
Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay

 , , , and
, , , and
Abstract
1. Introduction
2. Iron Supplementation in Erythropoietic Porphyrias
2.1. Erythropoietic Protoporphyrias
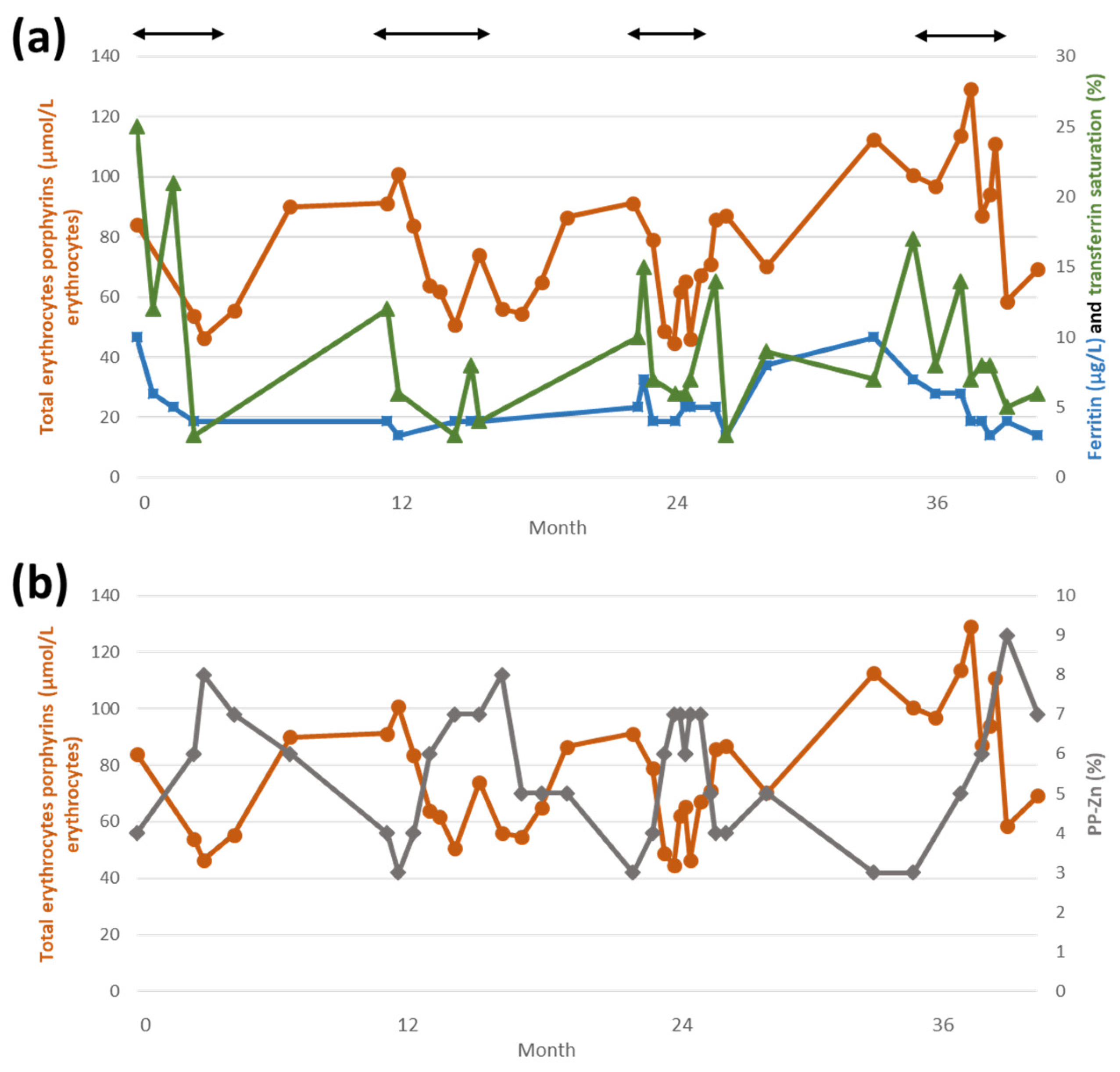
2.2. Iron Deficiency in CEP
3. Modulation of Heme Biosynthesis Pathway Activity by Iron Availability in the Erythron
3.1. IRE/IRP System
3.2. Iron-Sulfur Proteins
3.3. FECH and ALAS2 Regulation by Iron in EPP Patients
4. FECH Regulation at the Protein Level
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Erwin, A.L.; Desnick, R.J. Congenital erythropoietic porphyria: Recent advances. Mol. Genet. Metab. 2019, 128, 288–297. [Google Scholar] [CrossRef]
- Katugampola, R.P.; Badminton, M.N.; Finlay, A.Y.; Whatley, S.; Woolf, J.; Mason, N.; Deybach, J.C.; Puy, H.; Ged, C.; De Verneuil, H.; et al. Congenital erythropoietic porphyria: A single-observer clinical study of 29 cases. Br. J. Dermatol. 2012, 167, 901–913. [Google Scholar] [CrossRef]
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital erythropoietic porphyria due to a mutation in GATA1: The first trans-acting mutation causative for a human porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef] [PubMed]
- Gouya, L.; Puy, H.; Lamoril, J.; Da Silva, V.; Grandchamp, B.; Nordmann, Y.; Deybach, J.C. Inheritance in erythropoietic protoporphyria: A common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood 1999, 93, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Naik, H.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; Phillips, J.D.; Overbey, J.R.; Wang, B.; Singal, A.K.; et al. Clinical, biochemical, and genetic characterization of north American patients with erythropoietic protoporphyria and x-linked protoporphyria. JAMA Dermatol. 2017, 153, 789–796. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Taketani, S.; Adachi, Y.; Nakahashi, Y. Regulation of the expression of human ferrochelatase by intracellular iron levels. Eur. J. Biochem. 2000, 267, 4685–4692. [Google Scholar] [CrossRef]
- Egan, D.N.; Yang, Z.; Phillips, J.; Abkowitz, J.L. Inducing iron deficiency improves erythropoiesis and photosensitivity in congenital erythropoietic porphyria. Blood 2015, 126, 257–261. [Google Scholar] [CrossRef]
- Mirmiran, A.; Poli, A.; Ged, C.; Schmitt, C.; Lefebvre, T.; Manceau, H.; Daher, R.; Moulouel, B.; Peoc’h, K.; Simonin, S.; et al. Phlebotomy as an efficient long-term treatment of congenital erythropoietic porphyria. Haematologica 2020, 106, 913–917. [Google Scholar] [CrossRef]
- Blouin, J.-M.; Ged, C.; Bernardo-Seisdedos, G.; Cabantous, T.; Pinson, B.; Poli, A.; Puy, H.; Millet, O.; Gouya, L.; Morice-Picard, F.; et al. Identification of novel UROS mutations in a patient with congenital erythropoietic porphyria and efficient treatment by phlebotomy. Mol. Genet. Metab. Rep. 2021, 27, 100722. [Google Scholar] [CrossRef] [PubMed]
- Blouin, J.-M.; Ged, C.; Lalanne, M.; Lamrissi-Garcia, I.; Morice-Picard, F.; Costet, P.; Daher, R.; Moreau-Gaudry, F.; Bedel, A.; Puy, H.; et al. Iron chelation rescues hemolytic anemia and skin photosensitivity in congenital erythropoietic porphyria. Blood 2020, 136, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.P.; Meek, E.M. Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. Br. J. Haematol. 2013, 163, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Barman-Aksözen, J.; Minder, E.I.; Schubiger, C.; Biolcati, G.; Schneider-Yin, X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol. Dis. 2015, 54, 71–77. [Google Scholar] [CrossRef]
- Landefeld, C.; Kentouche, K.; Gruhn, B.; Stauch, T.; Rößler, S.; Schuppan, D.; Whatley, S.D.; Beck, J.F.; Stölzel, U. X-linked protoporphyria: Iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br. J. Haematol. 2016, 173, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Wahlin, S.; Floderus, Y.; Stål, P.; Harper, P. Erythropoietic protoporphyria in Sweden: Demographic, clinical, biochemical and genetic characteristics. J. Intern. Med. 2011, 269, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Reed, W.B. Erythropoietic protoporphyria. JAMA 1970, 214, 1060. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; Brittenham, G.M.; Hawkins, C.W.; Mukhtar, H.; Bickers, D.R. Iron therapy for hepatic dysfunction in erythropoietic protoporphyria. Ann. Intern. Med. 1986, 105, 27–31. [Google Scholar] [CrossRef]
- Graham-Brown, R.A.C.; Scheuer, P.J.; Sarkany, I. Histiocytosis X and erythropoietic protoporphyria. J. R. Soc. Med. 1984, 77, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Milligan, A.; Graham-Brown, R.A.C.; Sarkany, I.; Baker, H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br. J. Dermatol. 1988, 119, 63–66. [Google Scholar] [CrossRef]
- McClements, B.M.; Bingham, A.; Callender, M.E.; Trimble, E.R. Erythropoietic protoporphyria and iron therapy. Br. J. Dermatol. 1990, 122, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Todd, D.J.; Callender, M.E.; Mayne, E.E.; Walsh, M.; Burrows, D. Erythropoietic protoporphyria, transfusion therapy and liver disease. Br. J. Dermatol. 1992, 127, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Lyoumi, S.; Ducamp, S.; Martin-Schmitt, C.; Gouya, L.; Deybach, J.C.; Beaumont, C.; Puy, H. Excessive erythrocyte ppix influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Cell. Mol. Biol. 2009, 55, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Holme, S.A.; Thomas, C.L.; Whatley, S.D.; Bentley, D.P.; Anstey, A.V.; Badminton, M.N. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J. Am. Acad. Dermatol. 2007, 56, 1070–1072. [Google Scholar] [CrossRef]
- Bossi, K.; Lee, J.; Schmeltzer, P.; Holburton, E.; Groseclose, G.; Besur, S.; Hwang, S.; Bonkovsky, H.L. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur. J. Clin. Investig. 2015, 45, 1032–1041. [Google Scholar] [CrossRef]
- Bechtel, M.A.; Bertolone, S.J.; Hodge, S.J. Transfusion therapy in a patient with erythropoietic protoporphyria. Arch. Dermatol. 1981, 117, 99–101. [Google Scholar] [CrossRef]
- Dobozy, A.; Csató, M.; Siklósi, C.; Simon, N. Transfusion therapy for erythropoietic protoporphyria. Br. J. Dermatol. 1983, 109, 571–576. [Google Scholar] [CrossRef]
- Baker, H. Erythropoietic protoporphyria provoked by iron therapy. J. R. Soc. Med. 1971, 64, 610–611. [Google Scholar] [CrossRef]
- Weiss, Y.; Balwani, M.; Chen, B.; Yasuda, M.; Nazarenko, I.; Desnick, R.J. Congenital erythropoietic porphyria and erythropoietic protoporphyria: Identification of 7 uroporphyrinogen III synthase and 20 ferrochelatase novel mutations. Mol. Genet. Metab. 2019, 128, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Mleczko-Sanecka, K.; Silvestri, L. Cell-type-specific insights into iron regulatory processes. Am. J. Hematol. 2021, 96, 110–127. [Google Scholar] [CrossRef]
- Rouault, T.A. The indispensable role of mammalian iron sulfur proteins in function and regulation of multiple diverse metabolic pathways. BioMetals 2019, 32, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Jain, A.; Rouault, T.A. Mammalian iron–sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 2020, 55, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Puccio, H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum. Mol. Genet. 2010, 19, R103–R110. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Dailey, H.A.; Dailey, T.A.; Wu, C.K.; Medlock, A.E.; Rose, J.P.; Wang, K.F. Ferrochelatase at the millennium: Structures, mechanisms and [2Fe-2S] clusters. Cell. Mol. Life Sci. 2000, 57, 1909–1926. [Google Scholar] [CrossRef] [PubMed]
- Sellers, V.M.; Wang, K.F.; Johnson, M.K.; Daileyt, H.A. Evidence that the fourth ligand to the [2Fe-2S] cluster in animal ferrochelatase is a cysteine: Characterization of the enzyme from Drosophila melanogaster. J. Biol. Chem. 1998, 273, 22311–22316. [Google Scholar] [CrossRef]
- Crouse, B.R.; Sellers, V.M.; Finnegan, M.G.; Dailey, H.A.; Johnson, M.K. Site-directed mutagenesis and spectroscopic characterization of human ferrochelatase: Identification of residues coordinating the [2Fe-2S] cluster. Biochemistry 1996, 35, 16222–16229. [Google Scholar] [CrossRef]
- Schneider-Yin, X.; Gouya, L.; Dorsey, M.; Rüfenacht, U.; Deybach, J.C.; Ferreira, G.C. Mutations in the iron-sulfur cluster ligands of the human ferrochelatase lead to erythropoietic protoporphyria. Blood 2000, 96, 1545–1549. [Google Scholar] [CrossRef]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.-H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef]
- Barman-Aksözen, J.; Halloy, F.; Iyer, P.S.; Schümperli, D.; Minder, A.E.; Hall, J.; Minder, E.I.; Schneider-Yin, X. Delta-aminolevulinic acid synthase 2 expression in combination with iron as modifiers of disease severity in erythropoietic protoporphyria. Mol. Genet. Metab. 2019, 128, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Barman-Aksözen, J.; Béguin, C.; Dogar, A.M.; Schneider-Yin, X.; Minder, E.I. Iron availability modulates aberrant splicing of ferrochelatase through the iron- and 2-oxoglutarate dependent dioxygenase Jmjd6 and U2AF65. Blood Cells Mol. Dis. 2013, 51, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortès, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in apatient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030. [Google Scholar] [CrossRef] [PubMed]
- To-Figueras, J.; Ducamp, S.; Clayton, J.; Badenas, C.; Delaby, C.; Ged, C.; Lyoumi, S.; Gouya, L.; De Verneuil, H.; Beaumont, C.; et al. ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood 2011, 118, 1443–1451. [Google Scholar] [CrossRef]
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 Trial of an RNA interference therapy for acute intermittent porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Agarwal, S.; Simon, A.R.; Goel, V.; Habtemariam, B.A.; Clausen, V.A.; Kim, J.B.; Robbie, G.J. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid, givosiran, in patients with acute hepatic porphyria. Clin. Pharmacol. Ther. 2020, 108, 63–72. [Google Scholar] [CrossRef]
- Medlock, A.E.; Najahi-Missaoui, W.; Shiferaw, M.T.; Albetel, A.N.; Lanzilotta, W.N.; Dailey, H.A. Insight into the function of active site residues in the catalytic mechanism of human ferrochelatase. Biochem. J. 2021, 478, 3239–3252. [Google Scholar] [CrossRef]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H.; Kafina, M.D.; Coughlin, E.E.; Minogue, C.E.; Hebert, A.S.; et al. Erythropoietin signaling regulates heme biosynthesis. eLife 2017, 6, e24767. [Google Scholar] [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Taketani, S.; Kakimoto, K.; Ueta, H.; Masaki, R.; Furukawa, T. Involvement of ABC7 in the biosynthesis of heme in erythroid cells: Interaction of ABC7 with ferrochelatase. Blood 2003, 101, 3274–3280. [Google Scholar] [CrossRef]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef]
- Yien, Y.Y.; Shi, J.; Chen, C.; Cheung, J.T.M.; Grillo, A.S.; Shrestha, R.; Li, L.; Zhang, X.; Kafina, M.D.; Kingsley, P.D.; et al. FAM210B is an erythropoietin target and regulates erythroid heme synthesis by controlling mitochondrial iron import and ferrochelatase activity. J. Biol. Chem. 2018, 293, 19797–19811. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.; Farrell, C.; Wang, Y.; Singal, A.K.; Anderson, K.; Balwani, M.; Bissell, M.; Bonkovsky, H.; Seay, T.; Paw, B.; et al. Strong correlation of ferrochelatase enzymatic activity with Mitoferrin-1 mRNA in lymphoblasts of patients with protoporphyria. Mol. Genet. Metab. 2019, 128, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Langer, N.B.; Shaw, G.C.; Yang, G.; Li, L.; Kaplan, J.; Paw, B.H.; Bloomer, J.R. Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 2011, 39, 784–794. [Google Scholar] [CrossRef]
- Pandolfo, M.; Pastore, A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 2009, 256, 9–17. [Google Scholar] [CrossRef]
- Lesuisse, E.; Santos, R.; Matzanke, B.F.; Knight, S.A.B.; Camadro, J.M.; Dancis, A. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 2003, 12, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Bencze, K.Z.; Yoon, T.; Millán-Pacheco, C.; Bradley, P.B.; Pastor, N.; Cowan, J.A.; Stemmler, T.L. Human frataxin: Iron and ferrochelatase binding surface. Chem. Commun. 2007, 18, 1798–1800. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, S.; Gakh, O.; O’Neill, H.A.; Mangravita, A.; Nichol, H.; Ferreira, G.C.; Isaya, G. Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J. Biol. Chem. 2003, 278, 31340–31351. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Funk, J.; Körner, A.; Alberati, D.; Christen, F.; Schmitt, G.; Altmann, B.; Pospischil, A.; Singer, T. Effects of GlyT1 inhibition on erythropoiesis and iron homeostasis in rats. Exp. Hematol. 2016, 44, 964–974. [Google Scholar] [CrossRef]
- Matte, A.; Federti, E.; Winter, M.; Koerner, A.; Harmeier, A.; Mazer, N.; Tomka, T.; Di Paolo, M.L.; Defalco, L.; Andolfo, I.; et al. Bitopertin, a selective oral GLYT1 inhibitor, improves anemia in a mouse model of β-thalassemia. JCI Insight 2019, 4, e130111. [Google Scholar] [CrossRef]
- Taher, A.T.; Viprakasit, V.; Cappellini, M.D.; Kraus, D.; Cech, P.; Volz, D.; Winter, E.; Nave, S.; Dukart, J.; Khwaja, O.; et al. Haematological effects of oral administration of bitopertin, a glycine transport inhibitor, in patients with non-transfusion-dependent β-thalassaemia. Br. J. Haematol. 2021, 194, 474–477. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Authors | Year | Number of Patients | Sex | Type | Molecular Diagnosis | Biochemical Diagnosis | Intervention | Clinical Outcome | Biochemical Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Reed et al. | 1970 | 1 | F | NA | no | yes | oral iron therapy | worsening | NA |
| Baker et al. | 1971 | 1 | F | NA | no | yes | oral iron therapy | worsening | PPIX increase |
| Bechtel et al. | 1981 | 1 | M | NA | no | yes | repeated transfusions | improvement | PPIX decrease |
| Dobozy et al. | 1983 | 5 | 1 F and 4 M | NA | no | yes | repeated transfusions | improvement | PPIX decrease |
| Graham-Brown et al. | 1984 | 1 | F | NA | no | yes | oral iron therapy | worsening | PPIX decrease after iron therapy discontinuation |
| Gordeuk et al. | 1986 | 1 | F | NA | no | yes | oral iron therapy * | NA | PPIX decrease |
| Milligan et al. | 1988 | 2 ** | F | NA | no | yes | oral iron therapy | worsening | PPIX increase |
| McClements et al. | 1990 | 1 | F | NA | no | yes | oral iron therapy | worsening | PPIX decrease after iron therapy discontinuation |
| Todd et al. | 1992 | 1 | M | NA | no | yes | repeated transfusions | worsening | PPIX increase |
| Holme et al. | 2007 | 1 | M | EPP | yes | yes | oral iron therapy | improvement | stable PPIX |
| Whatley et al. | 2008 | 1 | M | XLPP | yes | yes | oral iron therapy | improvement | PPIX decrease |
| Whalin et al. | 2011 | 1 | F | EPP | yes | yes | oral iron therapy | worsening | stable PPIX |
| Bentley et al. | 2013 | 1 *** | M | EPP | yes | yes | IV iron therapy | improvement | PPIX decrease |
| Barman-Aksözen et al. | 2015 | 2 | F | EPP | yes | yes | IV or oral iron therapy | worsening | PPIX increase (one patient) stable PPIX (one patient) |
| Balwani et al. | 2017 | 8 | F | XLPP | yes | yes | oral iron therapy | improvement (7/8) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poli, A.; Schmitt, C.; Moulouel, B.; Mirmiran, A.; Puy, H.; Lefèbvre, T.; Gouya, L. Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay. Metabolites 2021, 11, 798. https://doi.org/10.3390/metabo11120798
Poli A, Schmitt C, Moulouel B, Mirmiran A, Puy H, Lefèbvre T, Gouya L. Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay. Metabolites. 2021; 11(12):798. https://doi.org/10.3390/metabo11120798
Chicago/Turabian StylePoli, Antoine, Caroline Schmitt, Boualem Moulouel, Arienne Mirmiran, Hervé Puy, Thibaud Lefèbvre, and Laurent Gouya. 2021. "Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay" Metabolites 11, no. 12: 798. https://doi.org/10.3390/metabo11120798
APA StylePoli, A., Schmitt, C., Moulouel, B., Mirmiran, A., Puy, H., Lefèbvre, T., & Gouya, L. (2021). Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay. Metabolites, 11(12), 798. https://doi.org/10.3390/metabo11120798