
Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions


Abstract
1. Introduction
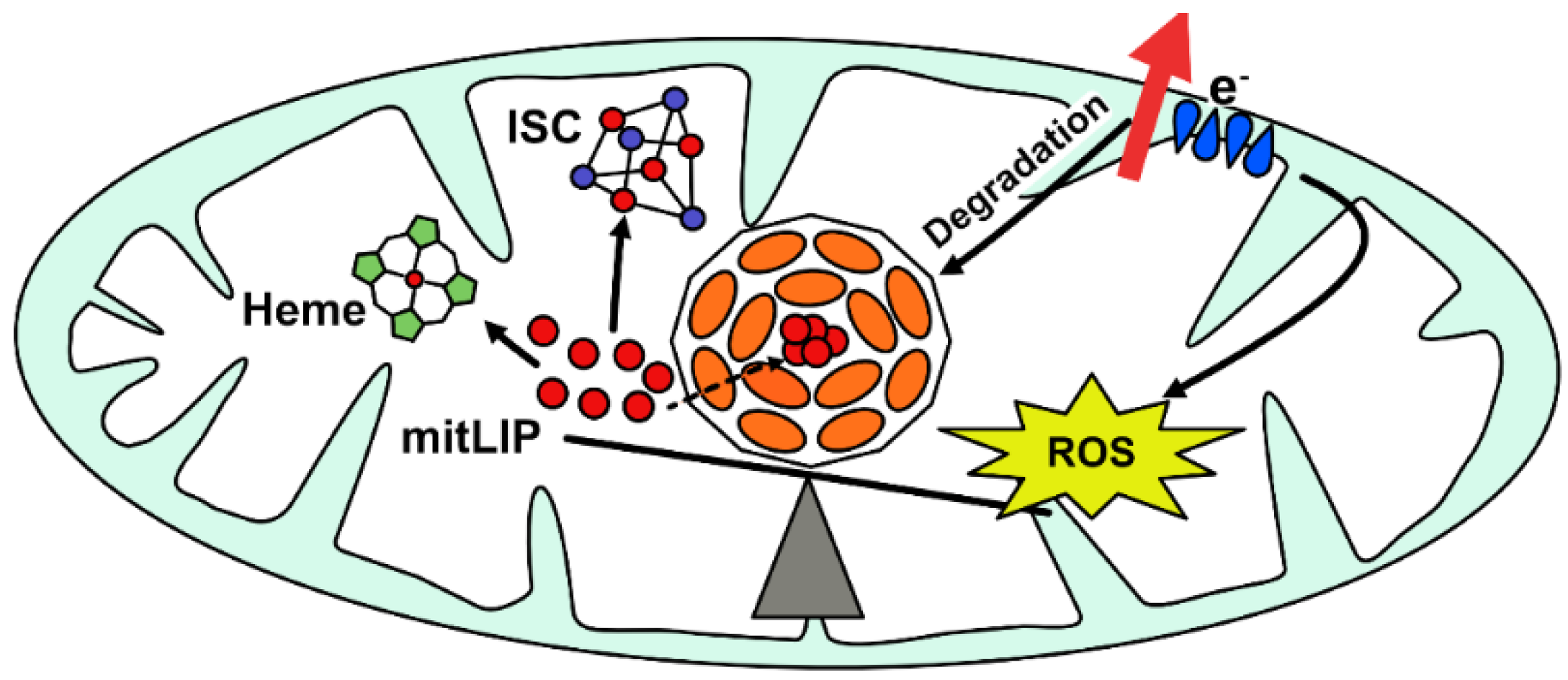
2. FtMt: Structural, Biochemical, Functional, and Regulatory Peculiarities
Regulation of FtMt Expression
3. Physiological Role of Mitochondrial Ferritin
4. Role of Mitochondrial Ferritin in Pathological Condition
4.1. Mitochondrial Ferritin in Sideroblastic Anemia
4.2. Role of Mitochondrial Ferritin in Friedreich’s Ataxia
4.3. Role of Mitochondrial Ferritin in Restless Legs Syndrome (RLS)
4.4. Role of Mitochondrial Ferritin in Neurodegenerative Diseases
4.5. Role of Mitochondrial Ferritin in Ischemic Stroke
4.6. Mitochondrial Ferritin as a Tumor Growth Inhibitor
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Ward, D.; Cloonan, S. Mitochondrial iron in human health and disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 2021, 513, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free. Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Lawson, D.M.; Artymiuk, P.J.; Yewdall, S.J.; Smith, J.M.A.; Livingstone, J.C.; Treffry, A.; Luzzago, A.; Levi, S.; Arosio, P.; Cesareni, G.; et al. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 1991, 349, 541–544. [Google Scholar] [CrossRef]
- Sun, S.; Chasteen, N.D.; Arosio, P.; Levi, S. Ferroxidase Kinetics of Human Liver Apoferritin, Recombinant H-Chain Apoferritin, and Site-Directed Mutants. Biochemistry 1993, 32, 9362–9369. [Google Scholar] [CrossRef]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial ferritin: A new player in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Ferritin: The protein nanocage and iron biomineral in health and in disease. Inorg. Chem. 2013, 52, 12223–12233. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A Human Mitochondrial Ferritin Encoded by an Intronless Gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef]
- Santambrogio, P.; Biasiotto, G.; Sanvito, F.; Olivieri, S.; Arosio, P.; Levi, S. Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 2007, 55, 1129–1137. [Google Scholar] [CrossRef]
- Levi, S.; Rovida, E. The role of iron in mitochondrial function. Biochim. Et Biophys. Acta Gen. Subj. 2009, 1790, 629–636. [Google Scholar] [CrossRef]
- Zancani, M.; Peresson, C.; Biroccio, A.; Federici, G.; Urbani, A.; Murgia, I.; Soave, C.; Micali, F.; Vianello, A.; Macrì, F. Evidence for the presence of ferritin in plant mitochondria. Eur. J. Biochem. 2004, 271, 3657–3664. [Google Scholar] [CrossRef]
- Missirlis, F.; Holmberg, S.; Georgieva, T.; Dunkov, B.C.; Rouault, T.A.; Law, J.H. Characterization of mitochondrial ferritin in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 5893–5898. [Google Scholar] [CrossRef] [PubMed]
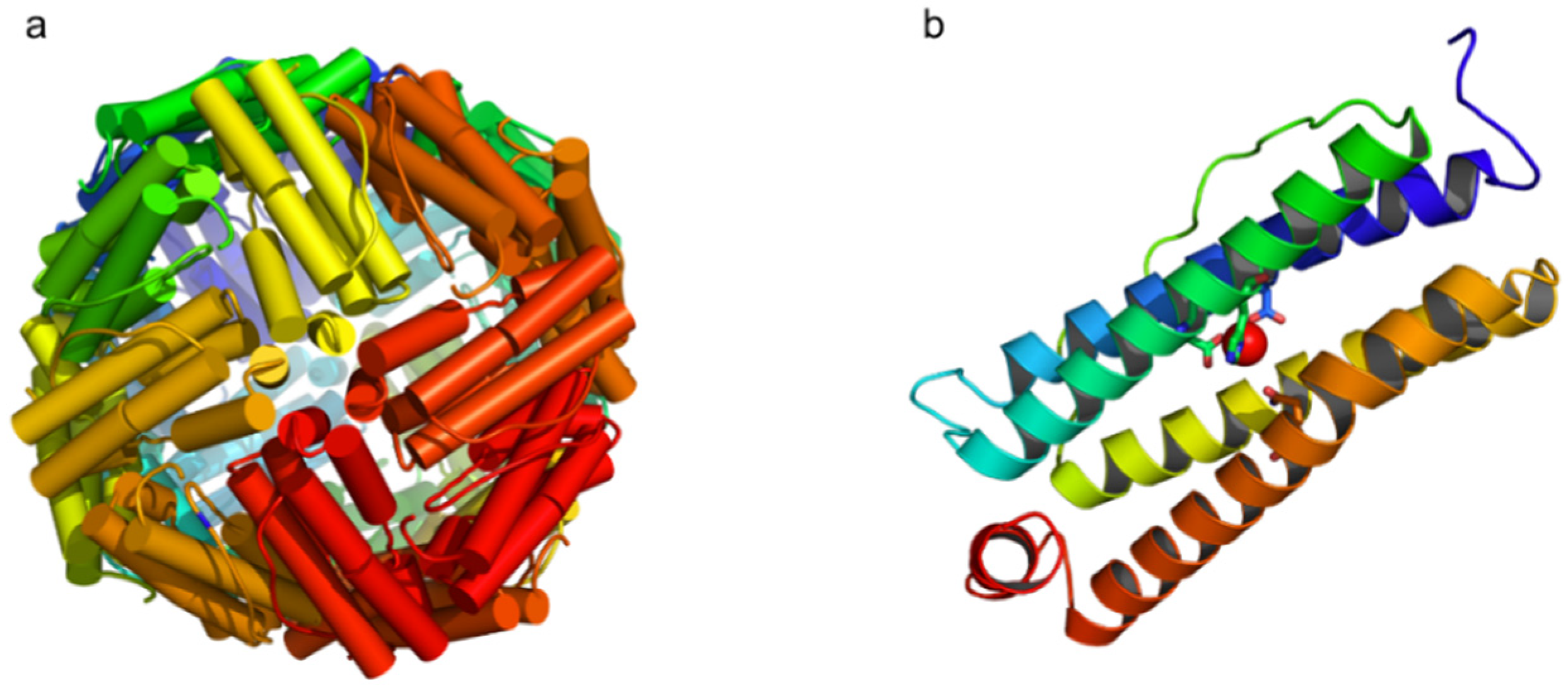
- D’Estaintot, B.L.; Santambrogio, P.; Granier, T.; Gallois, B.; Chevalier, J.M.; Précigoux, G.; Levi, S.; Arosio, P. Crystal structure and biochemical properties of the human mitochondrial ferritin and its mutant Ser144Ala. J. Mol. Biol. 2004, 340, 277–293. [Google Scholar]
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique iron binding and oxidation properties of human mitochondrial ferritin: A comparative analysis with human H-chain ferritin. J. Mol. Biol. 2005, 347, 543–554. [Google Scholar] [CrossRef]
- Corsi, B.; Cozzi, A.; Arosio, P.; Drysdale, J.; Santambrogio, P.; Campanella, A.; Biasiotto, G.; Albertini, A.; Levi, S. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J. Biol. Chem. 2002, 277, 22430–22437. [Google Scholar] [CrossRef] [PubMed]
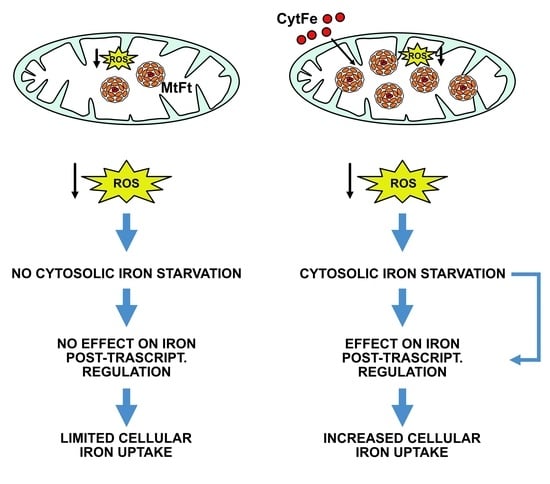
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167. [Google Scholar] [CrossRef]
- Behrouzi, B.; Kenigsberg, S.; Alladin, Z.; Swanson, S.; Zicherman, J.; Hong, S.H.; Moskovtsev, S.; Librach, C. Evaluation of potential protein biomarkers in patients with high sperm DNA damage. Syst. Biol. Reprod. Med. 2013, 59, 153–163. [Google Scholar] [CrossRef]
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000. [Google Scholar] [CrossRef]
- Hahn, P.; Dentchev, T.; Qian, Y.; Rouault, T.; Harris, Z.L.; Dunaief, J.L. Immunolocalization and regulation of iron handling proteins ferritin and ferroportin in the retina. Mol. Vis. 2004, 10, 598–607. [Google Scholar]
- Snyder, A.M.; Neely, E.B.; Levi, S.; Arosio, P.; Connor, J.R. Regional and cellular distribution of mitochondrial ferritin in the mouse brain. J. Neurosci. Res. 2010, 88, 3133–3143. [Google Scholar] [CrossRef]
- Levi, S.; Arosio, P. Mitochondrial ferritin. Int. J. Biochem. Cell Biol. 2004, 36, 1887–1889. [Google Scholar] [CrossRef]
- Wu, W.; Chang, S.; Wu, Q.; Xu, Z.; Wang, P.; Li, Y.; Yu, P.; Gao, G.; Shi, Z.; Duan, X.; et al. Mitochondrial ferritin protects the murine myocardium from acute exhaustive exercise injury. Cell Death Dis. 2016, 7, e2475. [Google Scholar] [CrossRef]
- Guaraldo, M.; Santambrogio, P.; Rovelli, E.; Savino, A.D.; Saglio, G.; Cittaro, D.; Roetto, A.; Levi, S. Characterization of human mitochondrial ferritin promoter: Identification of transcription factors and evidences of epigenetic control. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, J.; Yoshida, Y.; Kominato, Y.; Auron, P.E. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 2011, 54, 6–19. [Google Scholar] [CrossRef] [PubMed]
- González-Rubio, S.; López-Sánchez, L.; Muñoz-Castañeda, J.; Linares, C.I.; Aguilar-Melero, P.; Rodríguez-Perálvarez, M.; Sánchez-Sánchez, R.; Fernández-Álvarez, A.; Casado, M.; Montero-Álvarez, J.L.; et al. GCDCA down-regulates gene expression by increasing Sp1 binding to the NOS-3 promoter in an oxidative stress dependent manner. Biochem. Pharmacol. 2015, 96, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Erba, B.G.; Campanella, A.; Cozzi, A.; Causarano, V.; Cremonesi, L.; Gallì, A.; Della Porta, M.G.; Invernizzi, R.; Levi, S. Over-expression of mitochondrial ferritin affects the JAK2/STAT5 pathway in K562 cells and causes mitochondrial iron accumulation. Haematologica 2011, 96, 1424. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, R.; Travaglino, E.; Della Porta, M.G.; Gallì, A.; Malcovati, L.; Rosti, V.; Bergamaschi, G.; Erba, B.G.; Bellistri, F.; Bastia, R.; et al. Effects of mitochondrial ferritin overexpression in normal and sideroblastic erythroid progenitors. Br. J. Haematol. 2013, 161, 726–737. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.S.; Su, L.; Zheng, X.; Wu, W.Y.; Santambrogio, P.; Gou, Y.J.; Hao, Q.; Wang, P.N.; Li, Y.R.; et al. Mitochondrial Ferritin Is a Hypoxia-Inducible Factor 1α-Inducible Gene That Protects from Hypoxia-Induced Cell Death in Brain. Antioxid. Redox Signal. 2019, 30, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Isaya, G.; O’Neill, H.A.; Santambrogio, P.; Cozzi, A.; Arosio, P.; Levi, S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004, 13, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, N.; Wang, Y.Q.; Wu, Q.; Yu, P.; Shi, Z.H.; Duan, X.L.; Zhao, B.L.; Wu, W.S.; Chang, Y.Z. Mitochondrial ferritin protects hydrogen peroxide-induced neuronal cell damage. Aging Dis. 2017, 8, 458–470. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Lu, Z.; Nie, G.; Li, Y.; Soe-Lin, S.; Cao, T.; Zhang, Y.; Zhiyong, Y.; Liu, N.; Ponka, P.; Zhao, B. Overexpression of mitochondrial ferritin sensitizes cells to oxidative stress via an iron-mediated mechanism. Antioxid. Redox Signal. 2009, 11, 1791–1803. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Tanaka, A.; Kishi, F.; Lemasters, J.J.; Nishina, S.; Sasaki, K.; Hino, K. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bartnikas, T.B.; Campagna, D.R.; Antiochos, B.; Mulhern, H.; Pondarré, C.; Fleming, M.D. Characterization of mitochondrial ferritin-deficient mice. Am. J. Hematol. 2010, 85, 958–960. [Google Scholar] [CrossRef]
- Li, X.; Wang, P.; Wu, Q.; Xie, L.; Cui, Y.; Li, H.; Yu, P.; Chang, Y.Z. The construction and characterization of mitochondrial ferritin overexpressing mice. Int. J. Mol. Sci. 2017, 18, 1518. [Google Scholar] [CrossRef]
- Maccarinelli, F.; Gammella, E.; Asperti, M.; Regoni, M.; Biasiotto, G.; Turco, E.; Altruda, F.; Lonardi, S.; Cornaghi, L.; Donetti, E.; et al. Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. J. Mol. Med. 2014, 92, 859–869. [Google Scholar] [CrossRef]
- Maccarinelli, F.; Regoni, M.; Carmona, F.; Poli, M.; Meyron-Holtz, E.G.; Arosio, P. Mitochondrial ferritin deficiency reduces male fertility in mice. Reprod. Fertil. Dev. 2017, 29, 2005–2010. [Google Scholar] [CrossRef]
- Shi, Z.H.; Nie, G.; Duan, X.L.; Rouault, T.; Wu, W.S.; Ning, B.; Zhang, N.; Chang, Y.Z.; Zhao, B.L. Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: Implication for neuroprotection in parkinson’s disease. Antioxid. Redox Signal. 2010, 13, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Zhao, Y.S.; Shi, Z.H.; Chang, S.Y.; Nie, G.J.; Duan, X.L.; Zhao, S.M.; Wu, Q.; Yang, Z.L.; Zhao, B.L.; et al. Mitochondrial ferritin attenuates β-amyloid-induced neurotoxicity: Reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxid. Redox Signal. 2013, 18, 158–169. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Q.; Wu, W.; Li, H.; Guo, Y.; Yu, P.; Gao, G.; Shi, Z.; Zhao, B.; Chang, Y.Z. Mitochondrial Ferritin Deletion Exacerbates β-Amyloid-Induced Neurotoxicity in Mice. Oxidative Med. Cell. Longev. 2017, 2017, 1020357. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Fujikawa, K.; Kanda, M.; Fukuda, N.; Sasaki, K.; Takayama, T.; Kobune, M.; Takada, K.; Takimoto, R.; Hamada, H.; et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet. 2001, 69, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Bergamaschi, G.; Tonon, L.; Arbustini, E.; Grasso, M.; Vercesi, E.; Barosi, G.; Bianchi, P.E.; Cairo, G.; Arosio, P. Hereditary hyperferritinemia-cataract syndrome: Relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood 1997, 90, 814–821. [Google Scholar] [CrossRef]
- Cremonesi, L.; Cozzi, A.; Girelli, D.; Ferrari, F.; Fermo, I.; Foglieni, B.; Levi, S.; Bozzini, C.; Camparini, M.; Ferrari, M.; et al. Case report: A subject with a mutation in the ATG start codon of L-ferritin has no haematological or neurological symptoms. J. Med. Genet. 2004, 41, e81. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Santambrogio, P.; Ripamonti, M.; Rovida, E.; Levi, S.; Levi, S. Pathogenic mechanism and modeling of neuroferritinopathy. Cell. Mol. Life Sci. 2021, 78, 3355–3367. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, E.; Finazzi, D.; Goldwurm, S.; Levi, S.; Pezzoli, G.; Garavaglia, B.; Nardocci, N.; Malcovati, L.; Porta, M.G.D.; Gallì, A.; et al. Sequence variations in mitochondrial ferritin: Distribution in healthy controls and different types of patients. Genet. Test. Mol. Biomark. 2010, 14, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Stenirri, S.; Santambrogio, P.; Setaccioli, M.; Erba, B.G.; Manitto, M.P.; Rovida, E.; Ferrari, M.; Levi, S.; Cremonesi, L. Study of FTMT and ABCA4 genes in a patient affected by age-related macular degeneration: Identification and analysis of new mutations. Clin. Chem. Lab. Med. 2012, 50, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Della Porta, M.G.; Malcovati, L.; Invernizzi, R.; Travaglino, E.; Pascutto, C.; Maffioli, M.; Gallí, A.; Boggi, S.; Pietra, D.; Vanelli, L.; et al. Flow cytometry evaluation of erythroid dysplasia in patients with myelodysplastic syndrome. Leukemia 2006, 20, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.G.; Pickering, I.J.; George, G.N.; Nichol, H. The chemical form of mitochondrial iron in Friedreich’s ataxia. J. Inorg. Biochem. 2007, 101, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS ONE 2015, 10, e0116396. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.M.; Wang, X.; Patton, S.M.; Arosio, P.; Levi, S.; Earley, C.J.; Allen, R.P.; Connor, J.R. Mitochondrial ferritin in the substantia nigra in restless legs syndrome. J. Neuropathol. Exp. Neurol. 2009, 68, 1193–1199. [Google Scholar] [CrossRef]
- Wang, L.; Yang, H.; Zhao, S.; Sato, H.; Konishi, Y.; Beach, T.G.; Abdelalim, E.M.; Bisem, N.J.; Tooyama, I. Expression and localization of mitochondrial ferritin mRNA in Alzheimer’s disease cerebral cortex. PLoS ONE 2011, 6, e22325. [Google Scholar] [CrossRef]
- Tehranchi, R.; Invernizzi, R.; Grandien, A.; Zhivotovsky, B.; Fadeel, B.; Forsblom, A.M.; Travaglino, E.; Samuelsson, J.; Hast, R.; Nilsson, L.; et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood 2005, 106, 247–253. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Gerber, J.; Mühlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Zanella, I.; Derosas, M.; Corrado, M.; Cocco, E.; Cavadini, P.; Biasiotto, G.; Poli, M.; Verardi, R.; Arosio, P. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim. Et Biophys. Acta Mol. Basis Dis. 2008, 1782, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Soragni, E.; Wenyan, M.; Iudicello, M.; Jacoby, D.; Mercanti, S.M.D.; Clerico, M.; Longo, F.; Piga, A.; Ku, S.; Campau, E.; et al. Epigenetic Therapy for Friedreich Ataxia. Ann. Neurol. 2014, 76, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Hening, W.; Walters, A.S.; Allen, R.P.; Montplaisir, J.; Myers, A.; Ferini-Strambi, L. Impact, diagnosis and treatment of restless legs syndrome (RLS) in a primary care population: The REST (RLS epidemiology, symptoms, and treatment) primary care study. Sleep Med. 2004, 5, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Gamaldo, C.E.; Benbrook, A.R.; Allen, R.P.; Scott, J.A.; Henning, W.A.; Earley, C.J. Childhood and adult factors associated with restless legs syndrome (RLS) diagnosis. Sleep Med. 2007, 8, 716–722. [Google Scholar] [CrossRef]
- Schmidauer, C.; Sojer, M.; Seppi, K.; Stockner, H.; Högl, B.; Biedermann, B.; Brandauer, E.; Peralta, C.M.; Wenning, G.K.; Poewe, W. Transcranial ultrasound shows nigral hypoechogenicity in restless legs syndrome. Ann. Neurol. 2005, 58, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Wang, X.S.; Patton, S.M.; Menzies, S.L.; Troncoso, J.C.; Earley, C.J.; Allen, R.P. Decreased transferrin receptor expression by neuromelanin cells in restless legs syndrome. Neurology 2004, 62, 1563–1567. [Google Scholar] [CrossRef]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef]
- Altamura, S.; Muckenthaler, M.U. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimer’s Dis. 2009, 16, 879–895. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s disease: From pathogenesis to therapeutic implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Wu, W.-S.; Zhao, Y.-S.; Shi, Z.-H.; Chang, S.-Y.; Nie, G.-J.; Duan, X.-L.; Zhao, S.-M.; Wu, Q.; Yang, Z.-L.; Zhao, B.-L.; et al. Correction to Mitochondrial Ferritin Attenuates β-Amyloid-Induced Neurotoxicity: Reduction in Oxidative Damage Through the Erk/P38 Mitogen-Activated Protein Kinase Pathways, Authored by Wu WS, Zhao YS, Shi ZH, Chang SY, Nie GJ, Duan XL, Zhao SM, Wu Q, Yang ZL, Zhao BL, and Chang YZ (Antioxid Redox Signal 18: 158–169, 2013). Antioxid. Redox Signal. 2013, 18, 158–169. [Google Scholar] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Benkovic, S.A.; Connor, J.R. Ferritin, transferrin, and iron in selected regions of the adult and aged rat brain. J. Comp. Neurol. 1993, 338, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.I.; Wang, C.K.; Chen, J.J.; Chau, L.Y.; Lin, T.N. Differential regulation of H- and L-ferritin messenger RNA subunits, ferritin protein and iron following focal cerebral ischemia-reperfusion. Neuroscience 2000, 100, 475–484. [Google Scholar] [CrossRef]
- Ding, H.; Yan, C.Z.; Shi, H.; Zhao, Y.S.; Chang, S.Y.; Yu, P.; Wu, W.S.; Zhao, C.Y.; Chang, Y.Z.; Duan, X.L. Hepcidin is involved in iron regulation in the ischemic brain. PLoS ONE 2011, 6, e25324. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia-Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 1–16. [Google Scholar]
- Merlot, A.M.; Kalinowski, D.S.; Kovacevic, Z.; Jansson, P.J.; Sumit Sahni, M.L.-H.H.; Lane, D.J.R.; Lok, H.; Richardson, D.R. Exploiting Cancer Metal Metabolism using Anti-Cancer Metal-Binding Agents. Curr. Med. Chem. 2019, 26, 302–322. [Google Scholar] [CrossRef]
- Nie, G.; Chen, G.; Sheftel, A.D.; Pantopoulos, K.; Ponka, P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood 2006, 108, 2428–2434. [Google Scholar] [CrossRef]
- Shi, Z.H.; Shi, F.F.; Wang, Y.Q.; Sheftel, A.D.; Nie, G.; Zhao, Y.S.; You, L.H.; Gou, Y.J.; Duan, X.L.; Zhao, B.L.; et al. Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell. Mol. Life Sci. 2015, 72, 983–997. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| H Chain (FtH) | L Chain (FtL) | Mitochondrial Ferritin (FtMt) | |
|---|---|---|---|
| Chromosome | 11 | 19 | 5 |
| Introns | 3 | 3 | None |
| Length of the encoded protein | 175 amino acids | 183 amino acids | 242 amino acids |
| NH2 processing | N-terminal methionine cleavage, N-terminal block | N-terminal methionine cleavage, N-terminal block | N-terminal leader sequence cleavage |
| Structure of ferritin | Heteropolymers H/L | Heteropolymers H/L | Homopolymers |
| Catalytic site | Ferroxidase center | No ferroxidase center (facilitates iron nucleation) | Ferroxidase center |
| Fe-dependent regulation | IRE-dependent post-transcriptional regulation | IRE-dependent post-transcriptional regulation | No |
| Transcriptional regulation * | Dependent on the ARE element | Dependent on the ARE element | Dependent on the HRE element |
| Tissue expression | Ubiquitous | Ubiquitous | Cellular specific |
| Disease | FtMt Gene | FtMt Protein | FtMt Expression | Detected in * | Reference |
|---|---|---|---|---|---|
| Macular Degeneration (MD) | Mutated | Not Functional? | -- | -- | [50] |
| Sideroblastic Anemia (SD) | Wild Type | Normal | Upregulated | Sideroblasts | [20,51] |
| Friederich’s Ataxia (FA) | Wild Type | Normal | Upregulated | Cardiomyocytes, Fibroblasts, Heart | [52,53] |
| Restless Legs Syndrome (RLS) | Wild Type | Normal | Upregulated | Substantia Nigra | [54] |
| Alzheimer’s Disease (AD) | Wild Type | Normal | Upregulated | Temporal Cortex Neurons | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, S.; Ripamonti, M.; Dardi, M.; Cozzi, A.; Santambrogio, P. Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions. Cells 2021, 10, 1969. https://doi.org/10.3390/cells10081969
Levi S, Ripamonti M, Dardi M, Cozzi A, Santambrogio P. Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions. Cells. 2021; 10(8):1969. https://doi.org/10.3390/cells10081969
Chicago/Turabian StyleLevi, Sonia, Maddalena Ripamonti, Marko Dardi, Anna Cozzi, and Paolo Santambrogio. 2021. "Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions" Cells 10, no. 8: 1969. https://doi.org/10.3390/cells10081969
APA StyleLevi, S., Ripamonti, M., Dardi, M., Cozzi, A., & Santambrogio, P. (2021). Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions. Cells, 10(8), 1969. https://doi.org/10.3390/cells10081969