Nanoporous Gold Monolith for High Loading of Unmodified Doxorubicin and Sustained Co-Release of Doxorubicin-Rapamycin



Abstract
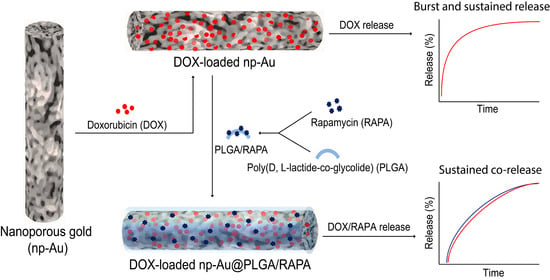
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
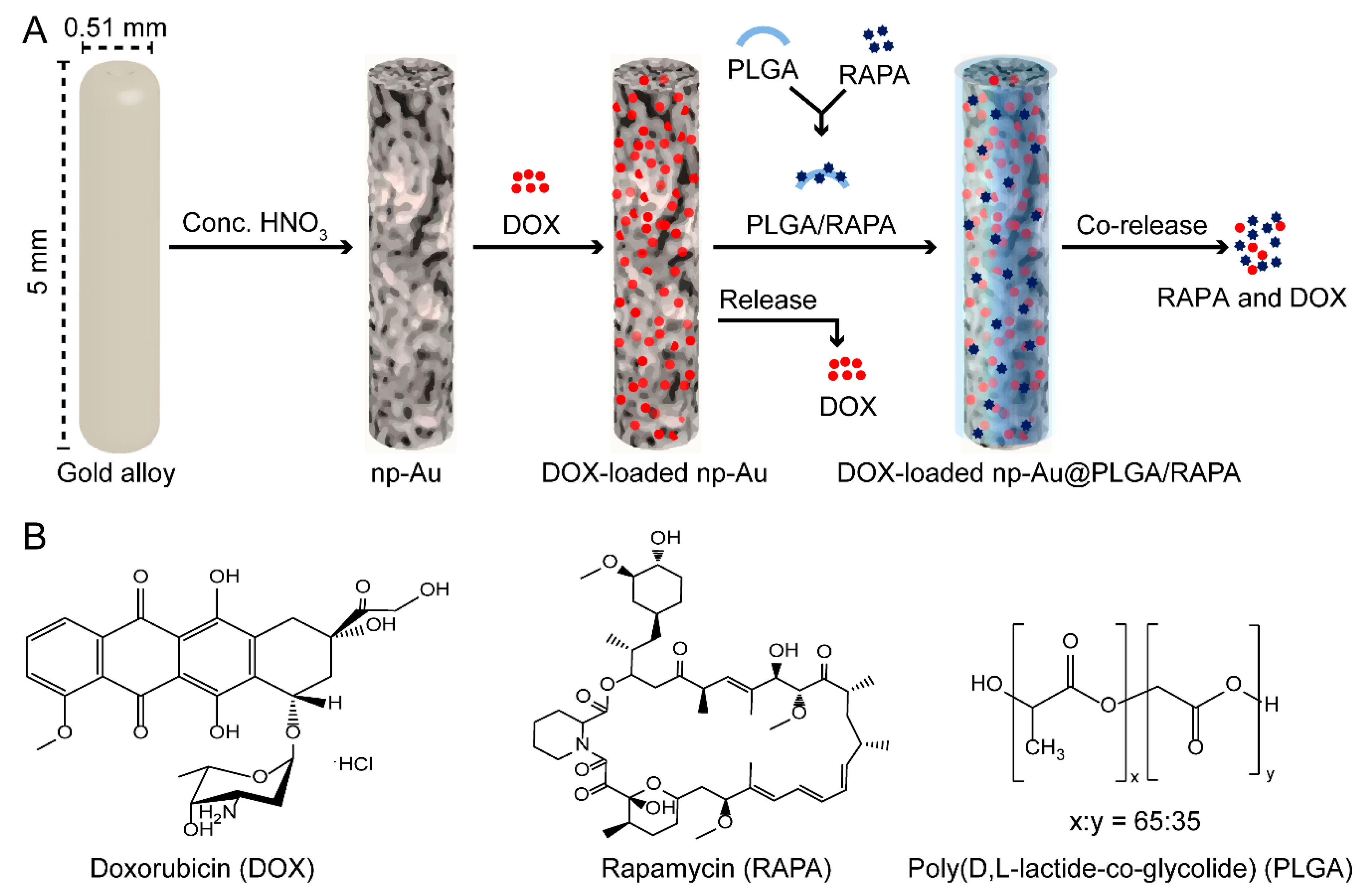
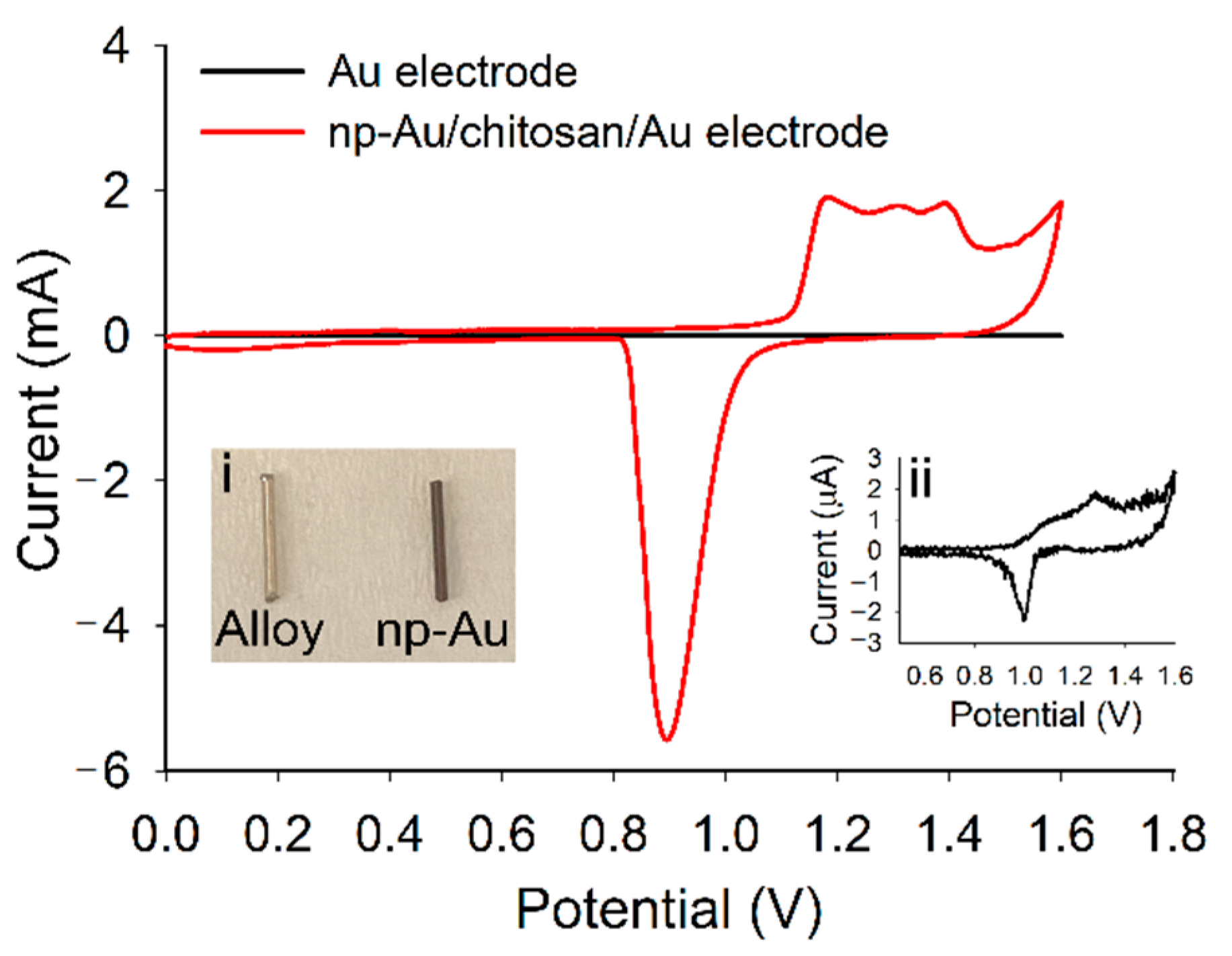
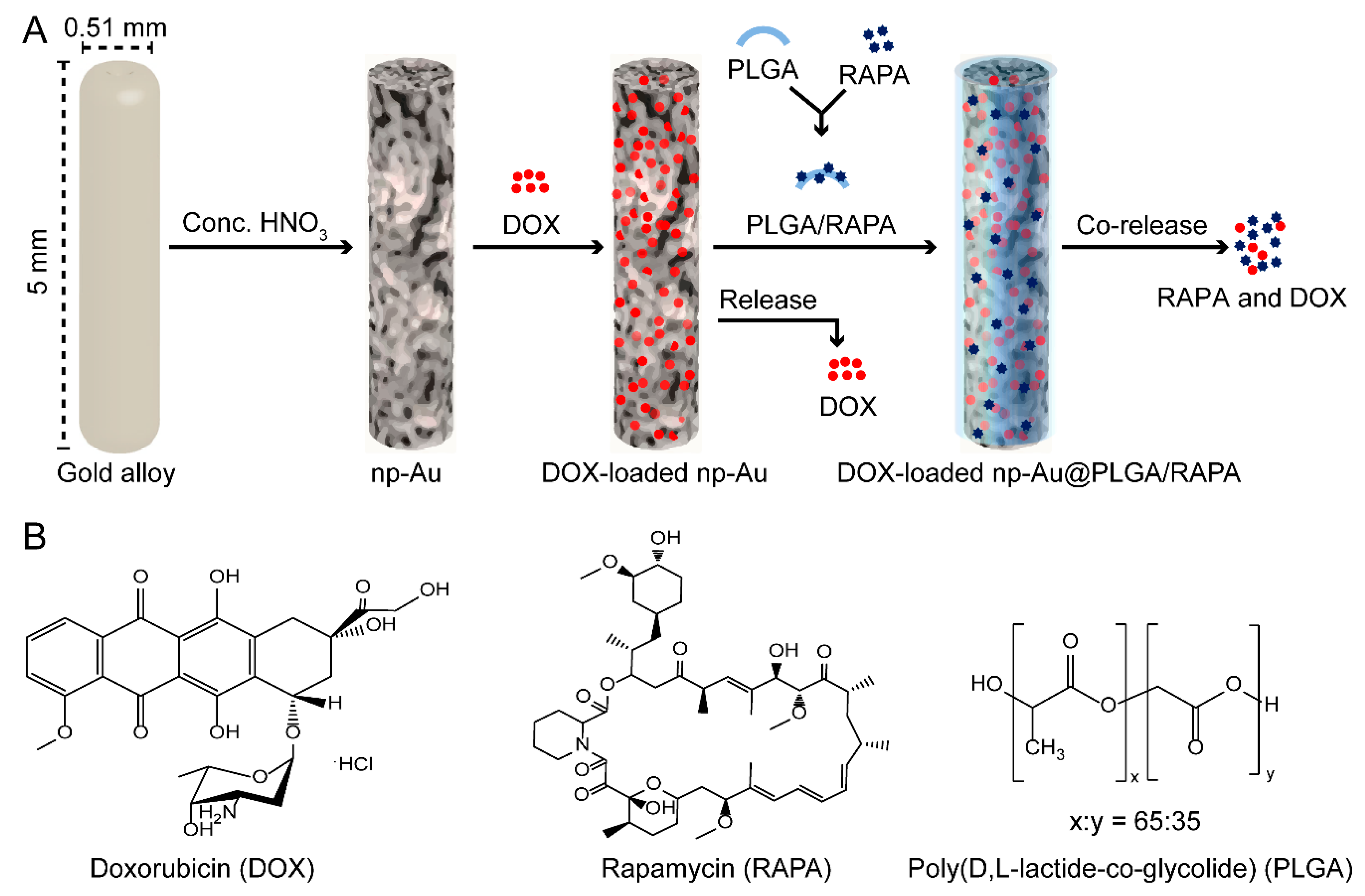
2.2. Preparation of np-Au Millirods
2.3. Surface Area Estimation of np-Au
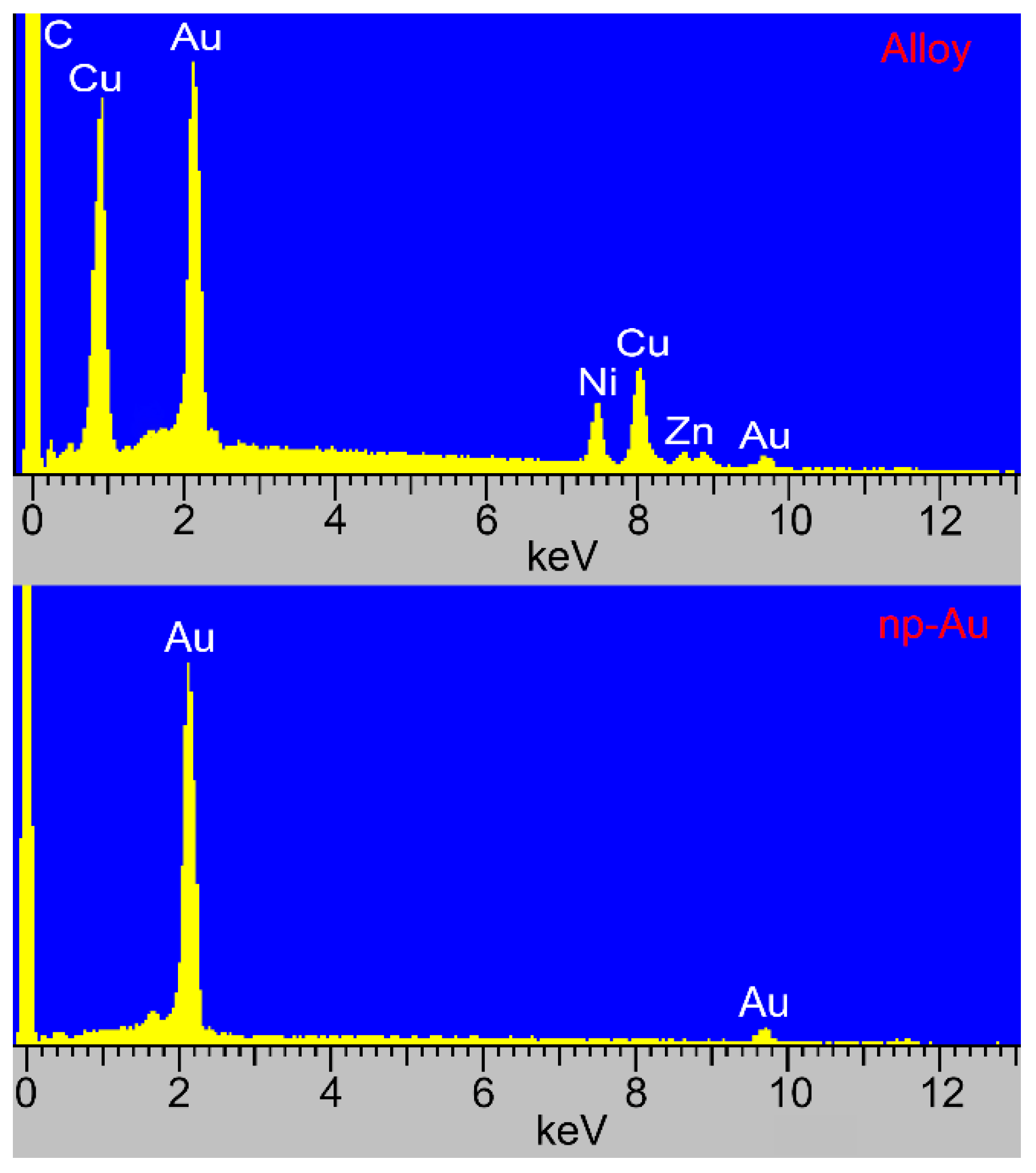
2.4. Surface Morphology and Composition of np-Au
2.5. Loading of DOX Inside np-Au
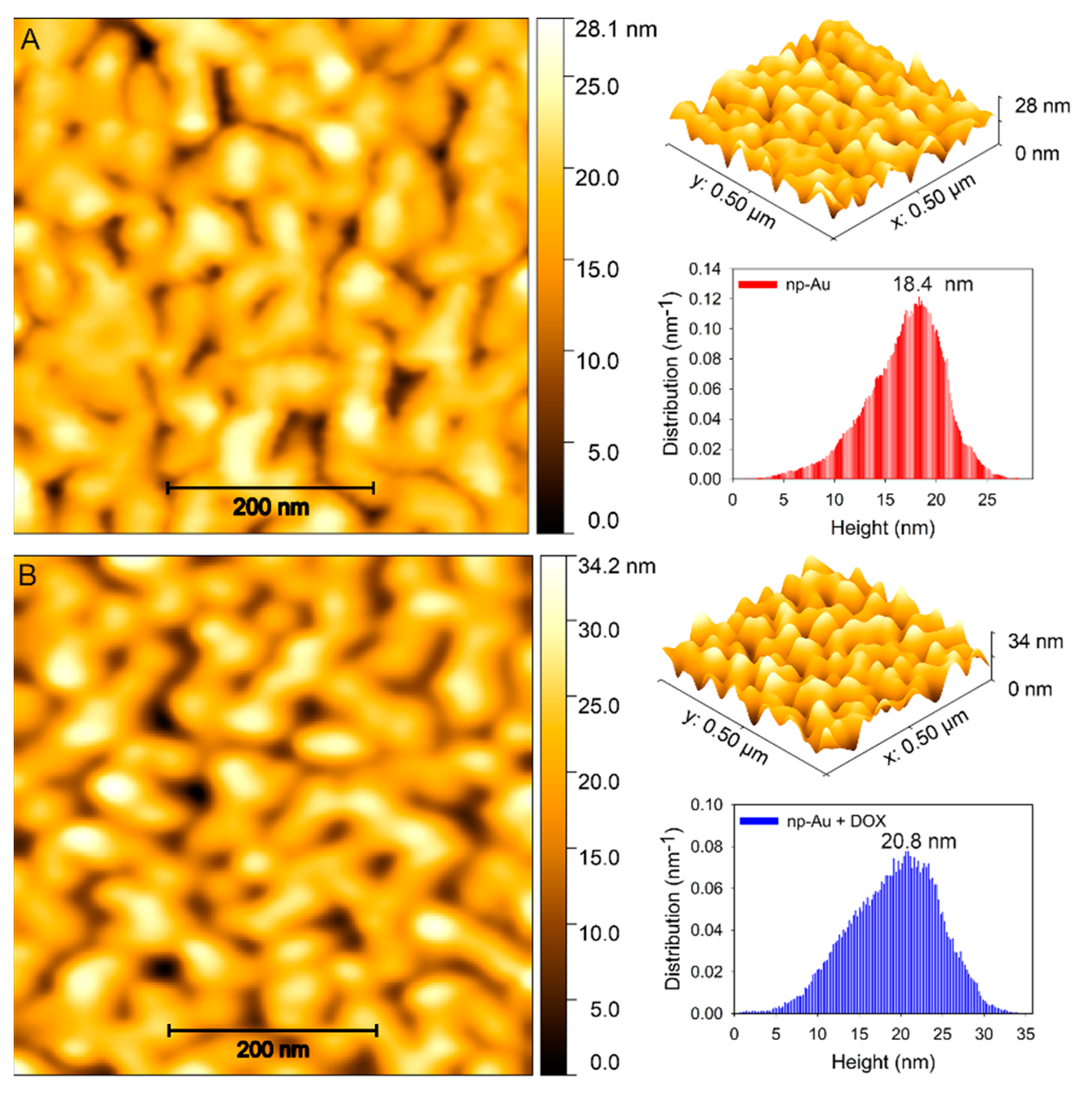
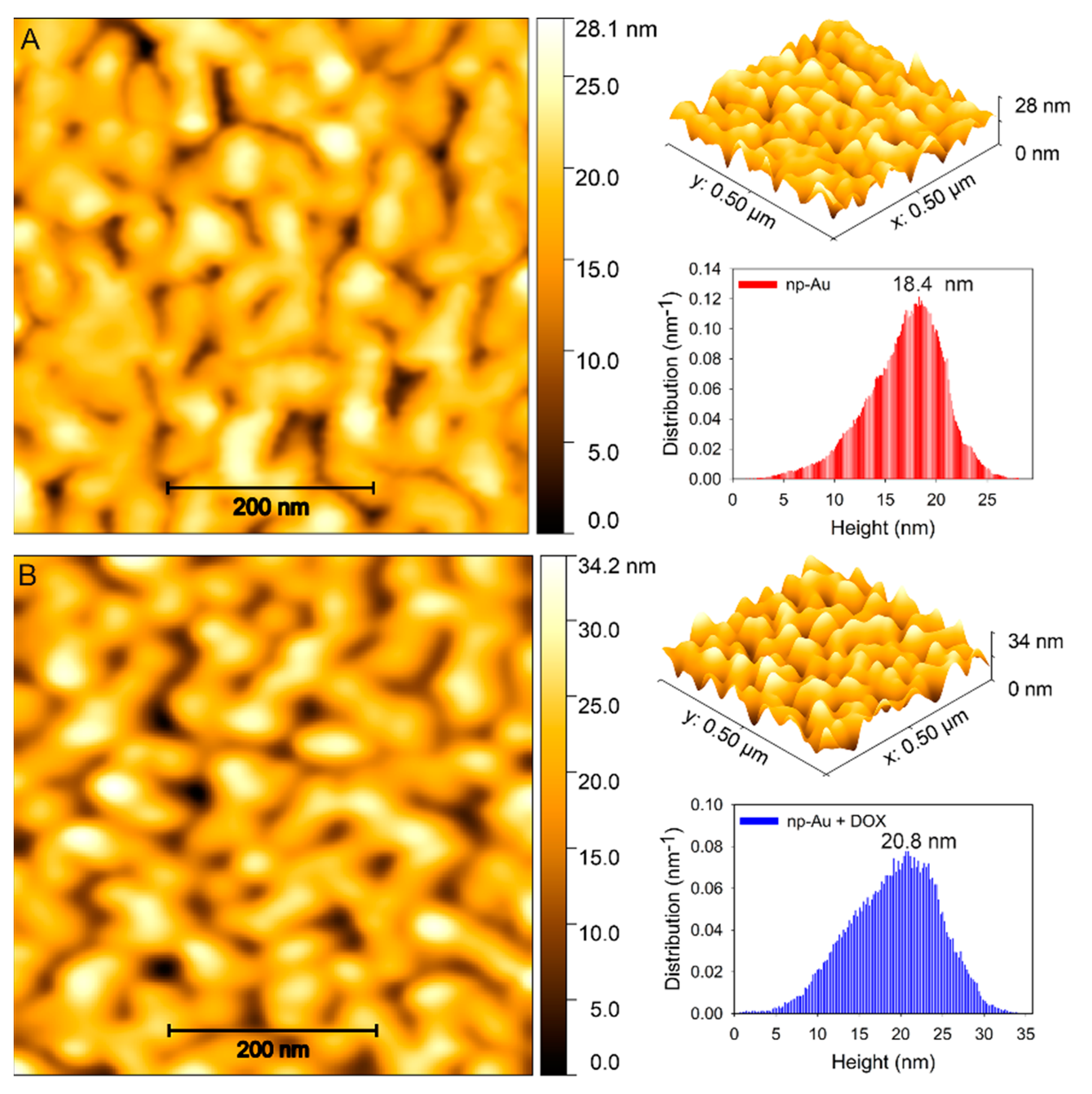
2.6. Atomic Force Microscopy (AFM) Imaging
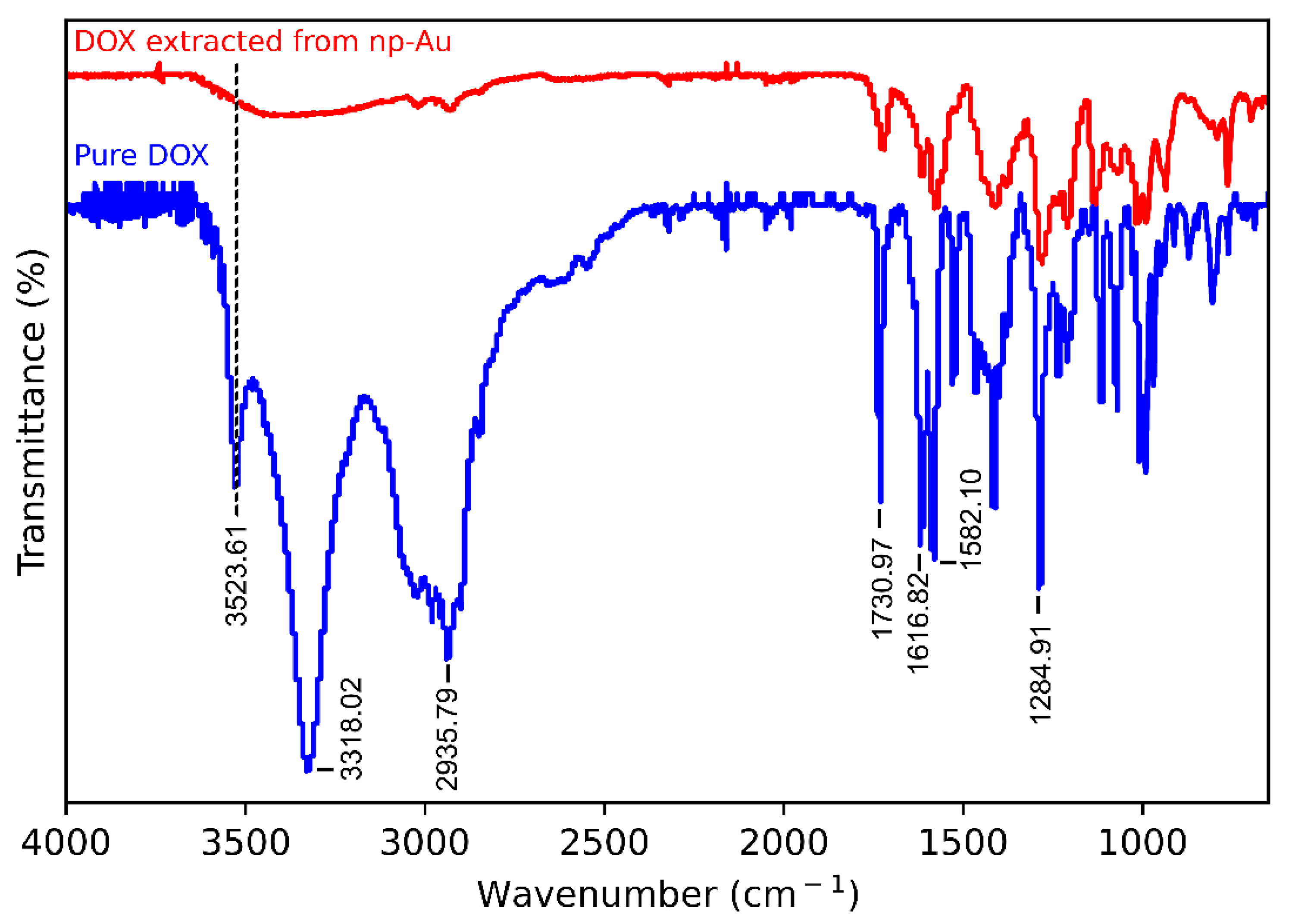
2.7. Fourier Transform Infrared (FTIR) Spectroscopy
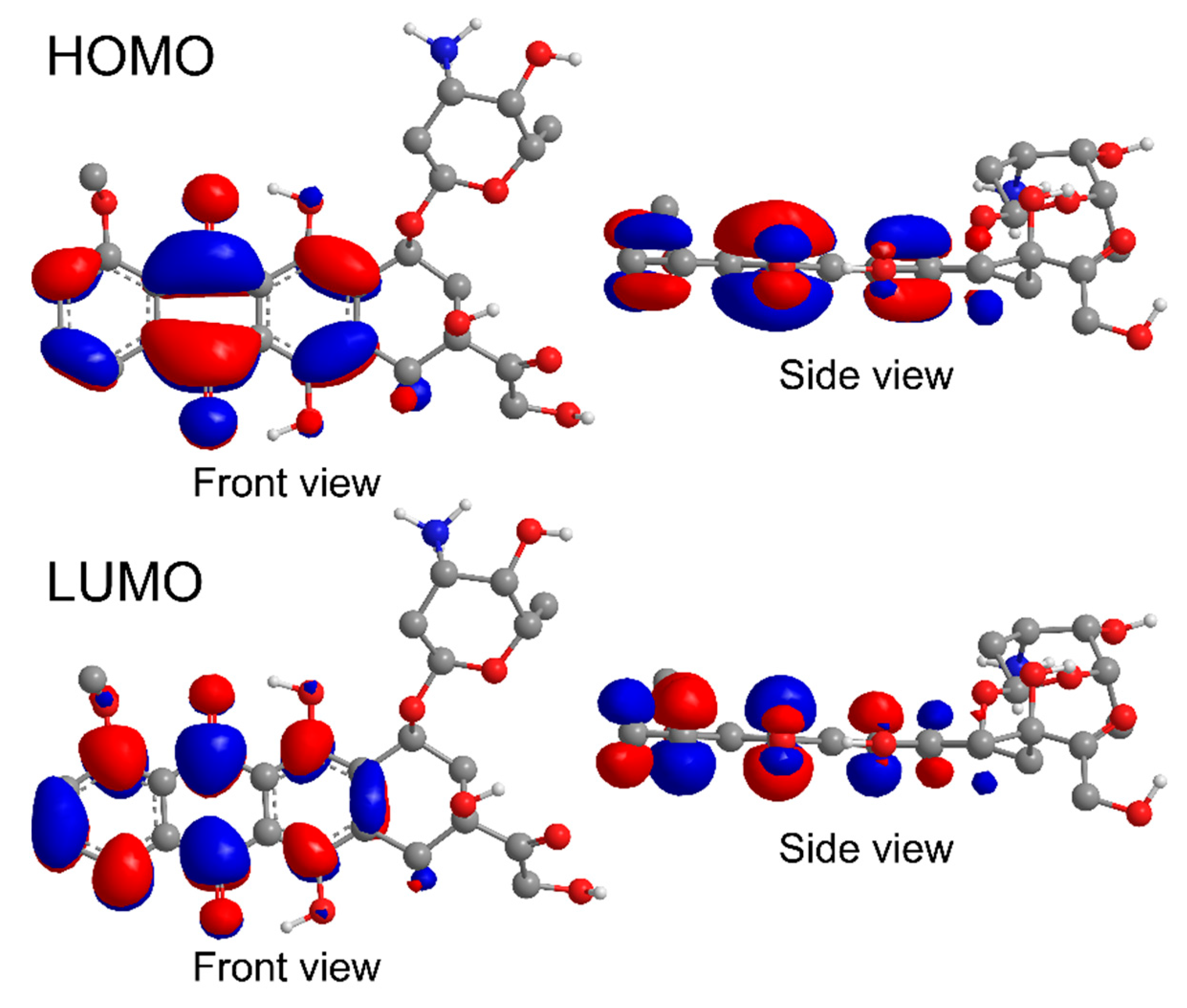
2.8. Computations
2.9. Coating of DOX-Loaded np-Au with PLGA/RAPA Film
2.10. In Vitro Drugs Release Kinetics
3. Results and Discussion
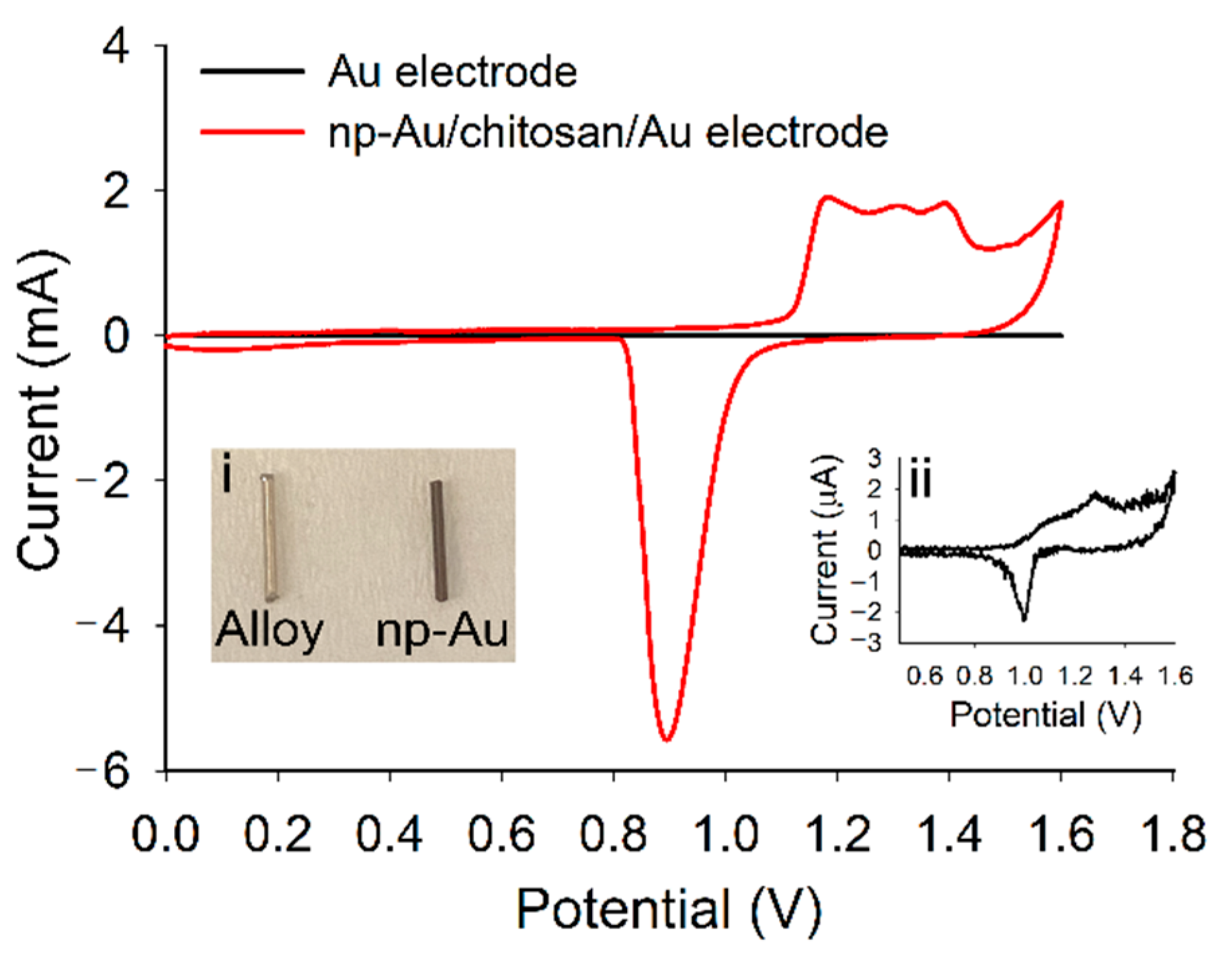
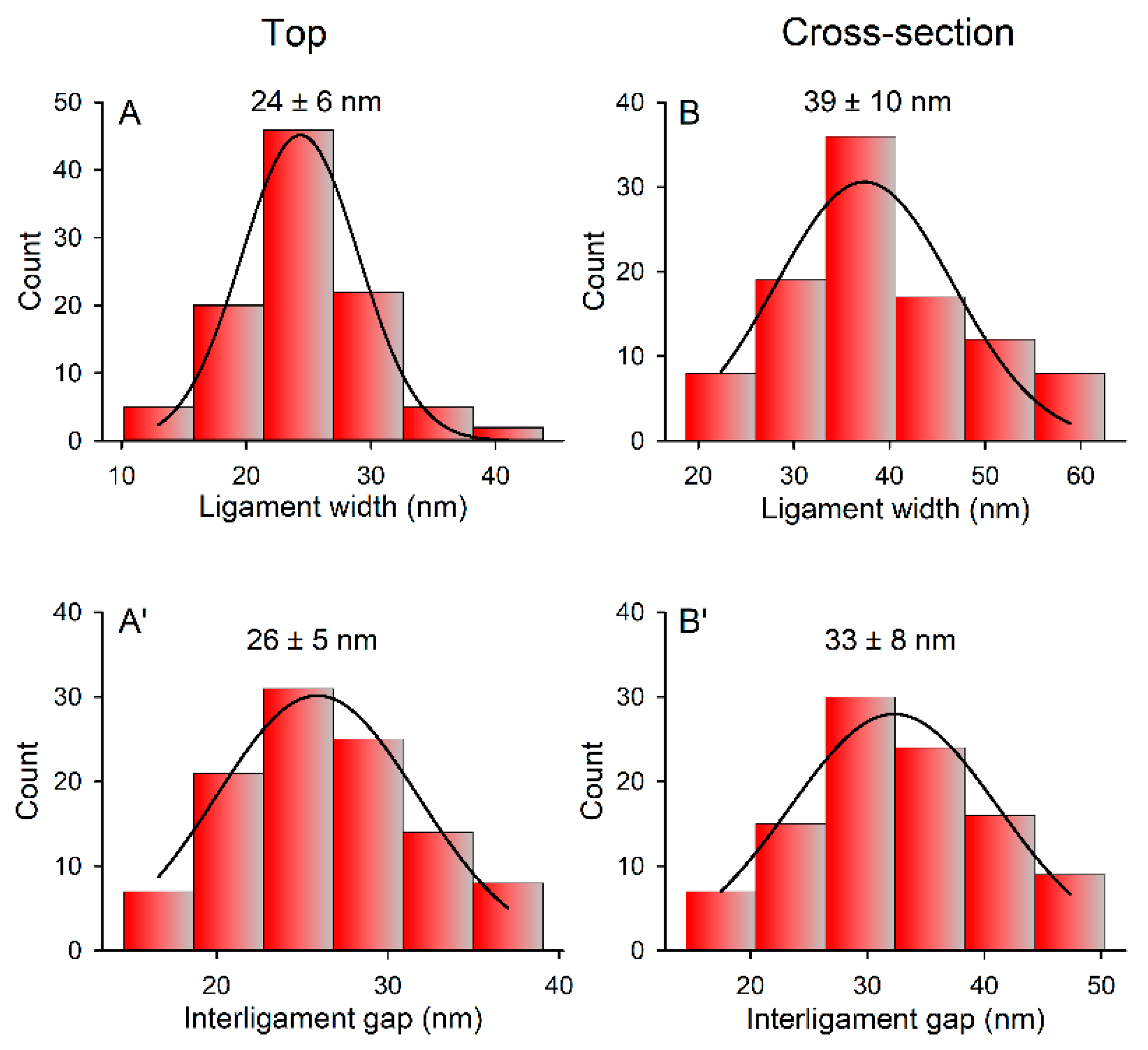
3.1. Fabrication and Characterization of np-Au Millrods
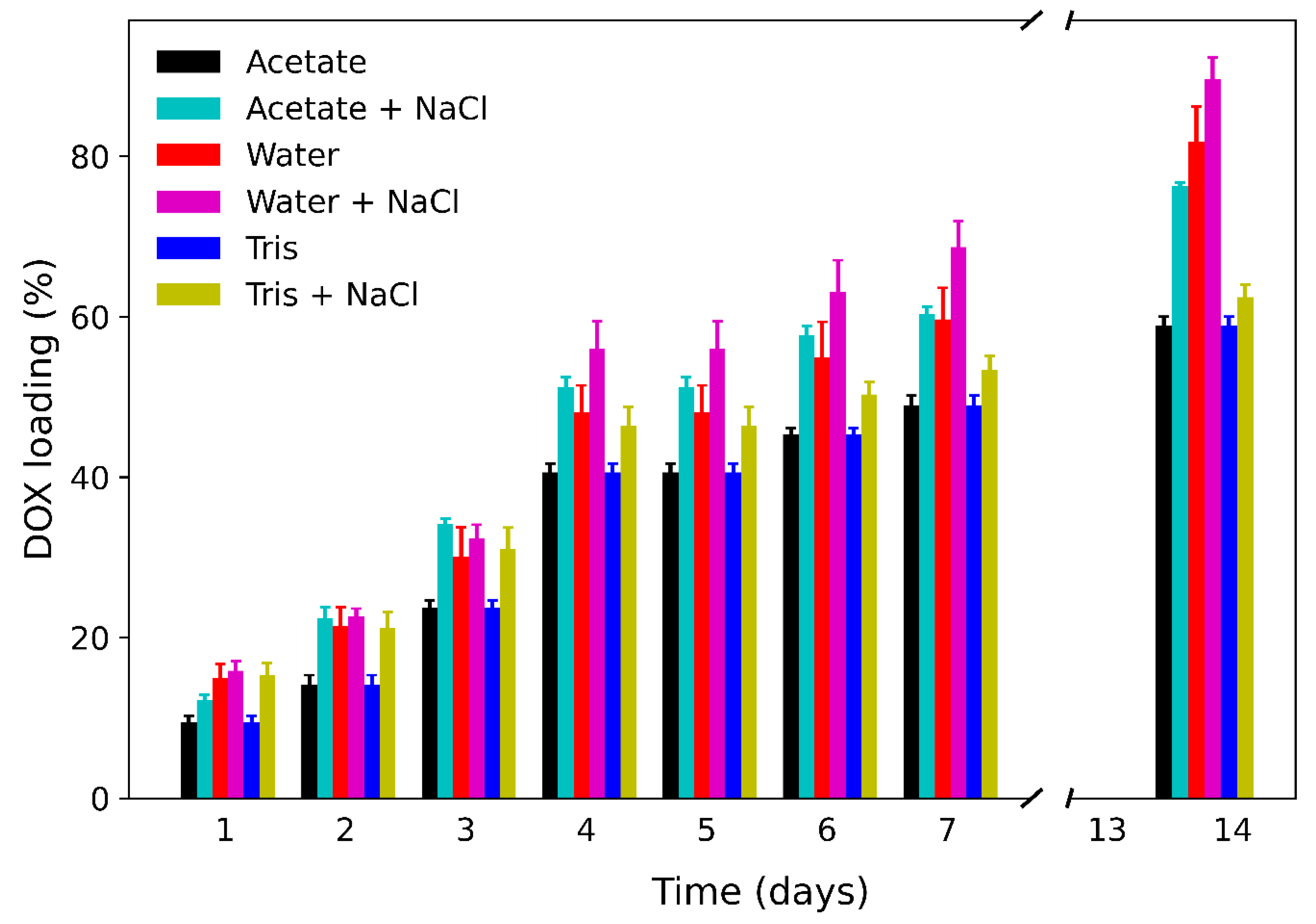
3.2. Loading of DOX at Low Concentration
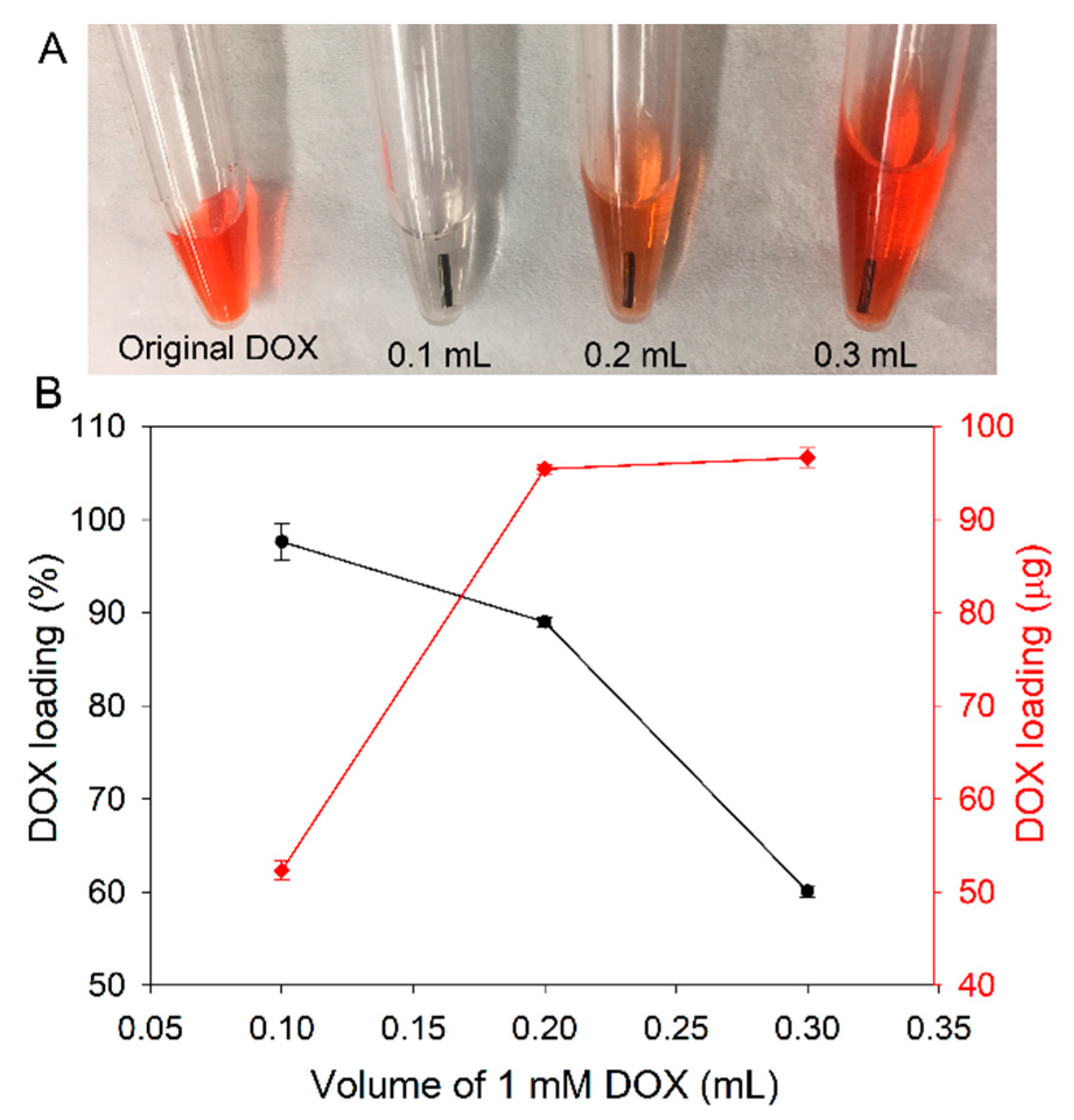
3.3. Loading of DOX at High Concentration
3.4. Effect of DMSO on Loading of DOX
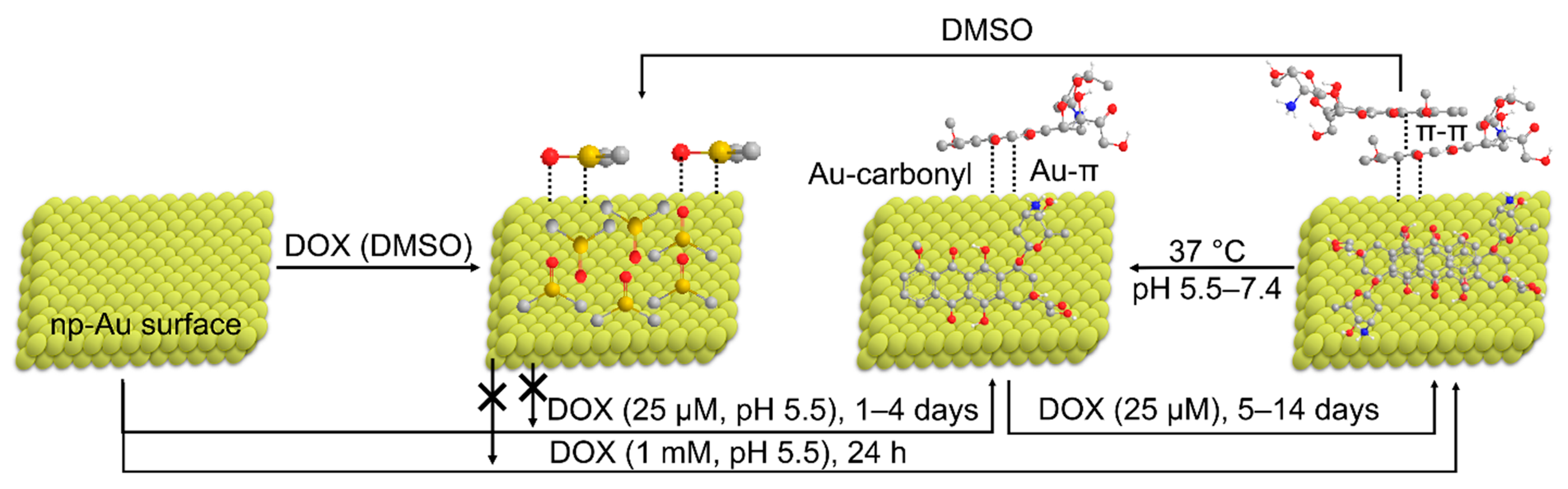
3.5. Mechanism of Loading of DOX on np-Au
3.6. PLGA/RAPA Coating
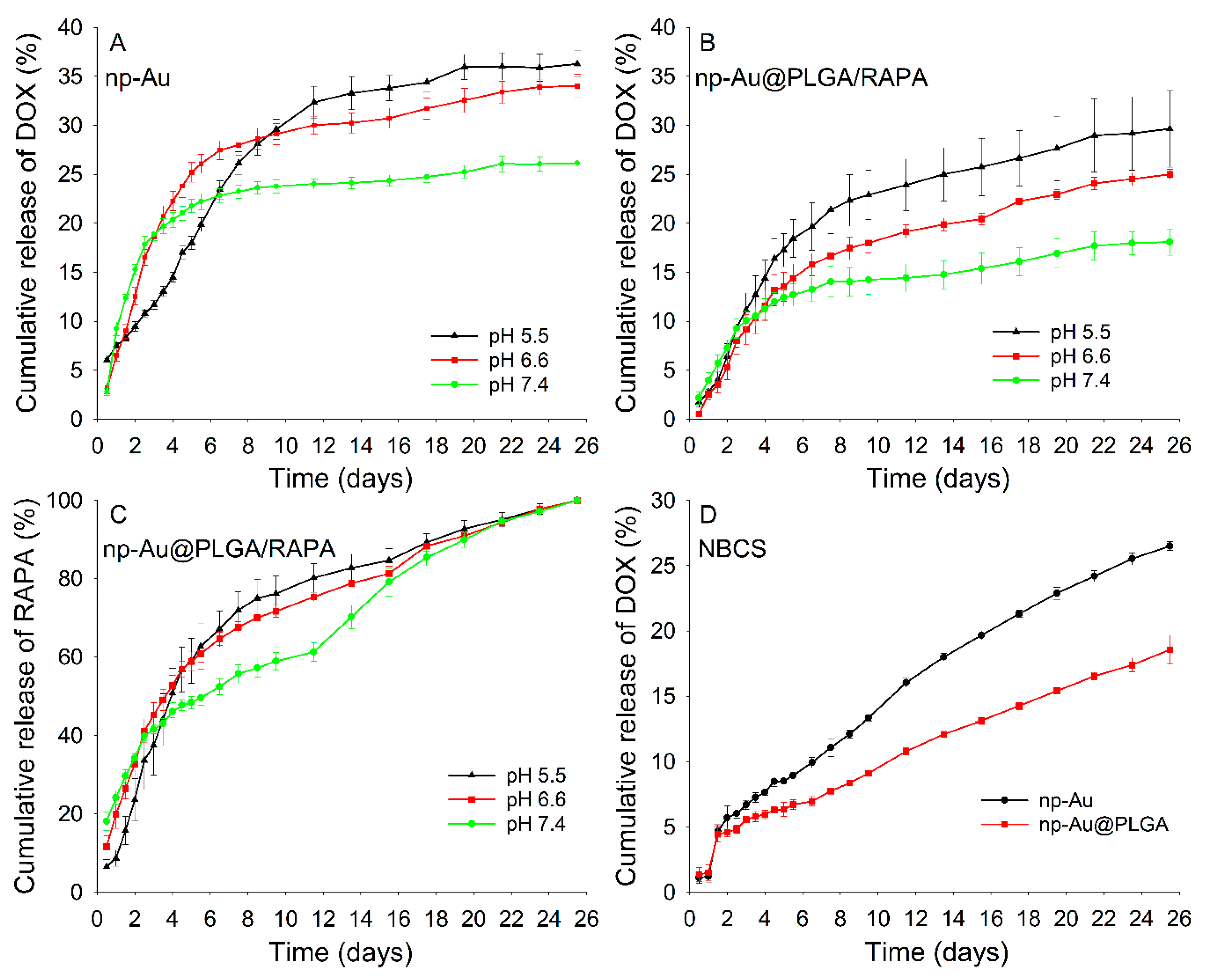
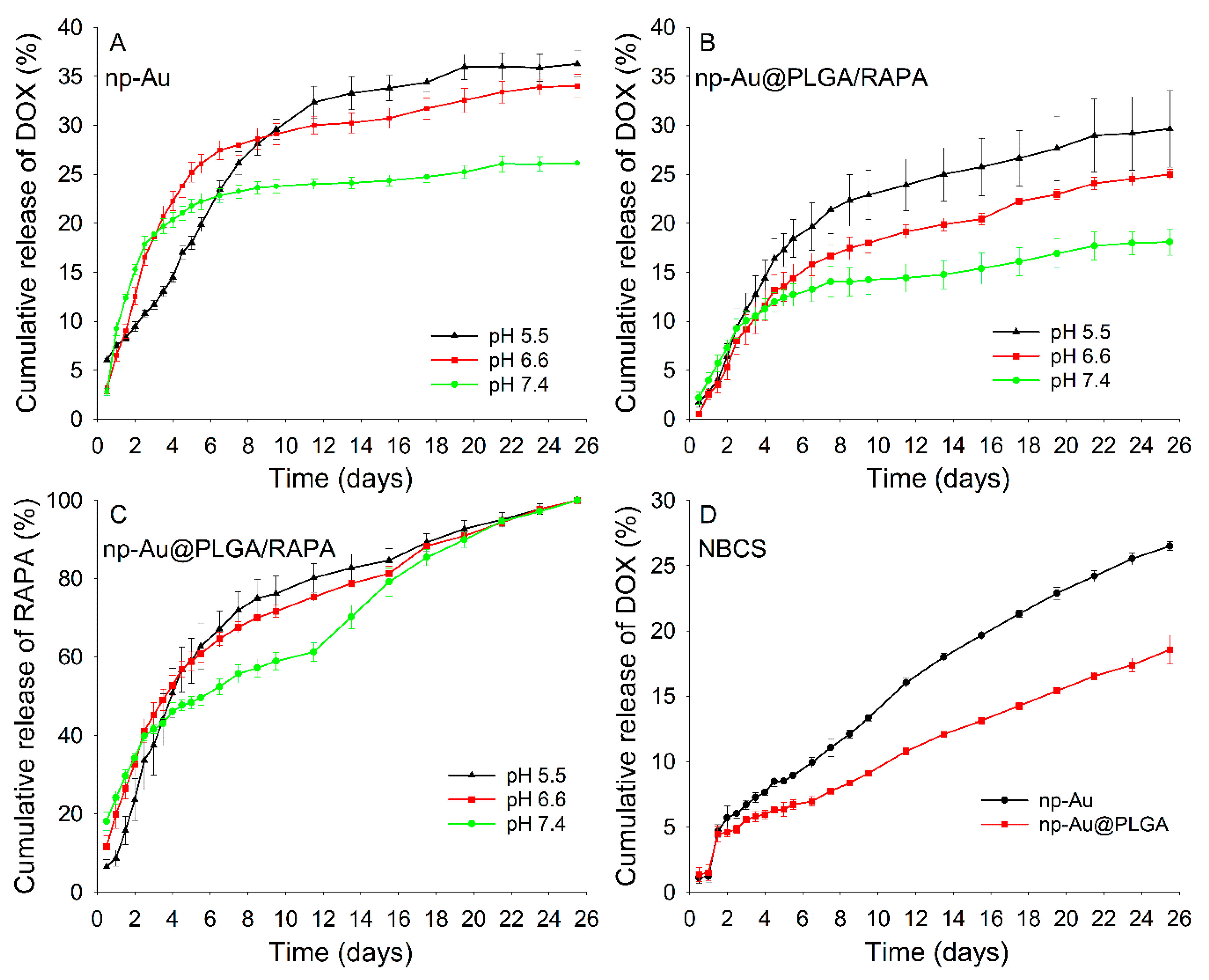
3.7. In Vitro Drug Release
3.8. Drug Release Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.; Tomczak, P.; Ackland, S. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX™/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef] [PubMed]
- A’hern, R.; Gore, M. Impact of doxorubicin on survival in advanced ovarian cancer. J. Clin. Oncol. 1995, 13, 726–732. [Google Scholar] [CrossRef] [PubMed]
- James, N.; Coker, R.; Tomlinson, D.; Harris, J.; Gompels, M.; Pinching, A.; Stewart, J. Liposomal doxorubicin (Doxil): An effective new treatment for Kaposi’s sarcoma in AIDS. Clin. Oncol. 1994, 6, 294–296. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Cai, X.; Luo, Y.; Zhang, W.; Du, D.; Lin, Y. pH-sensitive ZnO quantum dots-doxorubicin nanoparticles for lung cancer targeted drug delivery. ACS Appl. Mater. Interfaces 2016, 8, 22442–22450. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Sahlgren, C.; Linden, M. Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles—Opportunities & challenges. Nanoscale 2010, 2, 1870–1883. [Google Scholar]
- Dilnawaz, F.; Singh, A.; Mohanty, C.; Sahoo, S.K. Dual drug loaded superparamagnetic iron oxide nanoparticles for targeted cancer therapy. Biomaterials 2010, 31, 3694–3706. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, Y.; Leiser, Y.; Kachta, O.; El-Naaj, I.A. Does long-term treatment with Doxil® predispose patients to oral cancer? Int. J. Clin. Oncol. 2013, 18, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wang, P.; Li, Q.; Al-Khalaf, A.A.; Hozzein, W.N.; Zhang, F.; Li, X.; Zhao, D. Near-infrared triggered decomposition of nanocapsules with high tumor accumulation and stimuli responsive fast elimination. Angew. Chem. Int. Ed. 2018, 57, 2611–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Xia, Y.; Zou, Y.; Yang, W.; Zhang, J.; Zhong, Z.; Meng, F. ATN-161 peptide functionalized reversibly cross-linked polymersomes mediate targeted doxorubicin delivery into melanoma-bearing C57BL/6 mice. Mol. Pharm. 2017, 14, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles-based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J. Control. Release 2012, 159, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Aw, M.S.; Bariana, M.; Kumeria, T.; Wang, Y.; Losic, D. Drug-releasing implants: Current progress, challenges and perspectives. J. Mater. Chem. B 2014, 2, 6157–6182. [Google Scholar] [CrossRef]
- Whitmore, W.F.; Hilaris, B.; Grabstald, H. Retropubic implantation of iodine 125 in the treatment of prostatic cancer. J. Urol. 1972, 108, 918–920. [Google Scholar] [CrossRef]
- Giese, A.; Kucinski, T.; Knopp, U.; Goldbrunner, R.; Hamel, W.; Mehdorn, H.M.; Tonn, J.C.; Hilt, D.; Westphal, M. Pattern of recurrence following local chemotherapy with biodegradable carmustine (BCNU) implants in patients with glioblastoma. J. Neuro-Oncol. 2004, 66, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, V.; DeGraffenried, L.; Russel, D.; Friedrichs, W.E.; Ray, R.B.; Hidalgo, M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002, 62, 6141–6145. [Google Scholar] [PubMed]
- Romano, M.F.; Avellino, R.; Petrella, A.; Bisogni, R.; Romano, S.; Venuta, S. Rapamycin inhibits doxorubicin-induced NF-kappaB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur. J. Cancer 2004, 40, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Trape, A.P.; Katayama, M.L.; Roela, R.A.; Brentani, H.; Ravacci, G.R.; de Araujo Lima, L.; Brentani, M.M. Gene expression profile in response to doxorubicin-rapamycin combined treatment of HER-2-overexpressing human mammary epithelial cell lines. Mol. Cancer. Ther. 2012, 11, 464–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losic, D.; Simovic, S. Self-ordered nanopore and nanotube platforms for drug delivery applications. Expert Opin. Drug Deliv. 2009, 6, 1363–1381. [Google Scholar] [CrossRef]
- Aw, M.S.; Kurian, M.; Losic, D. Non-eroding drug-releasing implants with ordered nanoporous and nanotubular structures: Concepts for controlling drug release. Biomater. Sci. 2014, 2, 10–34. [Google Scholar]
- Jafari, S.; Mahyad, B.; Hashemzadeh, H.; Janfaza, S.; Gholikhani, T.; Tayebi, L. Biomedical applications of TiO2 nanostructures: Recent advances. Int. J. Nanomed. 2020, 15, 3447–3470. [Google Scholar] [CrossRef]
- Polat, O.; Seker, E. Halide-gated molecular release from nanoporous gold thin films. J. Phys. Chem. C 2015, 119, 24812–24818. [Google Scholar] [CrossRef] [Green Version]
- Kurtulus, O.; Daggumati, P.; Seker, E. Molecular release from patterned nanoporous gold thin films. Nanoscale 2014, 6, 7062–7071. [Google Scholar] [CrossRef] [Green Version]
- Seker, E.; Berdichevsky, Y.; Staley, K.J.; Yarmush, M.L. Microfabrication-compatible nanoporous gold foams as biomaterials for drug delivery. Adv. Healthc. Mater. 2012, 1, 172–176. [Google Scholar] [CrossRef]
- Santos, G.M.; Zhao, F.; Zeng, J.; Shih, W.-C. Characterization of nanoporous gold disks for photothermal light harvesting and light-gated molecular release. Nanoscale 2014, 6, 5718–5724. [Google Scholar] [CrossRef] [PubMed]
- Seker, E.; Shih, W.-C.; Stine, K.J. Nanoporous metals by alloy corrosion: Bioanalytical and biomedical applications. MRS Bull. 2018, 43, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, J.K.; Neupane, D.; Nepal, B.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Preparation, modification, characterization, and biosensing application of nanoporous gold using electrochemical techniques. Nanomaterials 2018, 8, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, J.K.; Neupane, D.; Nepal, B.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Structure and applications of gold in nanoporous form. In Noble and Precious Metals-Properties, Nanoscale Effects and Applications; Seehra, M.S., Bristow, A.D., Eds.; IntechOpen: London, UK, 2017; pp. 341–365. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Parmar, A.; Kori, S.; Sandhir, R. PLGA-based nanoparticles: A new paradigm in biomedical applications. TrAC-Trend. Anal. Chem. 2016, 80, 30–40. [Google Scholar] [CrossRef]
- Shukla, R.; Bansal, V.; Chaudhary, M.; Basu, A.; Bhonde, R.R.; Sastry, M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: A microscopic overview. Langmuir 2005, 21, 10644–10654. [Google Scholar] [CrossRef]
- Kang, M.S.; Lee, S.Y.; Kim, K.S.; Han, D.-W. State of the art biocompatible gold nanoparticles for cancer theragnosis. Pharmaceutics 2020, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Demann, E.T.K.; Stein, P.S.; Haubenreich, J.E. Gold as an implant in medicine and dentistry. J. Long Term Eff. Med. Implants 2005, 15, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.A.R.; Wang, L.; Chen, H.; Garrison, J.; Lein, P.J.; Seker, E. Nanoporous gold biointerfaces: Modifying nanostructure to control neural cell coverage and enhance electrophysiological recording performance. Adv. Funct. Mater. 2017, 27, 1604631. [Google Scholar] [CrossRef] [Green Version]
- Chapman, C.A.R.; Chen, H.; Stamou, M.; Biener, J.; Biener, M.M.; Lein, P.J.; Seker, E. Nanoporous gold as a neural interface coating: Effects of topography, surface chemistry, and feature size. ACS Appl. Mater. Interf. 2015, 7, 7093–7100. [Google Scholar] [CrossRef]
- Ding, S.; Cao, S.; Liu, Y.; Lian, Y.; Zhu, A.; Shi, G. Rational design of a stimuli-responsive polymer electrode interfacecoupled with in vivo microdialysis for measurement of sialic acid in live mouse brain in Alzheimer’s disease. ACS Sens. 2017, 2, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Jung, S.; Zhang, H.; Kang, T.; Yoo, Y.; Hong, J.-P.; Ahn, J.-P.; Kwak, J.; Jeon, D.; Kotov, N.; et al. Subcellular neural probes from single-crystal gold nanowires. ACS Nano 2014, 8, 8182–8189. [Google Scholar] [CrossRef] [PubMed]
- Alla, A.J.; d’Andrea, F.B.; Bhattarai, J.K.; Cooper, J.A.; Tan, Y.H.; Demchenko, A.V.; Stine, K.J. Selective capture of glycoproteins using lectin-modified nanoporous gold monolith. J. Chromatogr. A 2015, 1423, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Bhattarai, J.K.; Alla, A.J.; Demchenko, A.V.; Stine, K.J. Electrochemical annealing of nanoporous gold by application of cyclic potential sweeps. Nanotechnology 2015, 26, 085602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.H.; Davis, J.A.; Fujikawa, K.; Ganesh, N.V.; Demchenko, A.V.; Stine, K.J. Surface area and pore size characteristics of nanoporous gold subjected to thermal, mechanical, or surface modification studied using gas adsorption isotherms, cyclic voltammetry, thermogravimetric analysis, and scanning electron microscopy. J. Mater. Chem. 2012, 22, 6733–6745. [Google Scholar] [PubMed]
- Pandey, B.; Bhattarai, J.K.; Pornsuriyasak, P.; Fujikawa, K.; Catania, R.; Demchenko, A.V.; Stine, K.J. Square-wave voltammetry assays for glycoproteins on nanoporous gold. J. Electroanal. Chem. 2014, 717–718, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Yao, L.; An, M. Reversing the undesirable pH-profile of doxorubicin via activation of a di-substituted maleamic acid prodrug at tumor acidity. Chem. Commun. 2017, 53, 12826–12829. [Google Scholar]
- Giesbers, M.; Kleijn, J.M.; Cohen Stuart, M.A. The electrical double layer on gold probed by electrokinetic and surface force measurements. J. Coll. Interf. Sci. 2002, 248, 88–95. [Google Scholar] [CrossRef]
- Weinberg, B.D.; Ai, H.; Blanco, E.; Anderson, J.M.; Gao, J. Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods. J. Biomed. Mater. Res. A 2007, 81A, 161–170. [Google Scholar]
- Qian, L.H.; Chen, M.W. Ultrafine nanoporous gold by low temperature dealloying and kinetics of nanopore formation. Appl. Phys. Lett. 2007, 91, 083105. [Google Scholar]
- Zhai, S.; Hu, X.; Hu, Y.; Wu, B.; Xing, D. Visible light-induced crosslinking and physiological stabilization of diselenide-rich nanoparticles for redox-responsive drug release and combination chemotherapy. Biomaterials 2017, 121, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Swiech, O.; Mieczkowska, A.; Chmurski, K.; Bilewicz, R. Intermolecular interactions between doxorubicin and β-cyclodextrin 4-methoxyphenol conjugates. J. Phys. Chem. B 2012, 116, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, U.; Bukhari, N.I.; Rana, N.F.; Rehman, M.; Hussain, K.; Abbas, N.; Mehmood, A.; Raza, A. Doxorubicin-loaded quaternary ammonium palmitoyl glycol chitosan polymeric nanoformulation: Uptake by cells and organs. Int. J. Nanomed. 2019, 14, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, Y.-C.; Dou, S.; Xiong, M.-H.; Sun, T.-M.; Wang, J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano 2011, 5, 3679–3692. [Google Scholar] [CrossRef]
- Neupane, D.; Bhattarai, J.K.; Demchenko, A.V.; Stine, K.J. A pH sensitive thiolated β-cyclodextrin-modified nanoporous gold for controlled release of doxorubicin. J. Drug Deliv. Sci. Technol. 2020, 60, 101985. [Google Scholar] [CrossRef]
- You, J.; Zhang, G.; Li, C. Exceptionally high payload of doxorubicin in hollow gold nanospheres for near-infrared light-triggered drug release. ACS Nano 2010, 4, 1033–1041. [Google Scholar] [CrossRef] [Green Version]
- Curry, D.; Cameron, A.; MacDonald, B.; Nganou, C.; Scheller, H.; Marsh, J.; Beale, S.; Lu, M.; Shan, Z.; Kaliaperumal, R.; et al. Adsorption of doxorubicin on citrate-capped gold nanoparticles: Insights into engineering potent chemotherapeutic delivery systems. Nanoscale 2015, 7, 19611–19619. [Google Scholar] [CrossRef]
- Kumar, A.; Mandal, S.; Mathew, S.P.; Selvakannan, P.R.; Mandale, A.B.; Chaudhari, R.V.; Sastry, M. Benzene- and anthracene-mediated assembly of gold nanoparticles at the liquid−liquid interface. Langmuir 2002, 18, 6478–6483. [Google Scholar] [CrossRef]
- Tautz, F.S. Structure and bonding of large aromatic molecules on noble metal surfaces: The example of PTCDA. Prog. Surf. Sci. 2007, 82, 479–520. [Google Scholar] [CrossRef]
- Wagner, M.; Qvortrup, K.; Grier, K.E.; Ottosen, M.R.; Petersen, J.O.; Tanner, D.; Ulstrup, J.; Zhang, J. Gold–carbonyl group interactions in the electrochemistry of anthraquinone thiols self-assembled on Au(111)-surfaces. Chem. Sci. 2019, 10, 3927–3936. [Google Scholar] [CrossRef] [Green Version]
- Han, S.W.; Joo, S.W.; Ha, T.H.; Kim, Y.; Kim, K. Adsorption characteristics of anthraquinone-2-carboxylic acid on gold. J. Phys. Chem. B 2000, 104, 11987–11995. [Google Scholar] [CrossRef]
- Agrawal, P.; Barthwal, S.K.; Barthwal, R. Studies on self-aggregation of anthracycline drugs by restrained molecular dynamics approach using nuclear magnetic resonance spectroscopy supported by absorption, fluorescence, diffusion ordered spectroscopy and mass spectrometry. Eur. J. Med. Chem. 2009, 44, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, M.; Valentini, L.; Vannini, E.; Arcamone, F. Self-association of doxorubicin and related compounds in aqueous solution. J. Pharm. Sci. 1984, 73, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, W.; Duan, Y.; Qing, Z.; Huang, H.; Lu, X. Shaping gold nanocrystals in dimethyl sulfoxide: Toward trapezohedral and bipyramidal nanocrystals enclosed by {311} facets. J. Am. Chem. Soc. 2017, 139, 5817–5826. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.-Y. Phenomenology of the initial burst release of drugs from PLGA microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Wang, J.; Helder, L.; Shao, J.; Jansen, J.A.; Yang, M.; Yang, F. Encapsulation and release of doxycycline from electrospray-generated PLGA microspheres: Effect of polymer end groups. Int. J. Pharm. 2019, 564, 1–9. [Google Scholar] [CrossRef]
- Yoo, H.S.; Lee, K.H.; Oh, J.E.; Park, T.G. In vitro and in vivo anti-tumor activities of nanoparticles based on doxorubicin–PLGA conjugates. J. Control. Release 2000, 68, 419–431. [Google Scholar] [CrossRef]
- Feng, T.; Tian, H.; Xu, C.; Lin, L.; Xie, Z.; Lam, M.H.-W.; Liang, H.; Chen, X. Synergistic co-delivery of doxorubicin and paclitaxel by porous PLGA microspheres for pulmonary inhalation treatment. Eur. J. Pharm. Biopharm. 2014, 88, 1086–1093. [Google Scholar] [CrossRef]
- Lin, R.; Ng, L.S.; Wang, C.-H. In vitro study of anticancer drug doxorubicin in PLGA-based microparticles. Biomaterials 2005, 26, 4476–4485. [Google Scholar] [CrossRef]
- Lagreca, E.; Onesto, V.; Di Natale, C.; Manna, S.L.; Netti, P.A.; Vecchione, R. Recent advances in the formulation of PLGA microparticles for controlled drug delivery. Prog. Biomater. 2020, 9, 153–174. [Google Scholar] [PubMed]
- Al-Ahmady, Z.S.; Scudamore, C.L.; Kostarelos, K. Triggered doxorubicin release in solid tumors from thermosensitive liposome-peptide hybrids: Critical parameters and therapeutic efficacy. Int. J. Cancer 2015, 137, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Anyarambhatla, G.; Petros, W.P.; Braun, R.D.; Colvin, O.M.; Needham, D.; Dewhirst, M.W. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Res. 2000, 60, 6950–6957. [Google Scholar]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef]
- Solorio, L.; Patel, R.B.; Wu, H.; Krupka, T.; Exner, A.A. Advances in image-guided intratumoral drug delivery techniques. Ther. Deliv. 2010, 1, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Lang, X.; Qian, L.; Guan, P.; Zi, J.; Chen, M. Localized surface plasmon resonance of nanoporous gold. Appl. Phys. Lett. 2011, 98, 093701. [Google Scholar] [CrossRef]
- Song, J.; Yang, X.; Yang, Z.; Lin, L.; Liu, Y.; Zhou, Z.; Shen, Z.; Yu, G.; Dai, Y.; Jacobson, O. Rational design of branched nanoporous gold nanoshells with enhanced physico-optical properties for optical imaging and cancer therapy. ACS Nano 2017, 11, 6102–6113. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Release Medium | Drug | Implant | R2 | n | k |
|---|---|---|---|---|---|---|
| Korsmeyer-Peppas | pH 5.5 | DOX | np-Au | 0.942 | 0.47 | 1.66 |
| DOX | np-Au@PLGA/RAPA | 0.983 | 1.08 | 0.10 | ||
| RAPA | np-Au@PLGA/RAPA | 0.984 | 1.19 | 0.22 | ||
| pH 6.6 | DOX | np-Au | 0.997 | 0.98 | 0.28 | |
| DOX | np-Au@PLGA/RAPA | 0.988 | 1.13 | 0.07 | ||
| RAPA | np-Au@PLGA/RAPA | 0.991 | 0.69 | 2.28 | ||
| pH 7.4 | DOX | np-Au | 0.948 | 1.14 | 0.19 | |
| DOX | np-Au@PLGA/RAPA | 0.993 | 0.84 | 0.27 | ||
| RAPA | np-Au@PLGA/RAPA | 0.994 | 0.32 | 11.09 | ||
| Zero order | pH 7.4 (NBCS) | DOX | np-Au | 0.991 | NA | 0.04 |
| DOX | np-Au@PLGA/RAPA | 0.997 | NA | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattarai, J.K.; Neupane, D.; Nepal, B.; Demchenko, A.V.; Stine, K.J. Nanoporous Gold Monolith for High Loading of Unmodified Doxorubicin and Sustained Co-Release of Doxorubicin-Rapamycin. Nanomaterials 2021, 11, 208. https://doi.org/10.3390/nano11010208
Bhattarai JK, Neupane D, Nepal B, Demchenko AV, Stine KJ. Nanoporous Gold Monolith for High Loading of Unmodified Doxorubicin and Sustained Co-Release of Doxorubicin-Rapamycin. Nanomaterials. 2021; 11(1):208. https://doi.org/10.3390/nano11010208
Chicago/Turabian StyleBhattarai, Jay K., Dharmendra Neupane, Bishal Nepal, Alexei V. Demchenko, and Keith J. Stine. 2021. "Nanoporous Gold Monolith for High Loading of Unmodified Doxorubicin and Sustained Co-Release of Doxorubicin-Rapamycin" Nanomaterials 11, no. 1: 208. https://doi.org/10.3390/nano11010208