
Loosening ER–Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
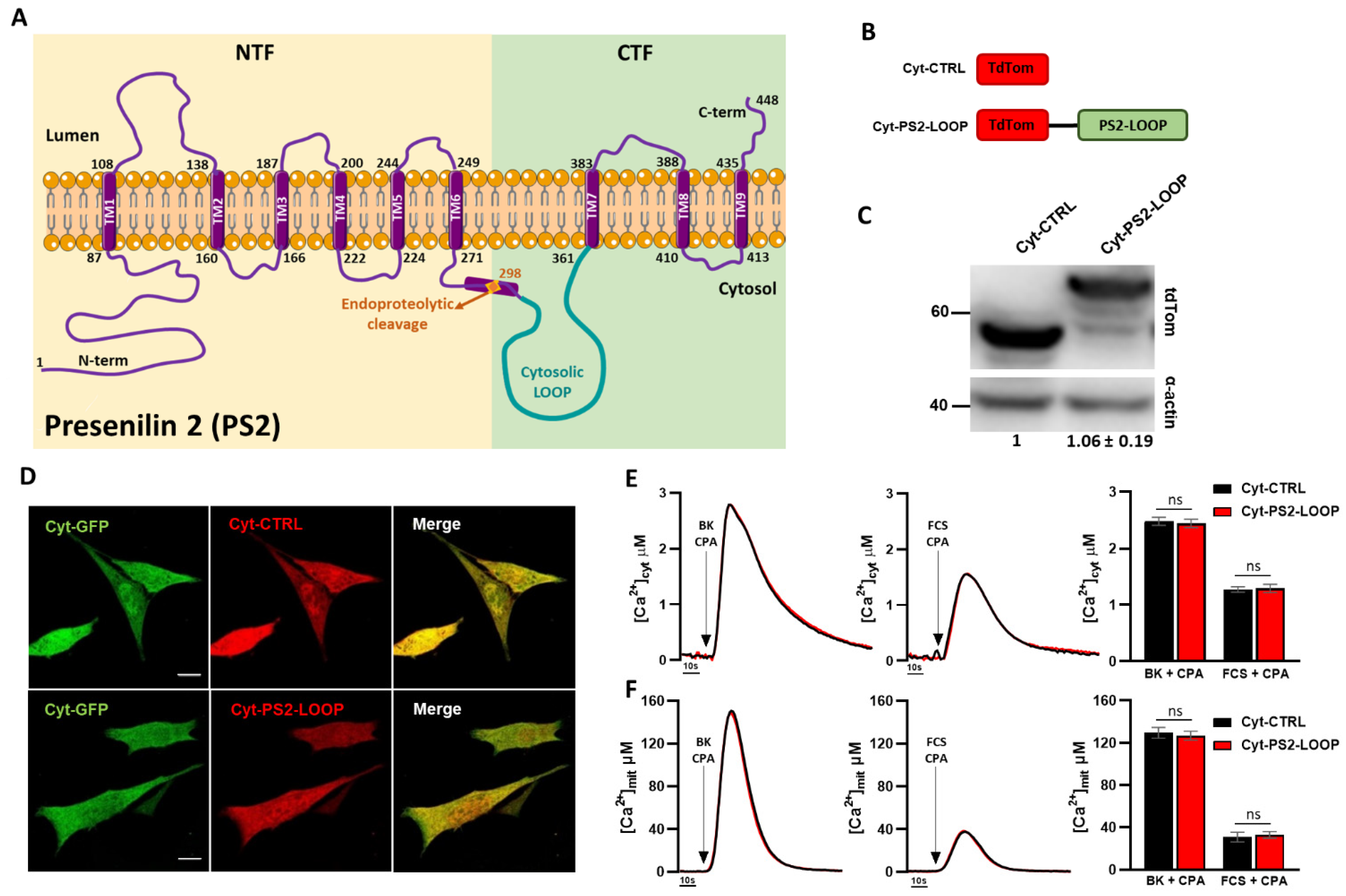
2.1. Plasmids
2.2. Cell Culture and Transfection
2.3. Protein Extraction and Western Blot Analysis
2.4. Immunoprecipitation
2.5. Aequorin Ca2+ Measurements
2.6. Immunofluorescence and Confocal Microscopy
2.7. Statistical Analysis
3. Results
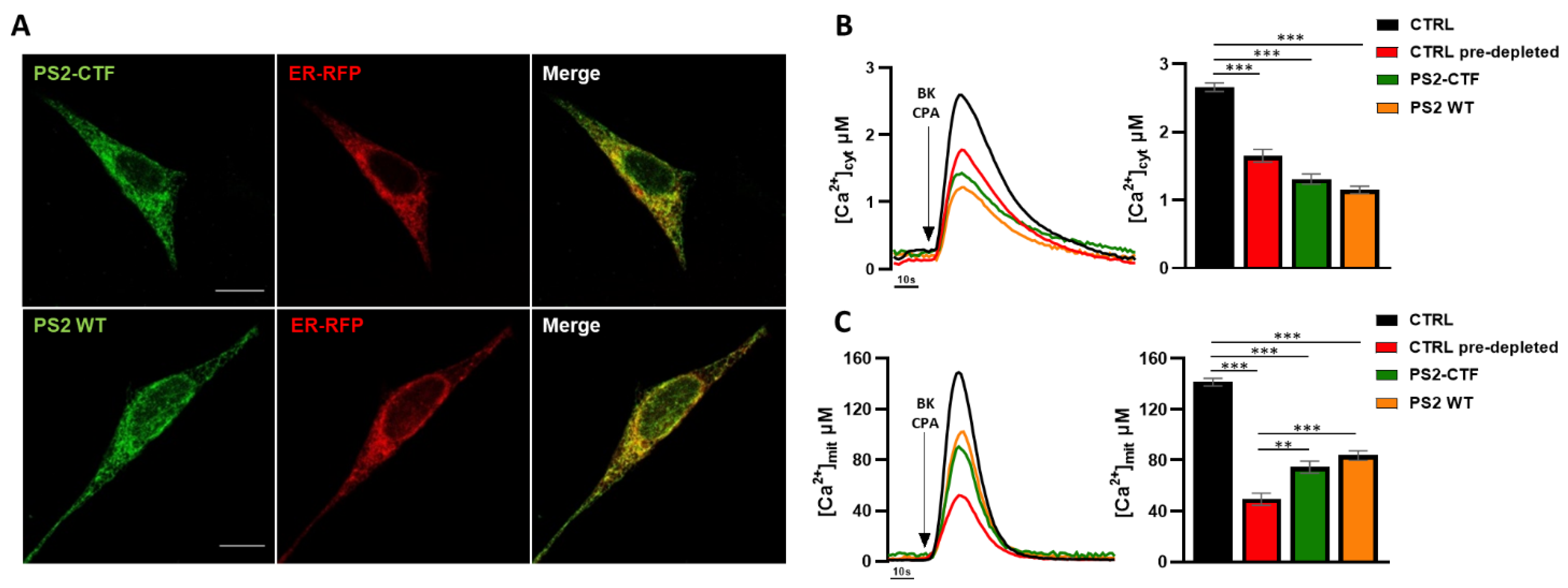
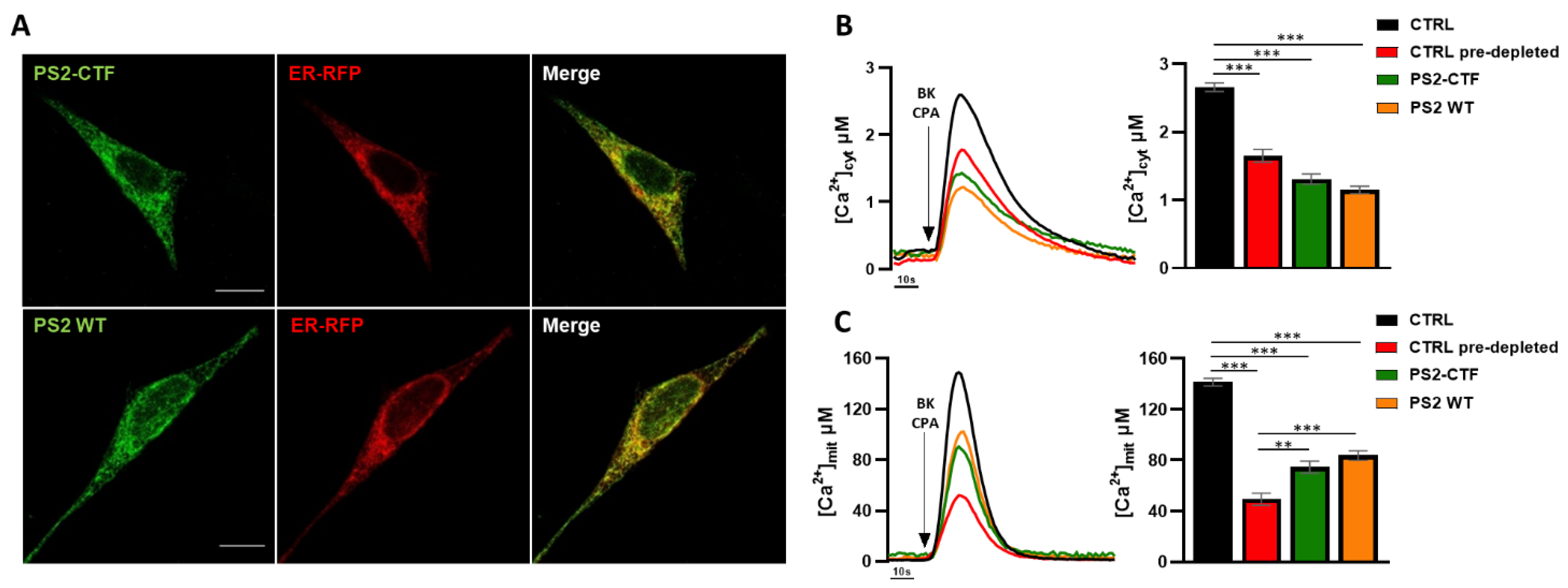
3.1. The PS2 C-Terminal Fragment, but Not Its Loop Domain, Retains the Effects of Full-Length PS2 on ER–Mitochondria Ca2+ Transfer
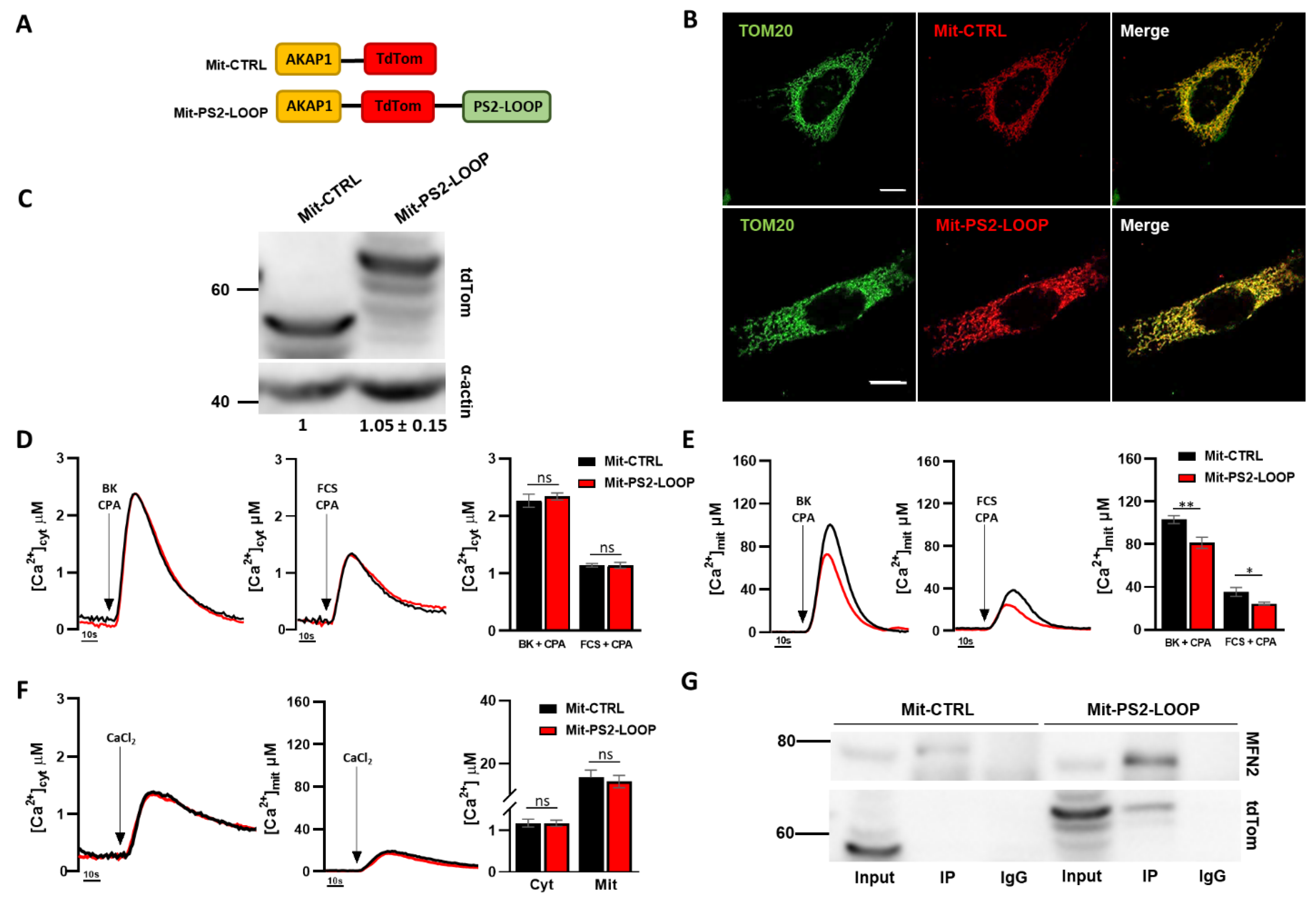
3.2. The Mitochondria-Targeted PS2 Loop Domain Exerts an Opposite Effect Compared to Full-Length PS2 on ER–Mitochondria Coupling
3.3. The Mitochondria-Targeted PS2 Loop Domain Normalizes ER–Mitochondria Tethering in FAD-PS2-N141I Patient-Derived Fibroblasts
3.4. The Mitochondria-Targeted PS2 Loop Domain Rectifies the Increased Lipid Droplet Formation Found in FAD-PS2-N141I Patient-Derived Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolfe, M.S. Probing mechanisms and therapeutic potential of γ-secretase in alzheimer’s disease. Molecules 2021, 26, 388. [Google Scholar] [CrossRef]
- Escamilla-Ayala, A.; Wouters, R.; Sannerud, R.; Annaert, W. Contribution of the Presenilins in the cell biology, structure and function of γ-secretase. Semin. Cell Dev. Biol. 2020, 105, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef]
- Duggan, S.P.; McCarthy, J.V. Beyond gamma-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell. Signal. 2016, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.; Ratan, R.R.; Shelanski, M.L.; Goldman, J.E. Altered response of fibroblasts from aged and Alzheimer donors to drugs that elevate cytosolic free calcium. Neurobiol. Aging 1988, 9, 261–266. [Google Scholar] [CrossRef]
- Agostini, M.; Fasolato, C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pendin, D.; Fasolato, C.; Basso, E.; Filadi, R.; Greotti, E.; Galla, L.; Gomiero, C.; Leparulo, A.; Redolfi, N.; Scremin, E.; et al. Familial Alzheimer’s disease presenilin-2 mutants affect Ca2+ homeostasis and brain network excitability. Aging Clin. Exp. Res. 2019, 33, 1705–1708. [Google Scholar] [CrossRef] [PubMed]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [Green Version]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef]
- Loncke, J.; Kaasik, A.; Bezprozvanny, I.; Parys, J.B.; Kerkhofs, M.; Bultynck, G. Balancing ER-Mitochondrial Ca2+ Fluxes in Health and Disease. Trends Cell Biol. 2021, 31, 598–612. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, T.; Rho, E.; Sample, V.; Akano, H.; Magari, M.; Ueno, T.; Gorshkov, K.; Chen, M.; Tokumitsu, H.; Zhang, J.; et al. Compartmentalized AMPK Signaling Illuminated by Genetically Encoded Molecular Sensors and Actuators. Cell Rep. 2015, 11, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Rimessi, A.; Romagnoli, A.; Prandini, A.; Rizzuto, R. Biosensors for the detection of calcium and pH. Methods Cell Biol. 2007, 80, 297–325. [Google Scholar]
- Xia, W. From Presenilinase to Υ-Secretase, Cleave to Capacitate. Curr. Alzheimer Res. 2008, 5, 172–178. [Google Scholar] [CrossRef]
- Herker, E.; Vieyres, G.; Beller, M.; Krahmer, N.; Bohnert, M. Lipid Droplet Contact Sites in Health and Disease. Trends Cell Biol. 2021, 31, 345–358. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid droplets and the management of cellular stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Venditti, R.; Wilson, C.; De Matteis, M.A. Regulation and physiology of membrane contact sites. Curr. Opin. Cell Biol. 2021, 71, 148–157. [Google Scholar] [CrossRef]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Petkovic, M.; O’Brien, C.E.; Jan, Y.N. Interorganelle communication, aging, and neurodegeneration. Genes Dev. 2021, 35, 449–469. [Google Scholar] [CrossRef] [PubMed]
- Simoes, I.C.M.; Morciano, G.; Lebiedzinska-Arciszewska, M.; Aguiari, G.; Pinton, P.; Potes, Y.; Wieckowski, M.R. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165834. [Google Scholar] [CrossRef]
- Rossini, M.; Filadi, R. Sarcoplasmic Reticulum-Mitochondria Kissing in Cardiomyocytes: Ca2+, ATP, and Undisclosed Secrets. Front. Cell Dev. Biol. 2020, 8, 532. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J. Role of Endoplasmic Reticulum-Mitochondria Communication in Type 2 Diabetes. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 997, pp. 171–186. [Google Scholar]
- Leal, N.S.; Martins, L.M. Mind the gap: Mitochondria and the endoplasmic reticulum in neurodegenerative diseases. Biomedicines 2021, 9, 227. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Naia, L.; Piras, A.; Graff, C.; Cattaneo, A.; Meli, G.; Hamasaki, M.; Nilsson, P.; et al. Amyloid Β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models. Cells 2020, 9, 2552. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.R.; Montesinos, J.; Larrea, D.; Area-Gomez, E.; Pera, M. The silence of the fats: A MAM’s story about Alzheimer. Neurobiol. Dis. 2020, 145, 105062. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.M.K.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef]
- Filadi, R.; Pizzo, P. Mitochondrial Calcium Handling and Neurodegeneration: When a good signal goes wrong. Curr. Opin. Physiol. 2020, 17, 224–233. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossini, M.; García-Casas, P.; Filadi, R.; Pizzo, P. Loosening ER–Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain. Cells 2021, 10, 1968. https://doi.org/10.3390/cells10081968
Rossini M, García-Casas P, Filadi R, Pizzo P. Loosening ER–Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain. Cells. 2021; 10(8):1968. https://doi.org/10.3390/cells10081968
Chicago/Turabian StyleRossini, Michela, Paloma García-Casas, Riccardo Filadi, and Paola Pizzo. 2021. "Loosening ER–Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain" Cells 10, no. 8: 1968. https://doi.org/10.3390/cells10081968
APA StyleRossini, M., García-Casas, P., Filadi, R., & Pizzo, P. (2021). Loosening ER–Mitochondria Coupling by the Expression of the Presenilin 2 Loop Domain. Cells, 10(8), 1968. https://doi.org/10.3390/cells10081968