of traditional Chinese medicines. J. Sep. Sci. 2010, 33, 410–421. [Google Scholar] [CrossRef]
- Grace, S.C.; Embry, S.; Luo, H. Haystack, a web-based tool for metabolomics research. BMC Bioinform. 2014, 15, S12. [Google Scholar] [CrossRef]
- Gorrochategui, E.; Jaumot, J.; Tauler, R. ROIMCR: A powerful analysis strategy for LC-MS metabolomic datasets. BMC Bioinform. 2019, 20, 256. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing value imputation approach for mass spectrometry-based metabolomics data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef]
- Smilde, A.K.; Van der Werf, M.J.; Bijlsma, S.; Van der Werff-van der Vat, B.J.C.; Jellema, R.H. Fusion of mass spectrometry-based metabolomics data. Anal. Chem. 2005, 77, 6729–6736. [Google Scholar] [CrossRef]
- Hrydziuszko, O.; Viant, M.R. Missing values in mass spectrometry based metabolomics: An undervalued step in the data processing pipeline. Metabolomics 2012, 8, 161–174. [Google Scholar] [CrossRef]
- Gromski, P.S.; Xu, Y.; Kotze, H.L.; Correa, E.; Ellis, D.I.; Armitage, E.G.; Turner, M.L.; Goodacre, R. Influence of missing values substitutes on multivariate analysis of metabolomics data. Metabolites 2014, 4, 433–452. [Google Scholar] [CrossRef]
- Do, K.T.; Wahl, S.; Raffler, J.; Molnos, S.; Laimighofer, M.; Adamski, J.; Suhre, K.; Strauch, K.; Peters, A.; Gieger, C.; et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 2018, 14, 128. [Google Scholar] [CrossRef]
- Parsons, H.M.; Ludwig, C.; Günther, U.L.; Viant, M.R. Improved classification accuracy in 1- and 2-dimensional NMR metabolomics data using the variance stabilising generalised logarithm transformation. BMC Bioinform. 2007, 8, 234. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, J.; Bearzatto, B.; Robert, A.; Govaerts, B.; Macq, B.; Gala, J.-L. Impact of the spotted microarray preprocessing method on fold-change compression and variance stability. BMC Bioinform. 2011, 12, 413. [Google Scholar] [CrossRef] [PubMed]
- Durbin, B.P.; Hardin, J.S.; Hawkins, D.M.; Rocke, D.M. A variance-stabilizing transformation for gene-expression microarray data. Bioinformatics 2002, 18, S105–S110. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate analysis in metabolomics. Curr. Metabolomics 2013, 1, 92–107. [Google Scholar]
- Rusilowicz, M.; Dickinson, M.; Charlton, A.; O’Keefe, S.; Wilson, J. A batch correction method for liquid chromatography-mass spectrometry data that does not depend on quality control samples. Metabolomics 2016, 12, 56. [Google Scholar] [CrossRef]
- Sánchez-Illana, Á.; Piñeiro-Ramos, J.D.; Sanjuan-Herráez, J.D.; Vento, M.; Quintás, G.; Kuligowski, J. Evaluation of batch effect elimination using quality control replicates in LC-MS metabolite profiling. Anal. Chim. Acta 2018, 1019, 38–48. [Google Scholar] [CrossRef]
- Rodríguez-Coira, J.; Delgado-Dolset, M.I.; Obeso, D.; Dolores-Hernández, M.; Quintás, G.; Angulo, S.; Barber, D.; Carrillo, T.; Escribese, M.M.; Villaseñor, A. Troubleshooting in large-scale LC-ToF-MS metabolomics analysis: Solving complex Issues in big cohorts. Metabolites 2019, 9, 247. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef]
- Mak, T.D.; Goudarzi, M.; Laiakis, E.C.; Stein, S.E. Disparate metabolomics data reassembler: A novel algorithm for agglomerating incongruent LC-MS metabolomics datasets. Anal. Chem. 2020, 92, 5231–5239. [Google Scholar] [CrossRef]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine learning applications for mass spectrometry-based metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Jannat, B.; Ghorbani, K.; Kouchaki, S.; Sadeghi, N.; Eslamifarsani, E.; Rabbani, F.; Beyramysoltan, S. Distinguishing tissue origin of bovine gelatin in processed products using LC/MS technique in combination with chemometrics tools. Food Chem. 2020, 319, 126302. [Google Scholar] [CrossRef]
- Barbosa, S.; Saurina, J.; Puignou, L.; Núñez, O. Classification and authentication of paprika by UHPLC-HRMS fingerprinting and multivariate calibration methods (PCA and PLS-DA). Foods 2020, 9, 486. [Google Scholar] [CrossRef]
- Mi, S.; Shang, K.; Li, X.; Zhang, C.-H.; Liu, J.-Q.; Huang, D.-Q. Characterization and discrimination of selected China’s domestic pork using an LC-MS-based lipidomics approach. Food Control 2019, 100, 305–314. [Google Scholar] [CrossRef]
- Bevilacqua, M.; Bro, R. Can we trust score plots? Metabolites 2020, 10, 278. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12, 253. [Google Scholar] [CrossRef]
- Chung, D.; Keles, S. Sparse partial least squares classification for high dimensional data. Stat. Appl. Genet. Mol. Biol. 2010, 9, 17. [Google Scholar] [CrossRef]
- Ruiz-Perez, D.; Guan, H.; Madhivanan, P.; Mathee, K.; Narasimhan, G. So you think you can PLS-DA? bioRxiv 2020, in press. [Google Scholar] [CrossRef]
- Senizza, B.; Rocchetti, G.; Ghisoni, S.; Busconi, M.; De Los Mozos Pascual, M.; Fernandez, J.A.; Lucini, L.; Trevisan, M. Identification of phenolic markers for saffron authenticity and origin: An untargeted metabolomics approach. Food Res. Int. 2019, 126, 108584. [Google Scholar] [CrossRef]
- Rocchetti, G.; Lucini, L.; Gallo, A.; Masoero, F.; Trevisan, M.; Giuberti, G. Untargeted metabolomics reveals differences in chemical fingerprints between PDO and non-PDO Grana Padano cheeses. Food Res. Int. 2018, 113, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Ma, Y.; Zhang, D.; Qian, L. Discrimination of conventional and organic rice using untargeted LC-MS-based metabolomics. J. Cereal Sci. 2018, 82, 73–81. [Google Scholar] [CrossRef]
- Cain, N.; Alka, O.; Segelke, T.; Von Wuthenau, K.; Kohlbacher, O.; Fischer, M. Food fingerprinting: Mass spectrometric determination of the cocoa shell content (Theobroma cacao L.) in cocoa products by HPLC-QTOF-MS. Food Chem. 2019, 298, 125013. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-l.; Dai, R. Metabolomic data processing based on mass spectrometry platforms. In Plant Metabolomics: Methods and Applications; Qi, X., Chen, X., Wang, Y., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 123–169. [Google Scholar]
- Zhu, M.; Xia, J.; Jin, X.; Yan, M.; Cai, G.; Yan, J.; Ning, G. Class weights random forest algorithm for processing class imbalanced medical data. IEEE Access 2018, 6, 4641–4652. [Google Scholar] [CrossRef]
- Weber, R.J.M.; Lawson, T.N.; Salek, R.M.; Ebbels, T.M.D.; Glen, R.C.; Goodacre, R.; Griffin, J.L.; Haug, K.; Koulman, A.; Moreno, P.; et al. Computational tools and workflows in metabolomics: An international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 2017, 13, 12. [Google Scholar] [CrossRef]
- Kalogiouri, N.P.; Aalizadeh, R.; Thomaidis, N.S. Application of an advanced and wide scope non-target screening workflow with LC-ESI-QTOF-MS and chemometrics for the classification of the Greek olive oil varieties. Food Chem. 2018, 256, 53–61. [Google Scholar] [CrossRef]
- Kalogiouri, N.P.; Aalizadeh, R.; Thomaidis, N.S. Investigating the organic and conventional production type of olive oil with target and suspect screening by LC-QTOF-MS, a novel semi-quantification method using chemical similarity and advanced chemometrics. Anal. Bioanal. Chem. 2017, 409, 5413–5426. [Google Scholar] [CrossRef]
- Ben-Hur, A.; Ong, C.S.; Sonnenburg, S.; Schölkopf, B.; Rätsch, G. Support vector machines and kernels for computational biology. PLoS Comput. Biol. 2008, 4, e1000173. [Google Scholar] [CrossRef]
- Xu, Y.; Zomer, S.; Brereton, R. Support vector machines: A recent method for classification in chemometrics. Crit. Rev. Anal. Chem. 2006, 36, 177–188. [Google Scholar] [CrossRef]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—a marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef]
- Chih-Wei, H.; Chih-Jen, L. A comparison of methods for multiclass support vector machines. IEEE Trans. Neural Netw. Learn. Syst. 2002, 13, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Mendez, K.M.; Reinke, S.N.; Broadhurst, D.I. A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics 2019, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid. Based Complement. Alternat. Med. 2013, 2013, 298183. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Series B, Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Bewick, V.; Cheek, L.; Ball, J. Statistics review 13: Receiver operating characteristic curves. Crit. Care 2004, 8, 508–512. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS Data in metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- FooDB. Available online: https://foodb.ca (accessed on 25 August 2020).
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [PubMed]
- MoNA—MassBank of North America. Available online: https://mona.fiehnlab.ucdavis.edu/ (accessed on 25 August 2020).
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 - PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Reports in Computational Chemistry; Wheeler, R.A., Spellmeyer, D.C., Eds.; Elsevier: Bethesda, MD, USA, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminf. 2016, 8, 3. [Google Scholar] [CrossRef]
- Ridder, L.; Van der Hooft, J.J.J.; Verhoeven, S.; De Vos, R.C.H.; Van Schaik, R.; Vervoort, J. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 2012, 26, 2461–2471. [Google Scholar] [CrossRef]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Witting, M.; Böcker, S. Current status of retention time prediction in metabolite identification. J. Sep. Sci. 2020, 43, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. MetaboLights: An Open-Access Database Repository for Metabolomics Data. Curr. Protoc. Bioinform. 2016, 53, 14. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2017, 46, D633–D639. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The Chemical Translation Service—a web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef]
- Kessler, N.; Neuweger, H.; Bonte, A.; Langenkämper, G.; Niehaus, K.; Nattkemper, T.W.; Goesmann, A. MeltDB 2.0-advances of the metabolomics software system. Bioinformatics 2013, 29, 2452–2459. [Google Scholar] [CrossRef]
- Xia, J. Computational strategies for biological interpretation of metabolomics Ddata. In Metabolomics: From Fundamentals to Clinical Applications; Sussulini, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 191–206. [Google Scholar]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633–651. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass Analyzer | Resolution | Mass Accuracy | Scan Rate | m/z Range | Linear Dynamic Range | Sensitivity | Quantitation | Handling | Cost Effort |
|---|---|---|---|---|---|---|---|---|---|
| FT-ICR-MS | +++++ | +++++ | ++ | ++++ | +++ | ++ | ++ | + | +++++ |
| Orbitrap | ++++ | +++++ | +++ | +++ | +++ | +++ | ++ | +++ | ++++ |
| ToF/QToF | +++ | ++++ | +++++ | +++++ | ++++ | ++++ | ++++ | +++ | +++ |
| QTrap | ++ | +++ | ++++ | ++ | +++ | +++++ | +++ | +++++ | ++ |
| QqQ | ++ | + | ++++ | ++ | +++++ | +++++ | +++++ | +++++ | + |
| Software | Provider | Access | Reference |
|---|---|---|---|
| Commercial programs for chemometric evaluation of LC-MS data | |||
| Compound Discoverer | ThermoFisher Scientific, Waltham, MA, USA | local installation required | [78] |
| DataAnalysis, ProfileAnalysis, MetaboScape | Bruker Daltonics, Bremen, Germany | local installation required | [79] |
| Mass Profiler Professional and various other modules that can be combined to design different workflows | Agilent Technologies, Santa Clara, CA, USA | local installation required | [80] |
| Progenesis QI | Progenesis QI Waters Corporation, Milford, MA, USA | local installation required | [81] |
| Freely available metabolomics tools | |||
| Galaxy-M | School of Biosciences, University of Birmingham, Birmingham, UK | local installation required | [82] |
| KnitMet | Department of Biochemistry and Cambridge Systems Biology Centre, University of Cambridge, Cambridge, UK | local installation required | [83] |
| MAVEN | Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NY, USA | local installation required | [84] |
| MetaboAnalyst | Xia Lab at McGill University, Montreal, QC, Canada | web-based | [44] |
| MZmine 2 | Okinawa Institute of Science and Technology (OIST), Onna, Okinawa, Japan / Quantitative Biology and Bioinformatics, VTT Technical Research Centre of Finland, Espoo, Finland | local installation required | [85] |
| OpenMS | Center for Integrative Bioinformatics (CIBI), University of Tübingen, Tübingen, Germany | local installation required | [86] |
| Workflow4Metabolomics | National Research Institute for Agriculture, Food and Environment, Paris, France | web-based | [87] |
| XCMS online | The Scripps Research Institute, La Jolla, CA, USA | web-based | [88] |
| Term | Explanation |
|---|---|
| Analysis of variance (ANOVA) | In contrast to the t-test, significant differences of more than two sample groups can be compared using ANOVA. |
| Bias | Random errors, which are based, for example, on inaccuracies in sample preparation, injection and fluctuations in the measuring instruments. |
| Bootstrap approach | Resampling method, which means that a sample can be used more than once. It can also be applied for non-normally distributed data [94]. |
| Correlation optimized warping (COW), dynamic time warping (DTW) and Parametric Time Warping (PTW) | Different warping algorithms that are used for retention time alignment by shifting, stretching or reducing the retention time axis. DTW [95] works point-wise, COW [96] segment-wise and PTW [97] is based on a polynomial transformation. |
| Cross validation (CV) | CV is an internal method for the validation of supervised models to check the predictive power and rule out overfitting. In this approach, a model is first calculated with the help of a training set, which is checked with a test data set. The process is repeated several times [98,99,100]. |
| Feature | In LC-MS analyses, a feature is defined based on retention time and m/z. |
| Mean Centering | Subtraction of the average of a feature from each measure of that feature so that the new average of that feature is zero. The interpretation of the data is made easier because the differences are in the foreground and an offset of the data is eliminated [101]. |
| Normalization | Ensures the comparability of the samples with each other by eliminating systematic errors, e.g., from different sample weights or dilutions [102,103,104,105,106,107,108,109,110]. |
| Null hypothesis (H0) | The null hypothesis is based on the assumption that there is no difference in various sample groups and should usually be rejected. This indirect approach is intended to prevent the likelihood of false positive results. |
| Out-of-bag (OOB) error | The OOB error is used to describe the predictive power of random forests models. |
| Over-representation analysis (ORA), functional class scoring (FCS), pathway topology (PT), mummichog, gene set enrichment analysis (GSEA) | Different algorithms for performing pathway analyses. The identification of metabolites is not necessary for the mummichog and GSEA algorithm [111,112,113,114,115]. |
| Overfitting | Overinterpretation of a data set. Correlations are recognized that are based on noise signals and not on real differences between the samples. |
| Permutation test | The class names are swapped randomly, and a new classification model is calculated on this basis. This new model should not be able to achieve a good separation of the different groups of samples [44]. |
| Principal component analysis (PCA) | Unsupervised method to show differences and similarities in various samples by orthogonal transformation. This approach is often used to get a first overview of the data [116,117,118,119]. |
| Partial least square discriminant analysis (PLS-DA), orthogonal PLS-DA (OPLS-DA), sparse PLS-DA (SPLS-DA) | Fast and simple supervised method, which sometimes tends to overfit. Therefore, careful validation should take place. OPLS-DA and SPLS-DA are extensions of a classical PLS-DA [98,99,100]. |
| R2 and Q2 | Parameters for the assessment of supervised methods to identify possible overfitting. R2 (goodness of fit) describes the proportion of the declared variance in the total variance. R2 can have a maximum value of 1. In the ideal case, a model should achieve the largest possible R2 value. Q2 (goodness of prediction) describes the prediction accuracy of a model and is obtained from a cross validation. Q2 can have a maximum of 1 [100,120,121]. |
| Regions of interest (ROI) | ROI describe a relevant measuring range that contains a supposed signal [122,123]. |
| Random forests (RF) | RF are based on decision trees, can also be used for very noisy data and small sample groups. Robust to overfitting and outliers, but equally large class sizes must be ensured. The visualization is quite complex, so VIP plots are often used to extract the most relevant features [124]. |
| Random oversampling (ROS) und synthetic minority over-sampling technique (SMOTE) | For some multivariate analysis methods, such as RF or SVM, class sizes must be the same. This requirement can either be achieved by excluding individual samples (undersampling) or by performing ROS. For example, by taking individual samples into account several times or calculating them synthetically. For the latter, the SMOTE algorithm is suitable. Briefly explained, the difference is calculated from a feature based on the intensities or peak areas found in two samples of the same class. The result is multiplied by a randomized number between 0 and 1. The lower feature value of the two samples is then added. A new value is obtained, which lies between the feature values of the two known samples [125]. |
| Scaling | Ensures the comparability of the different features with each other, since signals with strong intensities, compared to signals with lower intensities, otherwise have a greater influence [101]. |
| Support vector machines (SVM) | SVM is a kernel method. Robust to overfitting and outliers, sensitive to imbalance datasets. High calculation effort, can take some time with many samples and features [126]. |
| Transformation | Ensures that heteroscedasticity and skewness of the data are reduced to achieve an almost normal distribution of the data [101]. |
| Underfitting | The opposite of overfitting, which occurs when relevant features are not taken into account. |
| VIP | Variable importance in projection, reflects the influence of a feature on a model. Promising features have a VIP score >1. However, this limit should not be seen too narrowly. Features with a VIP score <0.5 are irrelevant for a model [127]. |
| Wavelet transformation | Transformation method developed by Morlet and Grossmann. In a way, an extension of a Fourier transform, which can also be used for signals with different lengths and frequencies, and which enables time and location to be resolved [128]. |
| Database | Provider | Availability of LC-MS/MS Reference Spectra | Reference |
|---|---|---|---|
| Chemspider | Royal Society of Chemistry, London, UK | Experimental LC-MS / MS spectra available for some compounds | [197] |
| FooDB | Canadian Institutes of Health Research, Canada Foundation for Innovation, Ottawa, Canada/The Metabolomics Innovation Centre, Edmonton, AB, Canada | Experimental LC-MS / MS spectra for numerous compounds are available where no original spectra are available, in-silco spectra can be used | [198] |
| HMDB (Human Metabolome Database) | Canadian Institutes of Health Research, Canada Foundation for Innovation, Ottawa, Canada/The Metabolomics Innovation Centre, Edmonton, AB, Canada | Experimental LC-MS / MS spectra for numerous compounds are available where no original spectra are available, in-silco spectra can be used | [199] |
| KNApSAcK | Nara Institute of Science and Technology, Nara, Japan | No, but helpful links to further primary literature | [200] |
| MoNA (Mass Bank of North America) | Fiehn Lab, Davis, CA, USA | Experimental LC-MS / MS spectra for numerous compounds are available | [201] |
| LipidMaps | Cardiff University, Cardiff UK/Babraham Institute, Cambridge, UK/University of California, San Diego, CA, USA | Spectra from other databases are partially embedded, and there is also the option of predicting MS / MS spectra for certain lipid classes | [202] |
| MassBank | Mass Spectrometry Society of Japan, Tokyo, Japan | Experimental LC-MS / MS spectra for numerous compounds are available | [203] |
| METLIN (Metabolite and Chemical Entity Database) | The Scripps Research Institute, Loa Jolla, CA, USA | Experimental LC-MS / MS spectra for numerous compounds are available | [204] |
| Pubchem | National Center for Biotechnology Information, Rockville Pike, MD, USA | Spectra from other databases are partially embedded | [205] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creydt, M.; Fischer, M. Food Phenotyping: Recording and Processing of Non-Targeted Liquid Chromatography Mass Spectrometry Data for Verifying Food Authenticity. Molecules 2020, 25, 3972. https://doi.org/10.3390/molecules25173972
Creydt M, Fischer M. Food Phenotyping: Recording and Processing of Non-Targeted Liquid Chromatography Mass Spectrometry Data for Verifying Food Authenticity. Molecules. 2020; 25(17):3972. https://doi.org/10.3390/molecules25173972
Chicago/Turabian StyleCreydt, Marina, and Markus Fischer. 2020. "Food Phenotyping: Recording and Processing of Non-Targeted Liquid Chromatography Mass Spectrometry Data for Verifying Food Authenticity" Molecules 25, no. 17: 3972. https://doi.org/10.3390/molecules25173972
APA StyleCreydt, M., & Fischer, M. (2020). Food Phenotyping: Recording and Processing of Non-Targeted Liquid Chromatography Mass Spectrometry Data for Verifying Food Authenticity. Molecules, 25(17), 3972. https://doi.org/10.3390/molecules25173972