
Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol Over Terra Nova Bay, Antarctica


 ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
2.1. PHg Concentrations in Antarctica
2.2. Size-Resolved Mercury Distribution
2.3. Seasonal Evolution of PHg in Antarctica
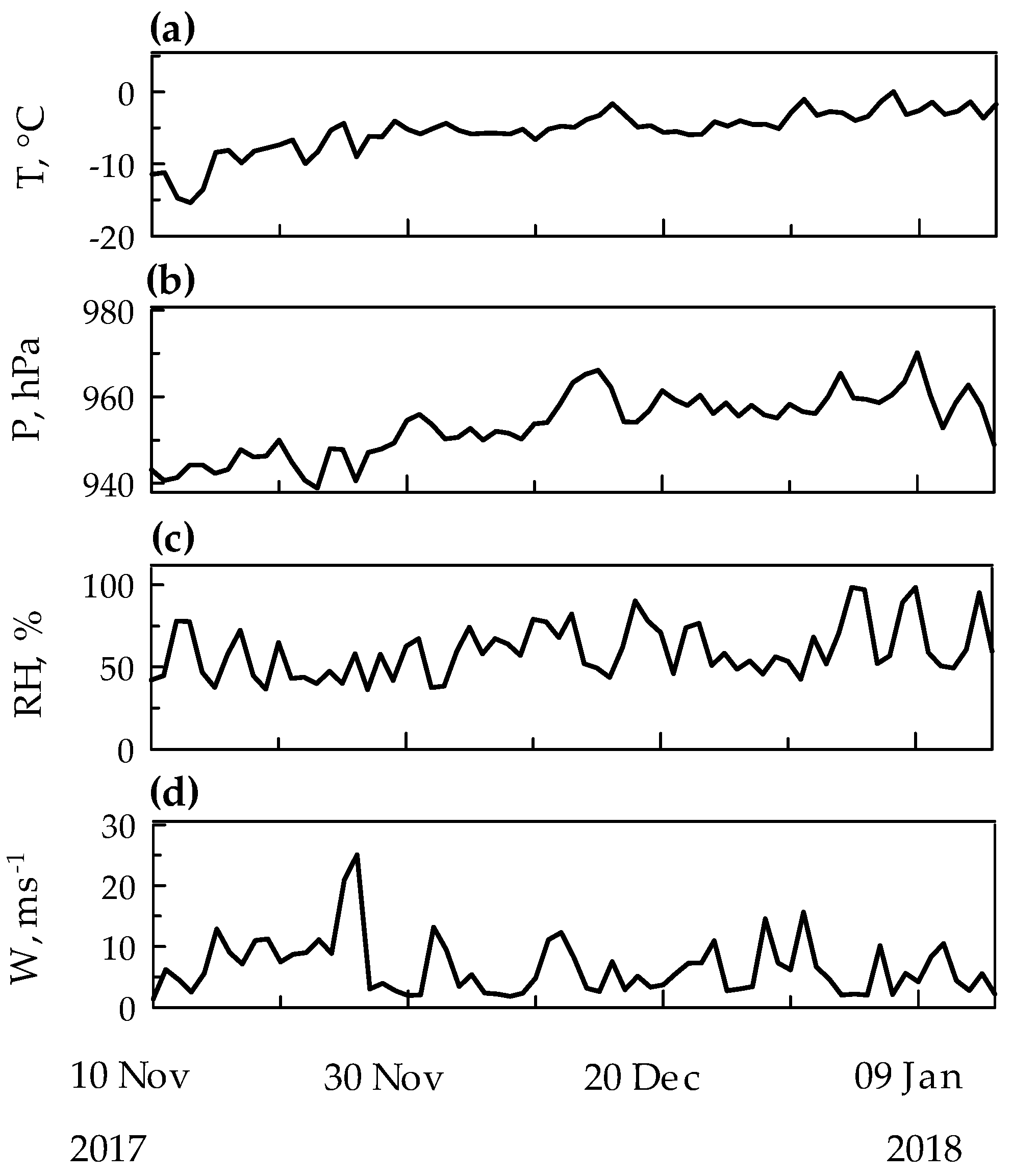
2.4. PHg and Meteorological Conditions
2.5. Estimation of PHg Dry Deposition Fluxes
3. Materials and Methods
3.1. The Study Area
3.2. Field Sampling
3.3. Mercury Analysis
3.4. Quality Control
3.5. Data Inversion and Statistical Analysis
3.6. Calculation of The Hg Dry Deposition Flux
3.7. Air Mass Origin
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pirrone, N.; Maffey, K.R. Where We Stand on Mercury Pollution and its Health Effects on Regional and Global Scales. In Dynamics of Mercury Pollution on Regional and Global Scales: Atmospheric Processes and Human Exposures around the World; Pirrone, N., Mahaffey, K.R., Eds.; Springer: Boston, MA, USA, 2005; Volume 1, pp. 1–21. [Google Scholar]
- Wangberg, I.; Munthe, J.; Pirrone, N.; Iverfeldt, A.; Bahlman, E.; Costa, P.; Ebinghaus, R.; Feng, X.; Ferrara, R.; Gardfeldt, K.; et al. Atmospheric mercury distribution in northern Europe and in the Mediterranean region. Atmos. Environ. 2001, 35, 3019–3025. [Google Scholar] [CrossRef]
- Dommergue, A.; Sprovieri, F.; Pirrone, N.; Ebinghaus, R.; Brooks, S.; Courteaud, J.; Ferrari, C.P. Overview of mercury measurements in the Antarctic troposphere. Atmos. Chem. Phys. 2010, 10, 3309–3319. [Google Scholar] [CrossRef]
- Pacyna, E.G.; Pacyna, J.M.; Steenhuisen, F.; Wilson, S. Global anthropogenic mercury emission inventory for 2000. Atmos. Environ. 2006, 40, 4048–4063. [Google Scholar] [CrossRef]
- Pacyna, E.G.; Pacyna, J.M.; Sundseth, K.; Munthe, J.; Kindbom, K.; Wilson, S.; Steenhuisen, F.; Maxson, P. Global emission of mercury to the atmosphere from anthropogenic sources in 2005 and projections to 2020. Atmos. Environ. 2010, 44, 2487–2499. [Google Scholar] [CrossRef]
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, R.B.; Friedli, H.R.; Leaner, J.; Mason, R.; Mukherjee, A.B.; Stracher, G.B.; Streets, D.G.; et al. Global mercury emissions to the atmosphere from anthropogenic and natural sources. Atmos. Chem. Phys. 2010, 10, 5951–5964. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Li, Y.; Li, T.; Mao, H.; Talbot, R.; Nie, X.; Wu, C.; Zhao, Y.; Hou, C.; et al. Characteristics and potential sources of atmospheric particulate mercury in Jinan, China. Sci. Total Environ. 2017, 574, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- UN Environment Programme, Chemicals and Health Branch Geneva, Switzerland. Global Mercury Assessment 2018; Narayana Press: Gylling, Denmark, 2019; p. 62. ISBN 978-92-807-3744-8. [Google Scholar]
- Lindberg, S.; Bullock, R.; Ebinghaus, R.; Engstrom, D.; Feng, X.; Fitzgerald, W.; Pirrone, N.; Prestbo, E.; Seigneur, C. A Synthesis of Progress and Uncertainties in Attributing the Sources of Mercury in Deposition. Ambio 2007, 36, 19–32. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Anlauf, K.G.; Barrie, L.A.; Lu, J.Y.; Steffen, A.; Schneeberger, D.R.; Berg, T. Arctic springtime depletion of mercury. Nature 1998, 394, 331–332. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pan, L.; Streets, D.G.; Shetty, S.K.; Jang, C.; Feng, X.; Chu, H.-W.; Ho, T.C. Estimating mercury emission outflow from East Asia using CMAQ-Hg. Atmos. Chem. Phys. 2010, 10, 1853–1864. [Google Scholar] [CrossRef]
- Steffen, A.; Lehnherr, I.; Cole, A.; Ariya, P.; Dastoor, A.; Durnford, D.; Kirk, J.; Pilote, M. Atmospheric mercury in the Canadian Arctic. Part I: A review of recent field measurements. Sci. Total Environ. 2015, 509–510, 3–15. [Google Scholar] [CrossRef]
- Xiu, G.L.; Jin, Q.; Zhang, D.; Shi, S.; Huang, X.; Zhang, W.; Bao, L.; Gao, P.; Chen, B. Characterization of size-fractionated particulate mercury in Shanghai ambient air. Atmos. Environ. 2005, 39, 419–427. [Google Scholar] [CrossRef]
- Shannon, J.D.; Voldner, E.C. Modeling atmospheric concentrations of mercury and deposition to the great lakes. Atmos. Environ. 1995, 29, 1649–1661. [Google Scholar] [CrossRef]
- Fang, F.; Wang, Q.; Li, J. Atmospheric particulate mercury concentration and its dry deposition flux in Changchun City, China. Sci. Total Environ. 2001, 281, 229–236. [Google Scholar] [CrossRef]
- Lehnherr, I. Methylmercury biogeochemistry: A review with special reference to Arctic aquatic ecosystems. Environ. Rev. 2014, 22, 229–243. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Carnevali, O.; Pignalosa, P.; Api, M.; Scarponi, G.; Illuminati, S. Determination of Hg in farmed and wild atlantic bluefin tuna (Thunnus thynnus L.) muscle. Molecules 2019, 24, 1273. [Google Scholar] [CrossRef] [PubMed]
- Muntean, M.; Janssens-Maenhout, G.; Song, S.; Selin, N.E.; Olivier, J.G.J.; Guizzardi, D.; Maas, R.; Dentener, F. Trend analysis from 1970 to 2008 and model evaluation of EDGARv4 global gridded anthropogenic mercury emissions. Sci. Total Environ. 2014, 494–495, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Douglas, T.; Amyot, M.; Ariya, P.; Aspmo, K.; Berg, T.; Bottenheim, J.; Brooks, S.; Cobbett, F.; Dastoor, A.; et al. A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow. Atmos. Chem. Phys. 2008, 8, 1445–1482. [Google Scholar] [CrossRef]
- Ebinghaus, R.; Kock, H.H.; Temme, C.; Einax, J.W.; Loewe, A.G.; Richter, A.; Burrows, J.P.; Schroeder, W.H. Antarctic Springtime Depletion of Atmospheric Mercury. Environ. Sci. Technol. 2002, 36, 1238–1244. [Google Scholar] [CrossRef]
- Sprovieri, F.; Pirrone, N.; Hedgecock, I.M.; Landis, M.S.; Stevens, R.K. Intensive atmospheric mercury measurements at Terra Nova Bay in Antarctica during November and December 2000. J. Geophys. Res. [Atmospheres] 2002, 107, ACH20/1–ACH20/8. [Google Scholar] [CrossRef]
- Angot, H.; Dion, I.; Vogel, N.; Legrand, M.; Magand, O.; Dommergue, A. Multi-year record of atmospheric mercury at Dumont d’Urville, East Antarctic coast: Continental outflow and oceanic influences. Atmos. Chem. Phys. 2016, 16, 8265–8279. [Google Scholar] [CrossRef]
- Brooks, S.; Lindberg, S.; Southworth, G.; Arimoto, R. Springtime atmospheric mercury speciation in the McMurdo, Antarctica coastal region. Atmos. Environ. 2008, 42, 2885–2893. [Google Scholar] [CrossRef]
- Sprovieri, F.; Pirrone, N. A preliminary assessment of mercury levels in the Antarctic and Arctic troposphere. J. Aerosol Sci. 2000, 31, 1999–2000. [Google Scholar] [CrossRef]
- Temme, C.; Einax, J.W.; Ebinghaus, R.; Schroeder, W.H. Measurements of Atmospheric Mercury Species at a Coastal Site in the Antarctic and over the South Atlantic Ocean during Polar Summer. Environ. Sci. Technol. 2003, 37, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, R.; Schloesslin, C.; Davis, D.; Hogan, A.; Grube, P.; Fitzgerald, W.; Lamborg, C. Lead and mercury in aerosol particles collected over the South Pole during ISCAT-2000. Atmos. Environ. 2004, 38, 5485–5491. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury-an overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, S.; Padro, J.; Barrie, L. A size-segregated particle dry deposition scheme for an atmospheric aerosol module. Atmos. Environ. 2001, 35, 549–560. [Google Scholar] [CrossRef]
- Kim, P.-R.; Han, Y.-J.; Holsen, T.M.; Yi, S.-M. Atmospheric particulate mercury: Concentrations and size distributions. Atmos. Environ. 2012, 61, 94–102. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, X.; Wang, X.; Feng, X. Measurements and distribution of atmospheric particulate-bound mercury: A review. Bull. Environ. Contam. Toxicol. 2019, 103, 48–54. [Google Scholar] [CrossRef]
- Munthe, J.; Wängberg, I.; Iverfeldt, Å.; Lindqvist, O.; Strömberg, D.; Sommar, J.; Gårdfeldt, K.; Petersen, G.; Ebinghaus, R.; Prestbo, E.; et al. Distribution of atmospheric mercury species in Northern Europe: Final results from the MOE project. Atmos. Environ. 2003, 37, 9–20. [Google Scholar] [CrossRef]
- Sprovieri, F.; Pirrone, N.; Ebinghaus, R.; Kock, H.; Dommergue, A. A review of worldwide atmospheric mercury measurements. Atmos. Chem. Phys. 2010, 10, 8245–8265. [Google Scholar] [CrossRef]
- Lamborg, C.H.; Fitzgerald, W.F.; Vandal, G.M.; Rolfhus, K.R. Atmospheric mercury in northern Wisconsin: Sources and species. Water. Air. Soil Pollut. 1995, 80, 189–198. [Google Scholar] [CrossRef]
- Liu, B.; Keeler, G.J.; Dvonch, J.T.; Barres, J.A.; Lynam, M.M.; Marsik, F.J.; Morgan, J.T. Temporal variability of mercury speciation in urban air. Atmos. Environ. 2007, 41, 1911–1923. [Google Scholar] [CrossRef]
- Cobbett, F.D.; Van Heyst, B.J. Measurements of GEM fluxes and atmospheric mercury concentrations (GEM, RGM and Hgp) from an agricultural field amended with biosolids in Southern Ont., Canada (October 2004–November 2004). Atmos. Environ. 2007, 41, 2270–2282. [Google Scholar] [CrossRef]
- Landis, M.S.; Vette, A.F.; Keeler, G.J. Atmospheric mercury in the Lake Michigan basin: Influence of the Chicago/Gary urban area. Environ. Sci. Technol. 2002, 36, 4508–4517. [Google Scholar] [CrossRef]
- Liu, B.; Keeler, G.J.; Dvonch, J.T.; Barres, J.A.; Lynam, M.M.; Marsik, F.J.; Morgan, J.T. Urban-rural differences in atmospheric mercury speciation. Atmos. Environ. 2010, 44, 2013–2023. [Google Scholar] [CrossRef]
- Rutter, A.P.; Snyder, D.C.; Stone, E.A.; Schauer, J.J.; Gonzalez-Abraham, R.; Molina, L.T.; Marquez, C.; Cardenas, B.; de Foy, B. In situ measurements of speciated atmospheric mercury and the identification of source regions in the Mexico City Metropolitan Area. Atmos. Chem. Phys. 2009, 9, 207–220. [Google Scholar] [CrossRef]
- Han, D.; Zhang, J.; Hu, Z.; Ma, Y.; Duan, Y.; Han, Y.; Chen, X.; Zhou, Y.; Cheng, J.; Wang, W. Particulate mercury in ambient air in Shanghai, China: Size-specific distribution, gas–particle partitioning, and association with carbonaceous composition. Environ. Pollut. 2018, 238, 543–553. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, T.; Talbot, R.; Mao, H.; Yang, X.; Fu, C.; Sun, J.; Zhuang, B.; Li, S.; Han, Y.; et al. Characteristics of atmospheric mercury deposition and size-fractionated particulate mercury in urban Nanjing, China. Atmos. Chem. Phys. 2014, 14, 2233–2244. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Brooks, S.; Lin, C.J.; Scott, K.J.; Landis, M.S.; Stevens, R.K.; Goodsite, M.; Richter, A. Dynamic oxidation of gaseous mercury in the arctic troposphere at polar sunrise. Environ. Sci. Technol. 2002, 36, 1245–1256. [Google Scholar] [CrossRef]
- Angot, H.; Magand, O.; Helmig, D.; Ricaud, P.; Quennehen, B.; Gallee, H.; Del Guasta, M.; Sprovieri, F.; Pirrone, N.; Savarino, J.; et al. New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale. Atmos. Chem. Phys. 2016, 16, 8249–8264. [Google Scholar] [CrossRef]
- Bau, S.; Witschger, O. A modular tool for analyzing cascade impactors data to improve exposure assessment to airborne nanomaterials. J. Phys. Conf. Ser. 2013, 429, 012002/1–012002/10. [Google Scholar] [CrossRef]
- Chen, S.J.; Lo, C.T.; Fang, G.C.; Huang, C.S. Particulate-Bound Mercury (Hg[p]) Size distributions in central Taiwan. Environ. Forensics 2012, 13, 98–104. [Google Scholar] [CrossRef]
- Fang, G.-C.; Tsai, J.-H.; Lin, Y.-H.; Chang, C.-Y. Dry deposition of atmospheric particle-bound mercury in the middle Taiwan. Aerosol Air Qual. Res. 2012, 12, 1298–1308. [Google Scholar] [CrossRef]
- Xu, L.; Chen, J.; Niu, Z.; Yin, L.; Chen, Y. Characterization of mercury in atmospheric particulate matter in the southeast coastal cities of China. Atmos. Pollut. Res. 2013, 4, 454–461. [Google Scholar] [CrossRef]
- Pfaffhuber, K.A.; Berg, T.; Hirdman, D.; Stohl, A. Atmospheric mercury observations from Antarctica: Seasonal variation and source and sink region calculations. Atmos. Chem. Phys. 2012, 12, 3241–3251. [Google Scholar] [CrossRef]
- Illuminati, S.; Bau, S.; Annibaldi, A.; Mantini, C.; Libani, G.; Truzzi, C.; Scarponi, G. Evolution of size-segregated aerosol mass concentration during the Antarctic summer at Northern Foothills, Victoria Land. Atmos. Environ. 2016, 125, 212–221. [Google Scholar] [CrossRef]
- Barbaro, E.; Zangrando, R.; Kirchgeorg, T.; Bazzano, A.; Illuminati, S.; Annibaldi, A.; Rella, S.; Truzzi, C.; Grotti, M.; Ceccarini, A.; et al. An integrated study of the chemical composition of Antarctic aerosol to investigate natural and anthropogenic sources. Environ. Chem. 2016, 13, 867–876. [Google Scholar] [CrossRef]
- Fattori, I.; Bellandi, S.; Benassai, S.; Innocenti, M.; Mannini, A.; Udisti, R. Ion Balances of Size Resolved Aerosol Samples from Terra Nova Bay and Dome C (Antarctica); Colacino, M., Ed.; Italian Physical Society: Bologna, Italy, 2004; Volume 10th Works, pp. 101–115. [Google Scholar]
- Wang, J.; Xie, Z.; Wang, F.; Kang, H. Gaseous elemental mercury in the marine boundary layer and air-sea flux in the Southern Ocean in austral summer. Sci. Total Environ. 2017, 603–604, 510–518. [Google Scholar] [CrossRef]
- Feddersen, D.M.; Talbot, R.; Mao, H.; Sive, B.C. Size distribution of particulate mercury in marine and coastal atmospheres. Atmos. Chem. Phys. 2012, 12, 10899–10909. [Google Scholar] [CrossRef]
- Xiu, G.; Cai, J.; Zhang, W.; Zhang, D.; Büeler, A.; Lee, S.; Shen, Y.; Xu, L.; Huang, X.; Zhang, P. Speciated mercury in size-fractionated particles in Shanghai ambient air. Atmos. Environ. 2009, 43, 3145–3154. [Google Scholar] [CrossRef]
- Lu, J.Y.; Schroeder, W.H.; Bartie, L.A.; Welch, E.; Martin, K.; Lockhart, L.; Hunt, R.V.; Boila, G. Magnification of atmospheric mercury deposition to polar regions in springtime’ the link to tropospheric ozone depletion chemistry. Geophys. Res. Lett. 2001, 28, 3219–3222. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics. From Air Pollution to Climate Change, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1998; ISBN 978-1-119-22117-3. [Google Scholar]
- Liu, S.; Nadim, F.; Perkins, C.; Carley, R.J.; Hoag, G.E.; Lin, Y.; Chen, L. Atmospheric mercury monitoring survey in Beijing, China. Chemosphere 2002, 48, 97–107. [Google Scholar] [CrossRef]
- Huang, J.; Kang, S.; Guo, J.; Zhang, Q.; Cong, Z.; Sillanpää, M.; Zhang, G.; Sun, S.; Tripathee, L. Atmospheric particulate mercury in Lhasa city, Tibetan Plateau. Atmos. Environ. 2016, 142, 433–441. [Google Scholar] [CrossRef]
- Zhang, L.; Wright, L.P.; Blanchard, P. A review of current knowledge concerning dry deposition of atmospheric mercury. Atmos. Environ. 2009, 43, 5853–5864. [Google Scholar] [CrossRef]
- Nho-Kim, E.Y.; Michou, M.; Peuch, V.H. Parameterization of size-dependent particle dry deposition velocities for global modeling. Atmos. Environ. 2004, 38, 1933–1942. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Chen, Z.; Zhang, Y. Mercury concentrations in size-fractionated airborne particles at urban and suburban sites in Beijing, China. Atmos. Environ. 2006, 40, 2194–2201. [Google Scholar] [CrossRef]
- Pirrone, N.; Keeler, G.J.; Warner, P.O. Trends of ambient concentrations and deposition fluxes of particulate trace metals in Detroit from 1982 to 1992. Sci. Total Environ. 1995, 162, 43–61. [Google Scholar] [CrossRef]
- Petersen, G.; Iverfeldt, Å.; Munthe, J. Atmospheric Mercury Species over Europe. Model Calculations and Comparison with Observations from the Nordic Air and Precipitation Network for 1987 and 1988. Atmos. Environ. 1995, 29, 47–67. [Google Scholar] [CrossRef]
- Capodaglio, G.; Toscano, G.; Scarponi, G.; Cescon, P. Lead speciation in the surface waters of the Ross Sea (Antarctica). Ann. Chim. (Rome Ital.) 1989, 79, 543–559. [Google Scholar]
- Scarponi, G.; Barbante, C.; Turetta, C.; Gambaro, A.; Cescon, P. Chemical contamination of Antarctic snow: The case of lead. Microchem. J. 1997, 55, 24–32. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Truzzi, C.; Libani, G.; Mantini, C.; Scarponi, G. Determination of water-soluble, acid-extractable and inert fractions of Cd, Pb and Cu in Antarctic aerosol by square wave anodic stripping voltammetry after sequential extraction and microwave digestion. J. Electroanal. Chem. 2015, 755, 182–196. [Google Scholar] [CrossRef]
- PNRA Meteo-Climatological Observatory of the Italian National Antarctic Research Programme (PNRA). Agenzia nazionale per le nuove tecnologie, l’energia e lo sviluppo economico sostenibile (ENEA), Rome, Italy. Available online: http://www.climantartide.it (accessed on 15 June 2020).
- Scarchilli, C.; Ciardini, V.; Grigioni, P.; Iaccarino, I.; De Silvestri, L.; Proposito, M.; Dolci, S.; Camporeale, G.; Schioppo, R.; Antonelli, A.; et al. Characterization of snowfall estimated by in situ and ground-based remote-sensing observations at Terra Nova Bay, Victoria Land, Antarctica. J. Glaciol. 2020, 1–18. [Google Scholar] [CrossRef]
- Parish, T.R.; Bromwich, D.H. The surface windfield over the Antarctic ice sheets. Nature 1987, 328, 51–54. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Scarponi, G. Direct gravimetric determination of aerosol mass concentration in central antarctica. Anal. Chem. 2011, 83, 143–151. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Bassotti, E.; Scarponi, G. Determination of water-soluble and insoluble (dilute-HCl-extractable) fractions of Cd, Pb and Cu in Antarctic aerosol by square wave anodic stripping voltammetry: Distribution and summer seasonal evolution at Terra Nova Bay (Victoria Land). Anal. Bioanal. Chem. 2007, 387, 977–998. [Google Scholar] [CrossRef]
- Truzzi, C.; Annibaldi, A.; Illuminati, S.; Mantini, C.; Scarponi, G. Chemical fractionation by sequential extraction of Cd, Pb, and Cu in Antarctic atmospheric particulate for the characterization of aerosol composition, sources, and summer evolution at Terra Nova Bay, Victoria Land. Air Qual. Atmos. Heal. 2017, 10, 783–798. [Google Scholar] [CrossRef]
- Melendez-Perez, J.J.; Fostier, A.H. Assessment of direct Mercury Analyzer® to quantify mercury in soils and leaf samples. J. Braz. Chem. Soc. 2013, 24, 1880–1886. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Bassotti, E.; Finale, C.; Scarponi, G. First systematic voltammetric measurements of Cd, Pb, and Cu in hydrofluoric acid-dissolved siliceous spicules of marine sponges: Application to Antarctic specimens. Anal. Lett. 2011, 44, 2792–2807. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Truzzi, C.; Scarponi, G. Heavy metal distribution in organic and siliceous marine sponge tissues measured by square wave anodic stripping voltammetry. Mar. Pollut. Bull. 2016, 111, 476–482. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Romagnoli, T.; Libani, G.; Antonucci, M.; Scarponi, G.; Totti, C.; Truzzi, C. Distribution of Cd, Pb and Cu between dissolved fraction, inorganic particulate and phytoplankton in seawater of Terra Nova Bay (Ross Sea, Antarctica) during austral summer 2011–12. Chemosphere 2017, 185, 1122–1135. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Truzzi, C.; Tercier-Waeber, M.-L.; Noel, S.; Braungardt, C.B.; Achterberg, E.P.; Howell, K.A.; Turner, D.; Marini, M.; et al. In-situ trace metal (Cd, Pb, Cu) speciation along the Po River plume (Northern Adriatic Sea) using submersible systems. Mar. Chem. 2019, 212, 47–63. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. Noaa’s hysplit atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Riley, M.; Leys, J.; Salter, D. Dust storm event of February 2019 in Central and East Coast of Australia and evidence of long-range transport to New Zealand and Antarctica. Atmosphere 2019, 10, 653. [Google Scholar] [CrossRef]
- Scarchilli, C.; Frezzotti, M.; Ruti, P.M. Snow precipitation at four ice core sites in East Antarctica: Provenance, seasonality and blocking factors. Clim. Dyn. 2011, 37, 2107–2125. [Google Scholar] [CrossRef]
- Mezgec, K.; Stenni, B.; Crosta, X.; Masson-delmotte, V.; Baroni, C.; Braida, M.; Ciardini, V.; Colizza, E.; Melis, R.; Salvatore, M.C.; et al. Holocene sea ice variability driven by wind and polynya efficiency in the Ross Sea. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Caiazzo, L.; Baccolo, G.; Barbante, C.; Becagli, S.; Bertò, M.; Ciardini, V.; Crotti, I.; Delmonte, B.; Dreossi, G.; Frezzotti, M.; et al. Prominent features in isotopic, chemical and dust stratigraphies from coastal East Antarctic ice sheet (Eastern Wilkes Land). Chemosphere 2017, 176, 273–287. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Period | Actual Air Volume (m3) | Standard Air Volume b (m3) | PHg Atmospheric Concentration (pg m−3) a | |
|---|---|---|---|---|
| Actual Air | Standard Air | |||
| 10/11/2017–20/11/2017 | 16,354 | 16,070 | 94 ± 8 | 95 ± 8 |
| 20/11/2017–30/11/2017 | 16,498 | 15,857 | 90 ± 6 | 94 ± 7 |
| 30/11/2017–10/12/2017 | 16,793 | 16,172 | 79 ± 6 | 82 ± 6 |
| 10/12/2014–20/12/2017 | 16,479 | 15,920 | 39 ± 3 | 40 ± 3 |
| 20/12/2017–28/12/2017 | 13,206 | 12,806 | 31 ± 2 | 32 ± 2 |
| 28/12/2017–05/01/2018 | 13,401 | 12,921 | 30 ± 2 | 31 ± 2 |
| 05/01/2018–13/01/2018 | 13,070 | 12,584 | 31 ± 1 | 32 ± 2 |
| Study Sites | Period | Mercury Concentrations Mean (Min–Max), pg m−3 | References |
|---|---|---|---|
| Terra Nova Bay | 2017–2018 | 51 ± 27 (30–90) | This work |
| Terra Nova Bay | 1999–2001 | 12 ± 6 (~ 4–20) | [21] |
| Neumayer station | 2000–2001 | NA (15–120) | [25] |
| South Pole | Nov 2000 | 224 ± 119 (71–660) | [26] |
| Dec 2001 | 166 ± 147 (11–827) | ||
| McMurdo | Oct–Nov 2003 | 49 ± 36 (5–182) | [23] |
| Sweden, EU | 1998–2004 | 8.1 ± 4.9 (1.7–19) | [2,31,32] |
| Germany, EU | 1998–2004 | 42 ± 29 (21–99) | [2,31,32] |
| France, EU | 1998–2004 | 108 ± 246 (1.0–662) | [2,31,32] |
| Italy, EU | 1998–2004 | 15 ± 14 (1.0–46) | [32] |
| Spain, EU | 1998–2004 | 30 ± 26 (9.1–86) | [32] |
| Barrow, Alaska | 1999–2004 | 24 ± 25 (3–116) | [23] |
| Alert, Canadian Arctic | 1995 to 2009 | 70 ± 65 (NA) | [12] |
| North America ruraland background areas | 1995 to 2004 | 9.2 ± 15 (1.6–42) | [30,33,34,35] |
| Toronto, Canada | 2003–2004 | NA (14.2–39.2) | [32] |
| Chicago, USA | 70 ± 67 (NA) | [36] | |
| Detroit, USA | 2004 | NA (1.0–1345) | [37] |
| Mexico City, Mexico | 2006 | 187 ± 300 (NA) | [38] |
| Shangai, China | 2017 | 318 ± 144 (99–611) | [39] |
| Tokyo, Japan | 98 ± 51 (NA) | [40] | |
| Seoul, Korea | 2010 | 6.8 ± 6.5 (1.1–18.5) | [29] |
| Global | 111 ± 99 (0.6–1180) | [30] |
| PHgtot | 1.00 | PHg2.5–10.0 | ||||||
| PHg10.0–2.5 | 0.03 | 1.00 | PHg1.0–2.5 | |||||
| PHg2.5–1.0 | 0.94 | −0.28 | 1.00 | PHg0.1–1.0 | ||||
| PHg1.0–0.1 | −0.70 | 0.17 | −0.78 | 1.00 | T | |||
| T | −0.81 | 0.33 | −0.87 | 0.83 | 1.00 | P | ||
| P | −0.96 | 0.18 | −0.98 | 0.80 | 0.92 | 1.00 | RU | |
| RH | −0.60 | 0.44 | −0.73 | 0.47 | 0.61 | 0.71 | 1.00 | Wind |
| Wind | 0.30 | −0.54 | 0.52 | −0.53 | −0.29 | −0.38 | −0.63 | 1.00 |
| Sampling Period | Dry Deposition Flux (ng m−2 d−1) | |||
|---|---|---|---|---|
| 0.1–1.0 µm | 1.0–2.5 µm | 2.5–10.0 µm | Total | |
| 10/11/2017–20/11/2017 | 0.12 | 111 | 0 | 111 |
| 20/11/2017–30/11/2017 | 0.19 | 104 | 28 | 132 |
| 30/11/2017–10/12/2017 | 0.34 | 48 | 103 | 151 |
| 10/12/2014–20/12/2017 | 0.22 | 30 | 33 | 63 |
| 20/12/2017–28/12/2017 | 0.41 | 12 | 33 | 45 |
| 28/12/2017–05/01/2018 | 0.55 | 11 | 34 | 48 |
| 05/01/2018–13/01/2018 | 0.71 | 13 | 28 | 42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illuminati, S.; Annibaldi, A.; Bau, S.; Scarchilli, C.; Ciardini, V.; Grigioni, P.; Girolametti, F.; Vagnoni, F.; Scarponi, G.; Truzzi, C. Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol Over Terra Nova Bay, Antarctica. Molecules 2020, 25, 3971. https://doi.org/10.3390/molecules25173971
Illuminati S, Annibaldi A, Bau S, Scarchilli C, Ciardini V, Grigioni P, Girolametti F, Vagnoni F, Scarponi G, Truzzi C. Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol Over Terra Nova Bay, Antarctica. Molecules. 2020; 25(17):3971. https://doi.org/10.3390/molecules25173971
Chicago/Turabian StyleIlluminati, Silvia, Anna Annibaldi, Sébastien Bau, Claudio Scarchilli, Virginia Ciardini, Paolo Grigioni, Federico Girolametti, Flavio Vagnoni, Giuseppe Scarponi, and Cristina Truzzi. 2020. "Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol Over Terra Nova Bay, Antarctica" Molecules 25, no. 17: 3971. https://doi.org/10.3390/molecules25173971
APA StyleIlluminati, S., Annibaldi, A., Bau, S., Scarchilli, C., Ciardini, V., Grigioni, P., Girolametti, F., Vagnoni, F., Scarponi, G., & Truzzi, C. (2020). Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol Over Terra Nova Bay, Antarctica. Molecules, 25(17), 3971. https://doi.org/10.3390/molecules25173971