Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
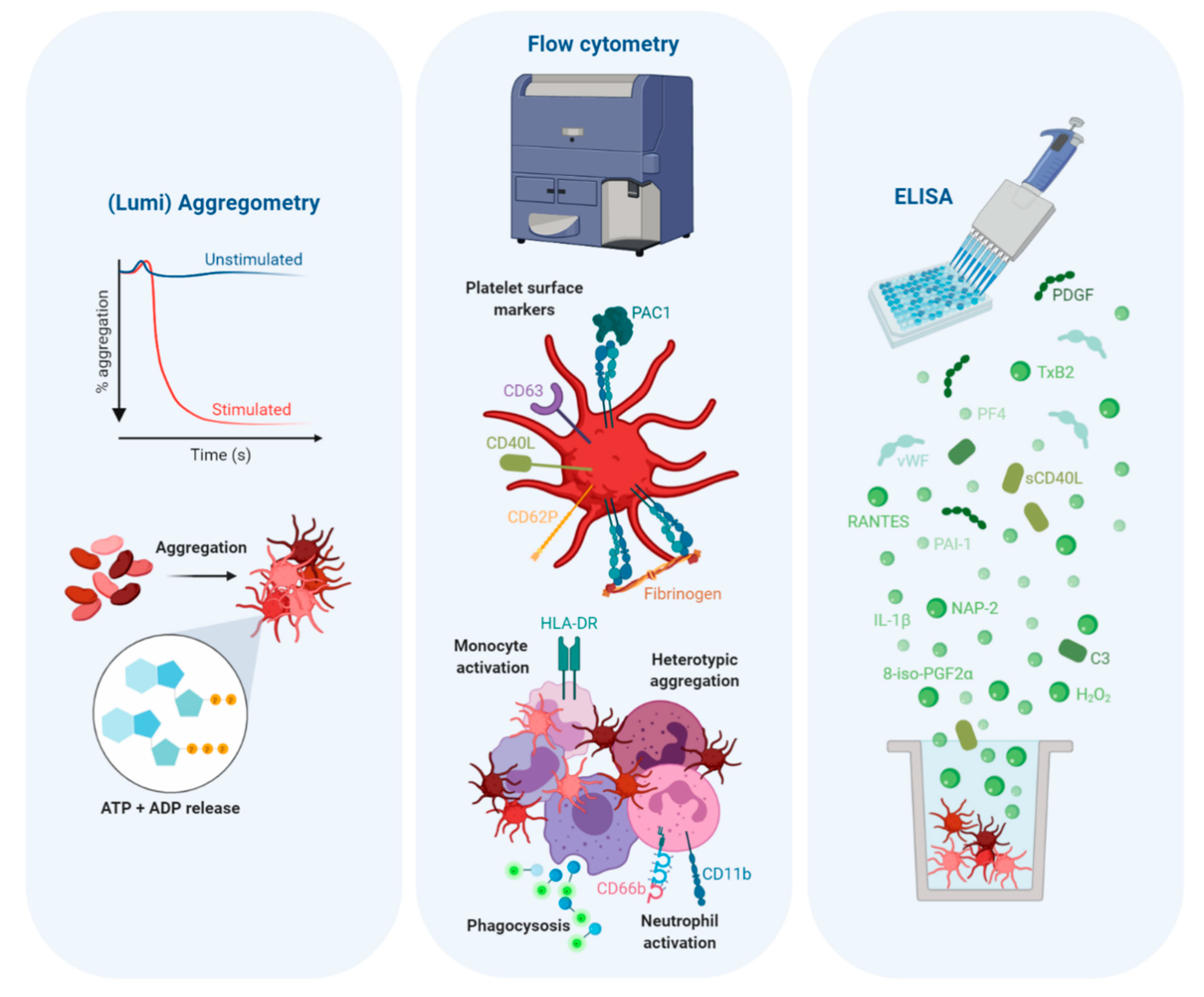
2. Platelets and Their Complex Role in Inflammation
2.1. Differential Granule Release
2.2. Adhesion Receptor Expression
2.3. Leukocyte-Mediated Inflammation
2.4. Platelets also Regulate Leukocyte-Mediated Inflammation
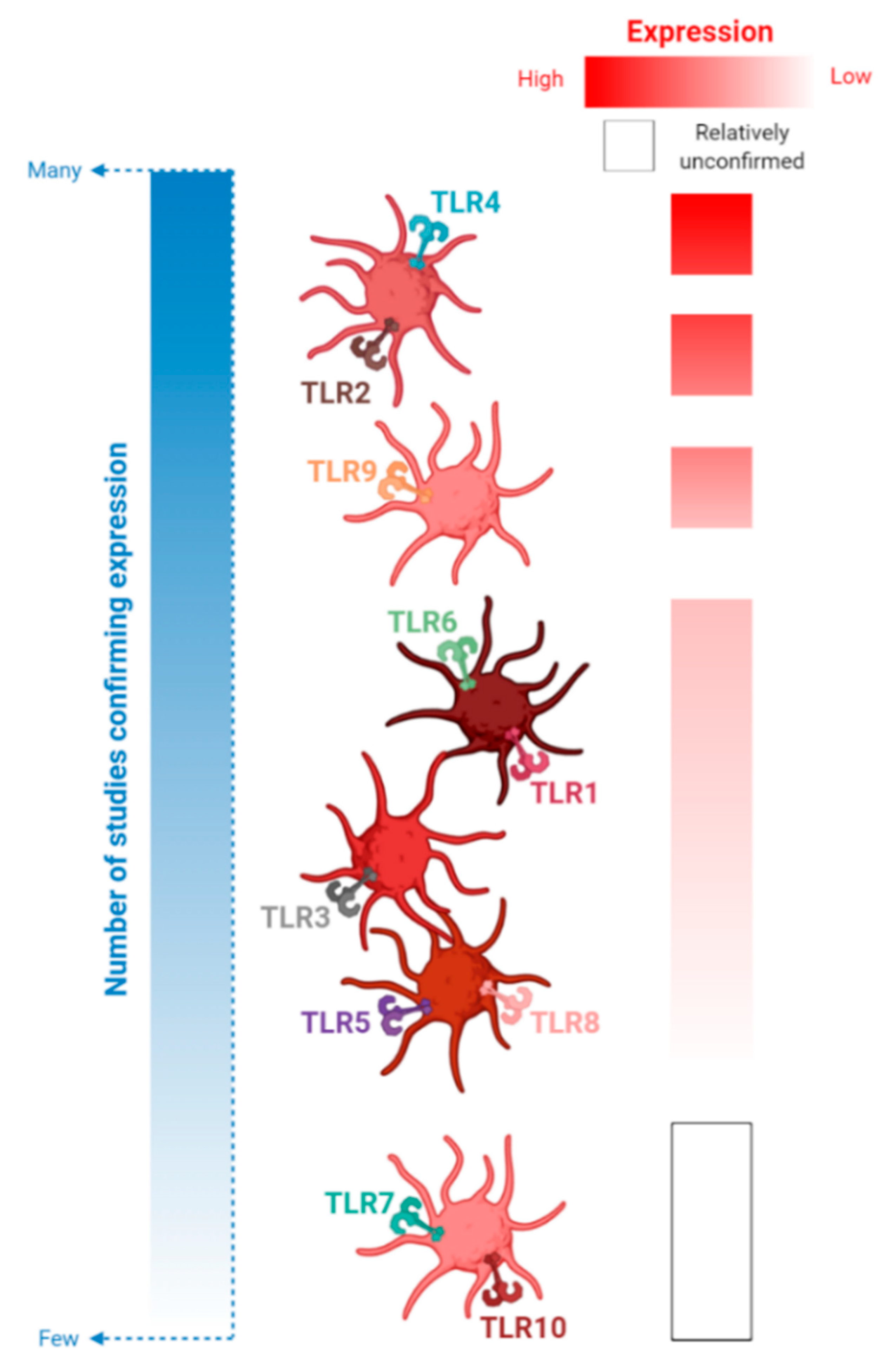
3. TLRs
4. An Update on Platelet-TLR Expression and TLR-Mediated Platelet Activation
4.1. TLR2 and Its Binding Partners, TLRs 1 and 6
4.1.1. Expression of Platelet-TLRs 1, 2 and 6 in Health and Disease
4.1.2. TLR2-Dependent Bacterial and Viral Activation
4.1.3. TLR2/1 Engagement with Pam3CSK4
4.1.4. Signaling Pathways Involved in Pam3CSK4-Mediated Platelet Activation
4.1.5. Platelet-TLR2/1 Engagement and Immunity
4.1.6. TLR2/6 Engagement
4.2. TLR3
4.3. TLR4
4.3.1. Expression of Platelet-TLR4 in Health and Disease
4.3.2. Engaging Platelet-TLR4 with Lipopolysaccharide (LPS)
4.3.3. Methodological Considerations for Examining Platelet Responses to LPS
4.4. TLR7
4.5. TLR9
4.6. TLRs 5, 8 and 10
5. Platelets are Capable of Releasing TLR-Stimulating DAMPs
5.1. mtDNA
5.2. HMGB1
5.3. Platelet DAMP Production during Storage of Platelet Transfusion Products
6. TLR Therapeutics: Potential for Targeting Platelet-TLRs?
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACS | Acute coronary syndrome |
| ADP | Adenosine diphosphate |
| AEs | Adverse events |
| AF | Atrial fibrillation |
| ATP | Adenosine triphosphate |
| C3 | Complement 3 |
| CAPs | Carboxy(alkylpyrrole) protein adducts |
| CD62P | P-selectin |
| cGMP | Cyclic guanosine monophosphate |
| CVD | Cardiovascular disease |
| DAMP | Damage-associated molecular pattern |
| DAPT | Dual anti-platelet therapy |
| FSL-1 | Fibroblast stimulating lipopeptide-1 |
| GP | Glycoprotein |
| HCMV | Human cytomegalovirus |
| HMGB1 | High Mobility Group Box 1 |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MALP-2 | Macrophage Activating Lipopeptide-2 |
| MIP | Macrophage inflammatory protein |
| MPO | Myeloperoxidase |
| MPs | Microparticles |
| mtDNA | Mitochondrial DNA |
| NET | Neutrophil extracellular trap |
| NF-κB | Nuclear factor-kappa B |
| oxPC | Oxidized phospholipids |
| PAMP | Pathogen-associated molecular pattern |
| PBMCs | Peripheral blood mononuclear cells |
| PC | Platelet components |
| PF4 | Platelet factor 4 |
| PGSL-1 | P-selectin glycoprotein ligand-1 |
| PRP | Platelet-rich plasma |
| PRR | Pattern recognition receptor |
| RAGE | Receptor for advanced glycation end products |
| RANTES | Regulated upon Activation, Normal T cell Expressed and Secreted |
| sCD40L | Soluble CD40L |
| TGF-β | Transforming growth factor-β |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor-alpha |
| TRAP | Thrombin receptor activator peptide |
| Treg | Regulatory T cells |
| TxA2 | Thromboxane A2 |
| VAMP | Vesicle-associated membrane protein |
| vWF | Von Willebrand factor |
| WPs | Washed platelets |
References
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2016, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and Immune Responses During Thromboinflammation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Wang, X.; Peter, K. Platelets in cardiac ischaemia/reperfusion injury: A promising therapeutic target. Cardiovasc. Res. 2019, 115, 1178–1188. [Google Scholar] [CrossRef]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Stocker, T.J.; Ishikawa-Ankerhold, H.; Massberg, S.; Schulz, C. Small but mighty: Platelets as central effectors of host defense. Thromb. Haemost. 2017, 117, 651–661. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Assinger, A.; Kral, J.B.; Yaiw, K.C.; Schrottmaier, W.C.; Kurzejamska, E.; Wang, Y.; Mohammad, A.A.; Religa, P.; Rahbar, A.; Schabbauer, G.; et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 801–809. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Badrnya, S.; Esfandeyari, A.; Volf, I. Periodontopathogens induce expression of CD40L on human platelets via TLR2 and TLR4. Thromb. Res. 2012, 130, e73–e78. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Schabbauer, G.; Hirschl, A.M.; Buchberger, E.; Binder, B.R.; Volf, I. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J. Thromb. Haemost. 2011, 9, 799–809. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, B.; Liang, Y.; Cao, S.H.; Liu, L.; Xu, X.N. Role of platelet TLR4 expression in pathogensis of septic thrombocytopenia. World J. Emerg. Med. 2011, 2, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Larkin, C.M.; Hante, N.K.; Breen, E.P.; Tomaszewski, K.A.; Eisele, S.; Radomski, M.W.; Ryan, T.A.; Santos-Martinez, M.J. Role of matrix metalloproteinases 2 and 9, toll-like receptor 4 and platelet-leukocyte aggregate formation in sepsis-associated thrombocytopenia. PLoS ONE 2018, 13, e0196478. [Google Scholar] [CrossRef]
- Gurses, K.M.; Kocyigit, D.; Yalcin, M.U.; Canpinar, H.; Oto, M.A.; Ozer, N.; Tokgozoglu, L.; Guc, D.; Aytemir, K. Enhanced Platelet Toll-like Receptor 2 and 4 Expression in Acute Coronary Syndrome and Stable Angina Pectoris. Am. J. Cardiol. 2015, 116, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Heger, L.A.; Hortmann, M.; Albrecht, M.; Colberg, C.; Peter, K.; Witsch, T.; Stallmann, D.; Zirlik, A.; Bode, C.; Duerschmied, D.; et al. Inflammation in acute coronary syndrome: Expression of TLR2 mRNA is increased in platelets of patients with ACS. PLoS ONE 2019, 14, e0224181. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction. Thromb. Res. 2017, 158, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Cognasse, F.; Aloui, C.; Anh Nguyen, K.; Hamzeh-Cognasse, H.; Fagan, J.; Arthaud, C.A.; Eyraud, M.A.; Sebban, M.; Fromont, E.; Pozzetto, B.; et al. Platelet components associated with adverse reactions: Predictive value of mitochondrial DNA relative to biological response modifiers. Transfusion 2016, 56, 497–504. [Google Scholar] [CrossRef]
- Cognasse, F.; Sut, C.; Hamzeh-Cognasse, H.; Garraud, O. Platelet-derived HMGB1: Critical mediator of SARs related to transfusion. Ann. Transl. Med. 2020, 8, 140. [Google Scholar] [CrossRef]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef]
- Maynard, D.M.; Heijnen, H.F.G.; Horne, M.K.; White, J.G.; Gahl, W.A. Proteomic analysis of platelet α-granules using mass spectrometry. J. Thromb. Haemost. 2007, 5, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, H.; Poon, M.-C.; Han, Z.; Gu, D.; Xu, M.; Jia, H.; Yang, R.; Han, Z.C. Abnormality of CD4+CD25+ regulatory T cells in idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2007, 78, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Heck, S.; Patel, V.; Levan, J.; Yu, Y.; Bussel, J.B.; Yazdanbakhsh, K. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood 2008, 112, 1325–1328. [Google Scholar] [CrossRef]
- Ren, Q.; Ye, S.; Whiteheart, S.W. The platelet release reaction: Just when you thought platelet secretion was simple. Curr. Opin. Hematol. 2008, 15, 537–541. [Google Scholar] [CrossRef]
- Sehgal, S.; Storrie, B. Evidence that differential packaging of the major platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. J. Thromb. Haemost. 2007, 5, 2009–2016. [Google Scholar] [CrossRef]
- Kamykowski, J.; Carlton, P.; Sehgal, S.; Storrie, B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet α-granules. Blood 2011, 118, 1370–1373. [Google Scholar] [CrossRef]
- Battinelli, E.M.; Thon, J.N.; Okazaki, R.; Peters, C.G.; Vijey, P.; Wilkie, A.R.; Noetzli, L.J.; Flaumenhaft, R.; Italiano, J.E., Jr. Megakaryocytes package contents into separate α-granules that are differentially distributed in platelets. Blood Adv. 2019, 3, 3092–3098. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet α granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef]
- Ma, L.; Perini, R.; McKnight, W.; Dicay, M.; Klein, A.; Hollenberg, M.D.; Wallace, J.L. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc. Natl. Acad. Sci. USA 2005, 102, 216–220. [Google Scholar] [CrossRef]
- Jonnalagadda, D.; Izu, L.T.; Whiteheart, S.W. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood 2012, 120, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Larsen, E.; Celi, A.; Gilbert, G.E.; Furie, B.C.; Erban, J.K.; Bonfanti, R.; Wagner, D.D.; Furie, B. PADGEM protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 1989, 59, 305–312. [Google Scholar] [CrossRef]
- Sheikh, S.; Parhar, R.S.; Bakheet, R.; Saleh, S.; Collison, K.; Al-Mohanna, F. Immobilization of rolling NK cells on platelet-borne P-selectin under flow by proinflammatory stimuli, interleukin-12, and leukotriene B4. J. Leukoc. Biol. 2004, 76, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Ulfman, L.H.; Joosten, D.P.; van Aalst, C.W.; Lammers, J.W.; van de Graaf, E.A.; Koenderman, L.; Zwaginga, J.J. Platelets promote eosinophil adhesion of patients with asthma to endothelium under flow conditions. Am. J. Respir. Cell Mol. Biol. 2003, 28, 512–519. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Ludwig, R.J.; Bergmann, P.; Garbaraviciene, J.; von Stebut, E.; Radeke, H.H.; Gille, J.; Diehl, S.; Hardt, K.; Henschler, R.; Kaufmann, R.; et al. Platelet, not endothelial, P-selectin expression contributes to generation of immunity in cutaneous contact hypersensitivity. Am. J. Pathol. 2010, 176, 1339–1345. [Google Scholar] [CrossRef]
- Pitchford, S.C.; Momi, S.; Giannini, S.; Casali, L.; Spina, D.; Page, C.P.; Gresele, P. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 2005, 105, 2074–2081. [Google Scholar] [CrossRef]
- Pervushina, O.; Scheuerer, B.; Reiling, N.; Behnke, L.; Schröder, J.M.; Kasper, B.; Brandt, E.; Bulfone-Paus, S.; Petersen, F. Platelet factor 4/CXCL4 induces phagocytosis and the generation of reactive oxygen metabolites in mononuclear phagocytes independently of Gi protein activation or intracellular calcium transients. J. Immunol. 2004, 173, 2060–2067. [Google Scholar] [CrossRef]
- Xia, C.Q.; Kao, K.J. Effect of CXC chemokine platelet factor 4 on differentiation and function of monocyte-derived dendritic cells. Int. Immunol. 2003, 15, 1007–1015. [Google Scholar] [CrossRef]
- Kasper, B.; Brandt, E.; Ernst, M.; Petersen, F. Neutrophil adhesion to endothelial cells induced by platelet factor 4 requires sequential activation of Ras, Syk, and JNK MAP kinases. Blood 2006, 107, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gomez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Cognasse, F.; Boussoulade, F.; Chavarin, P.; Acquart, S.; Fabrigli, P.; Lamy, B.; Garraud, O. Release of potential immunomodulatory factors during platelet storage. Transfusion 2006, 46, 1184–1189. [Google Scholar] [CrossRef]
- Reinboldt, S.; Wenzel, F.; Rauch, B.H.; Hohlfeld, T.; Grandoch, M.; Fischer, J.W.; Weber, A.-A. Preliminary evidence for a matrix metalloproteinase-2 (MMP-2)-dependent shedding of soluble CD40 ligand (sCD40L) from activated platelets. Platelets 2009, 20, 441–444. [Google Scholar] [CrossRef]
- André, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-Derived CD40L. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef]
- Danese, S.; Katz, J.A.; Saibeni, S.; Papa, A.; Gasbarrini, A.; Vecchi, M.; Fiocchi, C. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut 2003, 52, 1435–1441. [Google Scholar] [CrossRef]
- Viallard, J.-F.O.; Solanilla, A.; Gauthier, B.; Contin, C.c.; Déchanet, J.; Grosset, C.; Moreau, J.-F.O.; Praloran, V.; Nurden, P.; Pellegrin, J.-L.; et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood 2002, 99, 2612–2614. [Google Scholar] [CrossRef]
- Aloui, C.; Prigent, A.; Sut, C.; Tariket, S.; Hamzeh-Cognasse, H.; Pozzetto, B.; Richard, Y.; Cognasse, F.; Laradi, S.; Garraud, O. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int. J. Mol. Sci. 2014, 15, 22342–22364. [Google Scholar] [CrossRef]
- Léveillé, C.; Bouillon, M.; Guo, W.; Bolduc, J.; Sharif-Askari, E.; El-Fakhry, Y.; Reyes-Moreno, C.; Lapointe, R.; Merhi, Y.; Wilkins, J.A.; et al. CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J. Biol. Chem. 2007, 282, 5143–5151. [Google Scholar] [CrossRef]
- Gros, A.; Ollivier, V.; Ho-Tin-Noé, B. Platelets in inflammation: Regulation of leukocyte activities and vascular repair. Front. Immunol. 2015, 5, 678. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsdottir, S.; Hasselbalch, H.C.; Nielsen, C.H. Activated platelets enhance IL-10 secretion and reduce TNF-alpha secretion by monocytes. J. Immunol. 2013, 191, 4059–4067. [Google Scholar] [CrossRef]
- Nami, N.; Feci, L.; Napoliello, L.; Giordano, A.; Lorenzini, S.; Galeazzi, M.; Rubegni, P.; Fimiani, M. Crosstalk between platelets and PBMC: New evidence in wound healing. Platelets 2016, 27, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Tunjungputri, R.N.; van der Ven, A.J.; Riksen, N.; Rongen, G.; Tacke, S.; van den Berg, T.N.; Fijnheer, R.; Gomes, M.E.; Dinarello, C.A.; van de Veerdonk, F.L.; et al. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2- and TLR4-mediated inflammatory responses. Thromb. Haemost. 2015, 113, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Sadallah, S.; Eken, C.; Martin, P.J.; Schifferli, J.A. Microparticles (Ectosomes) Shed by Stored Human Platelets Downregulate Macrophages and Modify the Development of Dendritic Cells. J. Immunol. 2011, 186, 6543–6552. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, C.M.; Dunzendorfer, S.; Pechlaner, C.; Ricevuti, G.; Wiedermann, C.J. The inhibition of oxygen radical release from human neutrophils by resting platelets is reversed by administration of acetylsalicylic acid or clopidogrel. Free Radic. Res. 2001, 34, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Jancinová, V.; Drábiková, K.; Nosál, R.; Petríková, M.; Cíz, M.; Lojek, A.; Danihelová, E. Inhibition of FMLP-stimulated neutrophil chemiluminescence by blood platelets increased in the presence of the serotonin-liberating drug chloroquine. Thromb. Res. 2003, 109, 293–298. [Google Scholar] [CrossRef]
- Sahoo, M.; del Barrio, L.; Miller, M.A.; Re, F. Neutrophil Elastase Causes Tissue Damage That Decreases Host Tolerance to Lung Infection with Burkholderia Species. PLoS Pathog. 2014, 10, e1004327. [Google Scholar] [CrossRef]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The Balancing Act of Neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Lösche, W.; Temmler, U.; Redlich, H.; Vickers, J.; Krause, S.; Spangenberg, P. Inhibition of leukocyte chemiluminescence by platelets: Role of platelet-bound fibrinogen. Platelets 2001, 12, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Looney, M.R.; Nguyen, J.X.; Hu, Y.; Van Ziffle, J.A.; Lowell, C.A.; Matthay, M.A. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J. Clin. Investig. 2009, 119, 3450–3461. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Abdulnour, R.E.; Dalli, J.; Colby, J.K.; Krishnamoorthy, N.; Timmons, J.Y.; Tan, S.H.; Colas, R.A.; Petasis, N.A.; Serhan, C.N.; Levy, B.D. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc. Natl. Acad. Sci. USA 2014, 111, 16526–16531. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; Bird, G.K.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets modulate multiple markers of neutrophil function in response to in vitro Toll-like receptor stimulation. PLoS ONE 2019, 14, e0223444. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets regulate leucocyte responses to Toll-like receptor stimulation. Clin. Transl. Immunol. 2018, 7, e1036. [Google Scholar] [CrossRef]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef]
- Panigrahi, S.; Ma, Y.; Hong, L.; Gao, D.; West, X.Z.; Salomon, R.G.; Byzova, T.V.; Podrez, E.A. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ. Res. 2013, 112, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.L.; Casey, A.E.; Pollock, S.J.; Gertz, J.M.; McMillan, D.H.; Narasipura, S.D.; Mody, N.A.; King, M.R.; Maggirwar, S.B.; Francis, C.W.; et al. Platelets and Megakaryocytes Contain Functional Nuclear Factor-κB. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Malaver, E.; Romaniuk, M.A.; D’Atri, L.P.; Pozner, R.G.; Negrotto, S.; Benzadón, R.; Schattner, M. NF-kappaB inhibitors impair platelet activation responses. J. Thromb Haemost. 2009, 7, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.C.; Prince, L.R.; Sabroe, I. Translational mini-review series on Toll-like receptors: Networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin. Exp. Immunol. 2007, 147, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell. Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Shiraki, R.; Inoue, N.; Kawasaki, S.; Takei, A.; Kadotani, M.; Ohnishi, Y.; Ejiri, J.; Kobayashi, S.; Hirata, K.; Kawashima, S.; et al. Expression of Toll-like receptors on human platelets. Thromb. Res. 2004, 113, 379–385. [Google Scholar] [CrossRef]
- Koupenova, M.; Mick, E.; Mikhalev, E.; Benjamin, E.J.; Tanriverdi, K.; Freedman, J.E. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arter. Thromb. Vasc. Biol. 2015, 35, 1030–1037. [Google Scholar] [CrossRef]
- Gurses, K.M.; Kocyigit, D.; Yalcin, M.U.; Canpinar, H.; Evranos, B.; Canpolat, U.; Yorgun, H.; Sahiner, L.; Guc, D.; Aytemir, K. Platelet Toll-like receptor and its ligand HMGB-1 expression is increased in the left atrium of atrial fibrillation patients. Cytokine 2018, 103, 50–56. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van ‘t Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef]
- Marín Oyarzún, C.P.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Baroni Pietto, M.C.; Moiraghi, B.; Castro Ríos, M.A.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients with Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Nylander, M.; Lindahl, T.L.; Bengtsson, T.; Grenegard, M. The periodontal pathogen Porphyromonas gingivalis sensitises human blood platelets to epinephrine. Platelets 2008, 19, 352–358. [Google Scholar] [CrossRef]
- Paraskevas, S.; Huizinga, J.D.; Loos, B.G. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. J. Clin. Periodontol. 2008, 35, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Leishman, S.J.; Do, H.L.; Ford, P.J. Cardiovascular disease and the role of oral bacteria. J. Oral Microbiol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Keane, C.; Tilley, D.; Cunningham, A.; Smolenski, A.; Kadioglu, A.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J. Thromb. Haemost. 2010, 8, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- de Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; van der Poll, T.; Nieuwland, R.; van ‘t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, H.; Luo, X.; Zhang, P.; Gao, Y.; Xie, S.; Xu, K.; Chang, J.; Ma, L. Strains of Group B streptococci from septic patients induce platelet activation via Toll-like Receptor 2. Clin. Exp. Pharmacol. Physiol. 2017, 44, 335–343. [Google Scholar] [CrossRef]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies Up to 2016. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Spinelli, S.L.; Maggirwar, S.B.; Blumberg, N.; Phipps, R.P. Nuclear emancipation: A platelet tour de force. Sci. Signal. 2010, 3, pe37. [Google Scholar] [CrossRef]
- Lannan, K.L.; Sahler, J.; Kim, N.; Spinelli, S.L.; Maggirwar, S.B.; Garraud, O.; Cognasse, F.; Blumberg, N.; Phipps, R.P. Breaking the mold: Transcription factors in the anucleate platelet and platelet-derived microparticles. Front. Immunol. 2015, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Damien, P.; Cognasse, F.; Payrastre, B.; Spinelli, S.L.; Blumberg, N.; Arthaud, C.-A.; Eyraud, M.-A.; Phipps, R.P.; McNicol, A.; Pozzetto, B.; et al. NF-κB Links TLR2 and PAR1 to Soluble Immunomodulator Factor Secretion in Human Platelets. Front. Immunol. 2017, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Kalvegren, H.; Skoglund, C.; Helldahl, C.; Lerm, M.; Grenegard, M.; Bengtsson, T. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thromb. Haemost. 2010, 103, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Klarstrom Engstrom, K.; Brommesson, C.; Kalvegren, H.; Bengtsson, T. Toll like receptor 2/1 mediated platelet adhesion and activation on bacterial mimetic surfaces is dependent on src/Syk-signaling and purinergic receptor P2X1 and P2Y12 activation. Biointerphases 2014, 9, 041003. [Google Scholar] [CrossRef]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef]
- Biswas, S.; Zimman, A.; Gao, D.; Byzova, T.V.; Podrez, E.A. TLR2 Plays a Key Role in Platelet Hyperreactivity and Accelerated Thrombosis Associated with Hyperlipidemia. Circ. Res. 2017, 121, 951–962. [Google Scholar] [CrossRef]
- Koessler, J.; Niklaus, M.; Weber, K.; Koessler, A.; Kuhn, S.; Boeck, M.; Kobsar, A. The Role of Human Platelet Preparation for Toll-Like Receptors 2 and 4 Related Platelet Responsiveness. Th Open 2019, 3, e94–e102. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. The effects of aspirin and ticagrelor on Toll-like receptor (TLR)-mediated platelet activation: Results of a randomized, cross-over trial. Platelets 2019, 30, 599–607. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Weikert, C.; Pultar, J.; Lee, S.; Eichelberger, B.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Ticagrelor Inhibits Toll-Like and Protease-Activated Receptor Mediated Platelet Activation in Acute Coronary Syndromes. Cardiovasc. Drugs Ther. 2020, 34, 53–63. [Google Scholar] [CrossRef]
- Rex, S.; Beaulieu, L.M.; Perlman, D.H.; Vitseva, O.; Blair, P.S.; McComb, M.E.; Costello, C.E.; Freedman, J.E. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb. Haemost. 2009, 102, 97–110. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolas, V.S.; Sergio Roberto, A.R. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Stahl, A.L.; Svensson, M.; Morgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef]
- Nocella, C.; Carnevale, R.; Bartimoccia, S.; Novo, M.; Cangemi, R.; Pastori, D.; Calvieri, C.; Pignatelli, P.; Violi, F. Lipopolysaccharide as trigger of platelet aggregation via eicosanoid over-production. Thromb. Haemost. 2017, 117, 1558–1570. [Google Scholar] [CrossRef]
- Schmid, W.; Novacek, G.; Vogelsang, H.; Papay, P.; Primas, C.; Eser, A.; Panzer, S. Platelets Toll-like receptor-4 in Crohns disease. Eur. J. Clin. Investig. 2017, 47, 109–116. [Google Scholar] [CrossRef]
- Malhotra, R.; Priest, R.; Foster, M.R.; Bird, M.I. P-selectin binds to bacterial lipopolysaccharide. Eur. J. Immunol. 1998, 28, 983–988. [Google Scholar] [CrossRef]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef]
- Vallance, T.M.; Ravishankar, D.; Albadawi, D.A.I.; Layfield, H.; Sheard, J.; Vaiyapuri, R.; Dash, P.; Patel, K.; Widera, D.; Vaiyapuri, S. Effect of ultrapure lipopolysaccharides derived from diverse bacterial species on the modulation of platelet activation. Sci. Rep. 2019, 9, 18258. [Google Scholar] [CrossRef]
- Damien, P.; Cognasse, F.; Eyraud, M.A.; Arthaud, C.A.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-Ligand. BMC Immunol. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Arthaud, C.A.; Eyraud, M.A.; Zéni, F.; Pozzetto, B.; McNicol, A.; Garraud, O.; Cognasse, F. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin. Immunol. 2012, 145, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Matus, V.; Valenzuela, J.G.; Hidalgo, P.; Pozo, L.M.; Panes, O.; Wozniak, A.; Mezzano, D.; Pereira, J.; Saez, C.G. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS ONE 2017, 12, e0185431. [Google Scholar] [CrossRef]
- Kappelmayer, J.; Beke Debreceni, I.; Vida, A.; Antal-Szalmas, P.; Clemetson, K.J.; Nagy, B., Jr. Distinct effects of Re- and S-forms of LPS on modulating platelet activation. J. Thromb. Haemost. 2013, 11, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.R.; Bingle, L.; Judge, H.M.; Brown, S.B.; Storey, R.F.; Whyte, M.K.; Dower, S.K.; Buttle, D.J.; Sabroe, I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb. Haemost. 2005, 94, 831–838. [Google Scholar] [CrossRef]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef]
- Hagihara, M.; Higuchi, A.; Tamura, N.; Ueda, Y.; Hirabayashi, K.; Ikeda, Y.; Kato, S.; Sakamoto, S.; Hotta, T.; Handa, S.; et al. Platelets, after Exposure to a High Shear Stress, Induce IL-10-Producing, Mature Dendritic Cells In Vitro. J. Immunol. 2004, 172, 5297–5303. [Google Scholar] [CrossRef]
- Suzuki, J.; Hamada, E.; Shodai, T.; Kamoshida, G.; Kudo, S.; Itoh, S.; Koike, J.; Nagata, K.; Irimura, T.; Tsuji, T. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int. Arch. Allergy Immunol. 2013, 160, 152–160. [Google Scholar] [CrossRef]
- Weyrich, A.S.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef]
- Schoergenhofer, C.; Schwameis, M.; Hobl, E.L.; Ay, C.; Key, N.S.; Derhaschnig, U.; Jilma, B.; Spiel, A.O. Potent irreversible P2Y12 inhibition does not reduce LPS-induced coagulation activation in a randomized, double-blind, placebo-controlled trial. Clin. Sci. (Lond.) 2016, 130, 433–440. [Google Scholar] [CrossRef]
- Jerez-Dolz, D.; Torramade-Moix, S.; Palomo, M.; Moreno-Castano, A.; Lopez-Vilchez, I.; Hernandez, R.; Badimon, J.J.; Zafar, M.U.; Diaz-Ricart, M.; Escolar, G. Internalization of microparticles by platelets is partially mediated by toll-like receptor 4 and enhances platelet thrombogenicity. Atherosclerosis 2020, 294, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Nocella, C.; Petrozza, V.; Cammisotto, V.; Pacini, L.; Sorrentino, V.; Martinelli, O.; Irace, L.; Sciarretta, S.; Frati, G.; et al. Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci. Rep. 2018, 8, 3598. [Google Scholar] [CrossRef]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Bortolus, C.; Lecointe, K.; Parny, M.; Charlet, R.; Sendid, B.; Jawhara, S. Fungal Chitin Reduces Platelet Activation Mediated via TLR8 Stimulation. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE 2013, 8, e59989. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Fürnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; De Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1–nucleosome complexes: Implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock (Augustaga.) 2006, 26, 174–179. [Google Scholar] [CrossRef]
- He, M.; Bianchi, M.E.; Coleman, T.R.; Tracey, K.J.; Al-Abed, Y. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol. Med. 2018, 24, 21. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- Fan, J.; Li, Y.; Levy, R.M.; Fan, J.J.; Hackam, D.J.; Vodovotz, Y.; Yang, H.; Tracey, K.J.; Billiar, T.R.; Wilson, M.A. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: Role of HMGB1-TLR4 signaling. J. Immunol. 2007, 178, 6573–6580. [Google Scholar] [CrossRef]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef]
- Tadie, J.M.; Bae, H.B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L342–l349. [Google Scholar] [CrossRef]
- Vogel, S.; Rath, D.; Borst, O.; Mack, A.; Loughran, P.; Lotze, M.T.; Neal, M.D.; Billiar, T.R.; Gawaz, M. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem. Biophys. Res. Commun. 2016, 478, 143–148. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Deng, M.; Liu, Y.; Yang, C.; Hoffman, R.; Zhou, J.; Loughran, P.A.; Scott, M.J.; Neal, M.D.; Billiar, T.R. Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv. 2018, 2, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018, 2, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.P.; Kaufman, J.; Blumberg, N. Platelet derived CD154 (CD40 ligand) and febrile responses to transfusion. Lancet 2001, 357, 2023–2024. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh-Cognasse, H.; Lafarge, S.; Acquart, S.; Chavarin, P.; Courbil, R.; Fabrigli, P.; Garraud, O. Donor platelets stored for at least 3 days can elicit activation marker expression by the recipient’s blood mononuclear cells: An in vitro study. Transfusion 2009, 49, 91–98. [Google Scholar] [CrossRef]
- Cognasse, F.; Laradi, S.; Berthelot, P.; Bourlet, T.; Marotte, H.; Mismetti, P.; Garraud, O.; Hamzeh-Cognasse, H. Platelet Inflammatory Response to Stress. Front. Immunol. 2019, 10, 1478. [Google Scholar] [CrossRef]
- Stolla, M.; Refaai, M.A.; Heal, J.M.; Spinelli, S.L.; Garraud, O.; Phipps, R.P.; Blumberg, N. Platelet transfusion—The new immunology of an old therapy. Front. Immunol. 2015, 6, 28. [Google Scholar] [CrossRef]
- Sut, C.; Tariket, S.; Aubron, C.; Aloui, C.; Hamzeh-Cognasse, H.; Berthelot, P.; Laradi, S.; Greinacher, A.; Garraud, O.; Cognasse, F. The Non-Hemostatic Aspects of Transfused Platelets. Front. Med. (Lausanne) 2018, 5, 42. [Google Scholar] [CrossRef]
- Marcoux, G.; Magron, A.; Sut, C.; Laroche, A.; Laradi, S.; Hamzeh-Cognasse, H.; Allaeys, I.; Cabon, O.; Julien, A.S.; Garraud, O.; et al. Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. Transfusion 2019, 59, 2403–2414. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Xin, G.; Wei, Z.; Ji, C.; Zheng, H.; Gu, J.; Ma, L.; Huang, W.; Morris-Natschke, S.L.; Yeh, J.-L.; Zhang, R.; et al. Metformin Uniquely Prevents Thrombosis by Inhibiting Platelet Activation and mtDNA Release. Sci. Rep. 2016, 6, 36222. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Yasmeen, F.; Choi, S. Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods. Pharmaceutics 2019, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Batool, M.; Choi, S. TLR4-Targeting Therapeutics: Structural Basis and Computer-Aided Drug Discovery Approaches. Molecules 2020, 25, 627. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef]
- Rossignol, D.P.; Lynn, M. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J. Endotoxin Res. 2002, 8, 483–488. [Google Scholar] [CrossRef]
- Mullarkey, M.; Rose, J.R.; Bristol, J.; Kawata, T.; Kimura, A.; Kobayashi, S.; Przetak, M.; Chow, J.; Gusovsky, F.; Christ, W.J.; et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharm. Exp. 2003, 304, 1093–1102. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.-F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.-P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of Eritoran, an Antagonist of MD2-TLR4, on Mortality in Patients with Severe Sepsis: The ACCESS Randomized Trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef]
- Younan, P.; Ramanathan, P.; Graber, J.; Gusovsky, F.; Bukreyev, A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice from Lethal Filovirus Challenge. mBio 2017, 8, e00226-17. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.A.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Nakajima, T.; Kuba, K.; Kimura, A. Losartan inhibits LPS-induced inflammatory signaling through a PPARgamma-dependent mechanism in human THP-1 macrophages. Hypertens. Res. 2010, 33, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct Angiotensin II Type 2 Receptor Stimulation Acts Anti-Inflammatory Through Epoxyeicosatrienoic Acid and Inhibition of Nuclear Factor κB. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef]
- Sato, Y.; Fujii, S.; Imagawa, S.; Ohmura, K.; Ohmura, Y.; Andoh, Y.; Dong, J.; Ishimori, N.; Furumoto, T.; Tsutsui, H. Platelet aggregability in patients with hypertension treated with angiotensin II type 1 receptor blockers. J. Atheroscler. Thromb. 2007, 14, 31–35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guerra-Cuesta, J.I.; Montón, M.; Rodríguez-Feo, J.A.; Jiménez, A.M.; González-Fernández, F.; Rico, L.A.; García, R.; Gómez, J.; Farré, J.; Casado, S.; et al. Effect of losartan on human platelet activation. J. Hypertens. 1999, 17, 447–452. [Google Scholar] [CrossRef]
- Schwemmer, M.; Sommer, O.; Bassenge, E. Angiotensin receptor blocker losartan suppresses platelet activity by interfering with thromboxane signaling. Cardiovasc. Drugs 2001, 15, 301–307. [Google Scholar] [CrossRef]
- Grothusen, C.; Umbreen, S.; Konrad, I.; Stellos, K.; Schulz, C.; Schmidt, B.; Kremmer, E.; Teebken, O.; Massberg, S.; Luchtefeld, M.; et al. EXP3179 Inhibits Collagen-Dependent Platelet Activation via Glycoprotein Receptor-VI Independent of AT1-Receptor Antagonism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1184–1190. [Google Scholar] [CrossRef]
- Jiang, P.; Loyau, S.; Tchitchinadze, M.; Ropers, J.; Jondeau, G.; Jandrot-Perrus, M. Inhibition of Glycoprotein VI Clustering by Collagen as a Mechanism of Inhibiting Collagen-Induced Platelet Responses: The Example of Losartan. PLoS ONE 2015, 10, e0128744. [Google Scholar] [CrossRef][Green Version]
- Murphy, M.B.; Medvedev, A.E. Long noncoding RNAs as regulators of Toll-like receptor signaling and innate immunity. J. Leukoc. Biol. 2016, 99, 839–850. [Google Scholar] [CrossRef]
- Bayraktar, R.; Bertilaccio, M.T.S.; Calin, G.A. The Interaction Between Two Worlds: MicroRNAs and Toll-Like Receptors. Front. Immunol. 2019, 10, 1053. [Google Scholar] [CrossRef]
- Arenas-Padilla, M.; Mata-Haro, V. Regulation of TLR signaling pathways by microRNAs: Implications in inflammatory diseases. Cent. Eur. J. Immunol. 2018, 43, 482–489. [Google Scholar] [CrossRef]
- Zhou, W.; Pal, A.S.; Hsu, A.Y.-H.; Gurol, T.; Zhu, X.; Wirbisky-Hershberger, S.E.; Freeman, J.L.; Kasinski, A.L.; Deng, Q. MicroRNA-223 Suppresses the Canonical NF-κB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell Rep. 2018, 22, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, H.; Liu, Y.; Song, Y.; Lai, L.; Han, Q.; Cao, X.; Wang, Q. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS ONE 2012, 7, e42971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fu, L.; Bu, Y.; Yao, Y.; Wang, Y. Downregulated expression of miR-223 promotes Toll-like receptor-activated inflammatory responses in macrophages by targeting RhoB. Mol. Immunol. 2017, 91, 42–48. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Feng, D.; Li, M.; Gao, Y.; Ramirez, T.; Cao, H.; Kim, S.J.; Yang, Y.; Cai, Y.; Ju, C.; et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 2017, 66, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Merkerova, M.; Belickova, M.; Bruchova, H. Differential expression of microRNAs in hematopoietic cell lineages. Eur. J. Haematol. 2008, 81, 304–310. [Google Scholar] [CrossRef]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef]
- Elgheznawy, A.; Shi, L.; Hu, J.; Wittig, I.; Laban, H.; Pircher, J.; Mann, A.; Provost, P.; Randriamboavonjy, V.; Fleming, I. Dicer Cleavage by Calpain Determines Platelet microRNA Levels and Function in Diabetes. Circ. Res. 2015, 117, 157–165. [Google Scholar] [CrossRef]
- Liu, J.; Qin, L.; Wang, Z.; Peng, L.; Liu, J.; Wang, X.; Du, R.; Zou, Y.; Wu, Y.; Yin, T. Platelet-derived miRNAs as determinants of the antiplatelet response in clopidogrel-treated patients with ACS. Thromb. Res. 2020, 186, 71–74. [Google Scholar] [CrossRef]
- Chyrchel, B.; Totoń-Żurańska, J.; Kruszelnicka, O.; Chyrchel, M.; Mielecki, W.; Kołton-Wróż, M.; Wołkow, P.; Surdacki, A. Association of plasma miR-223 and platelet reactivity in patients with coronary artery disease on dual antiplatelet therapy: A preliminary report. Platelets 2015, 26, 593–597. [Google Scholar] [CrossRef]
- Shi, R.; Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.Y.; Zeng, S.; Liu, X.; Zhao, J.H.; Zhang, W.C.; et al. Decreased platelet miR-223 expression is associated with high on-clopidogrel platelet reactivity. Thromb. Res. 2013, 131, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhou, X.; Ji, W.J.; Shi, R.; Lu, R.Y.; Li, J.L.; Yang, G.H.; Luo, T.; Zhang, J.Q.; Zhao, J.H.; et al. Decreased circulating microRNA-223 level predicts high on-treatment platelet reactivity in patients with troponin-negative non-ST elevation acute coronary syndrome. J. Thromb. Thrombolysis 2014, 38, 65–72. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. https://doi.org/10.3390/ijms21176150
Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, Cognasse F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. International Journal of Molecular Sciences. 2020; 21(17):6150. https://doi.org/10.3390/ijms21176150
Chicago/Turabian StyleHally, Kathryn, Sebastien Fauteux-Daniel, Hind Hamzeh-Cognasse, Peter Larsen, and Fabrice Cognasse. 2020. "Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis" International Journal of Molecular Sciences 21, no. 17: 6150. https://doi.org/10.3390/ijms21176150
APA StyleHally, K., Fauteux-Daniel, S., Hamzeh-Cognasse, H., Larsen, P., & Cognasse, F. (2020). Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. International Journal of Molecular Sciences, 21(17), 6150. https://doi.org/10.3390/ijms21176150