Trans-Cinnamaldehyde Alleviates Amyloid-Beta Pathogenesis via the SIRT1-PGC1α-PPARγ Pathway in 5XFAD Transgenic Mice

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
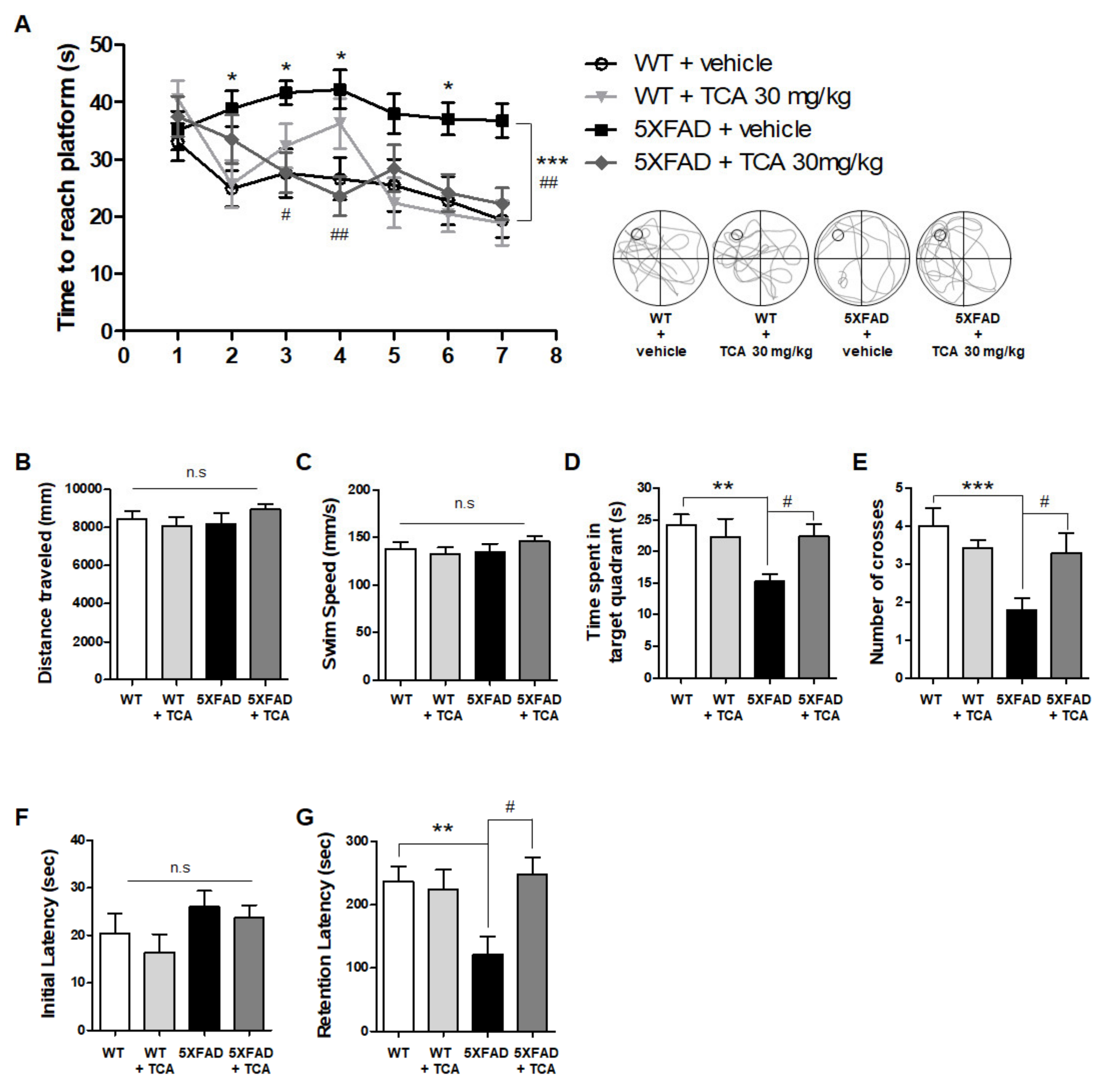
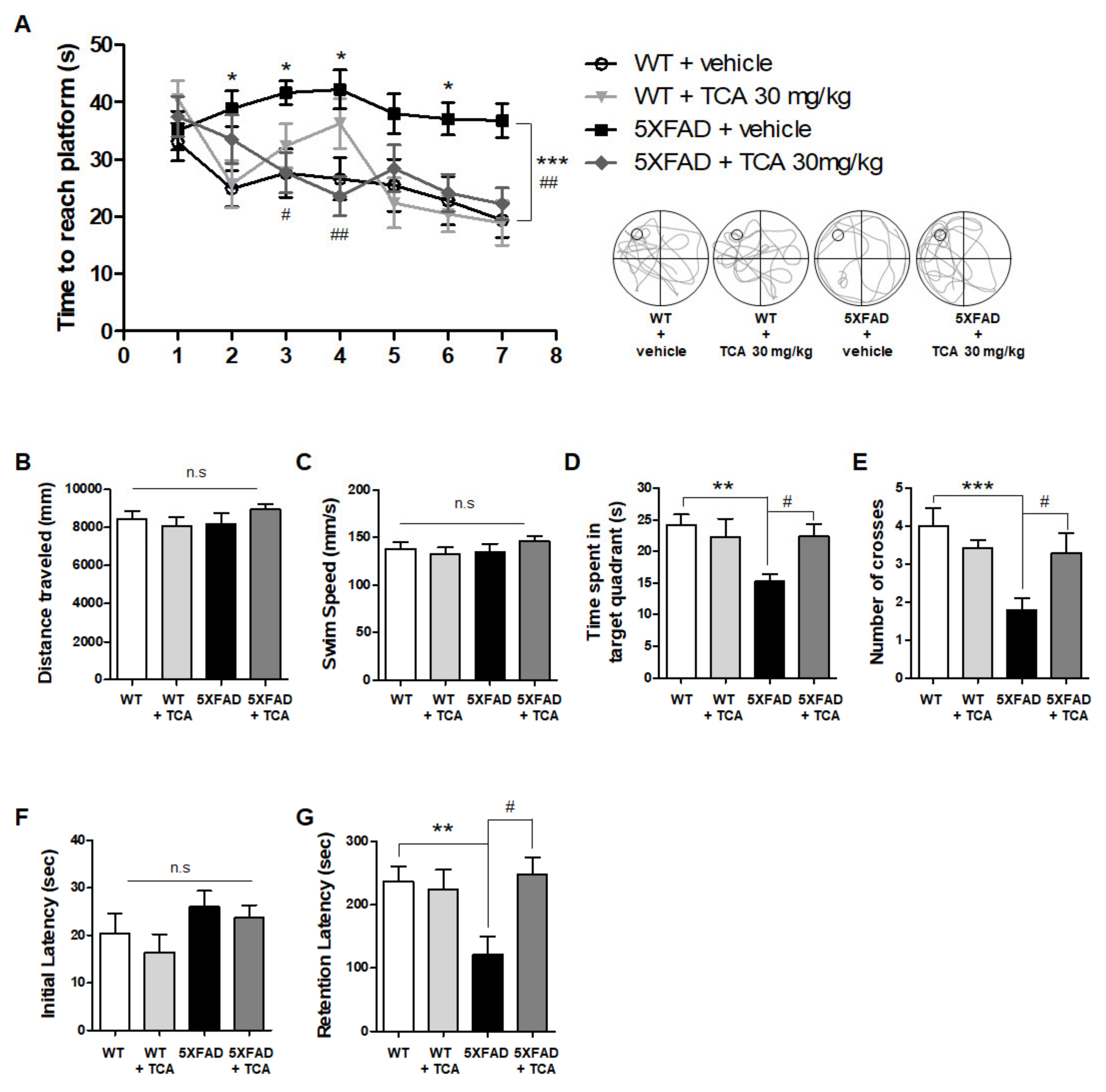
2.1. TCA Improves Cognitive Performance in 5XFAD Mouse Model of AD
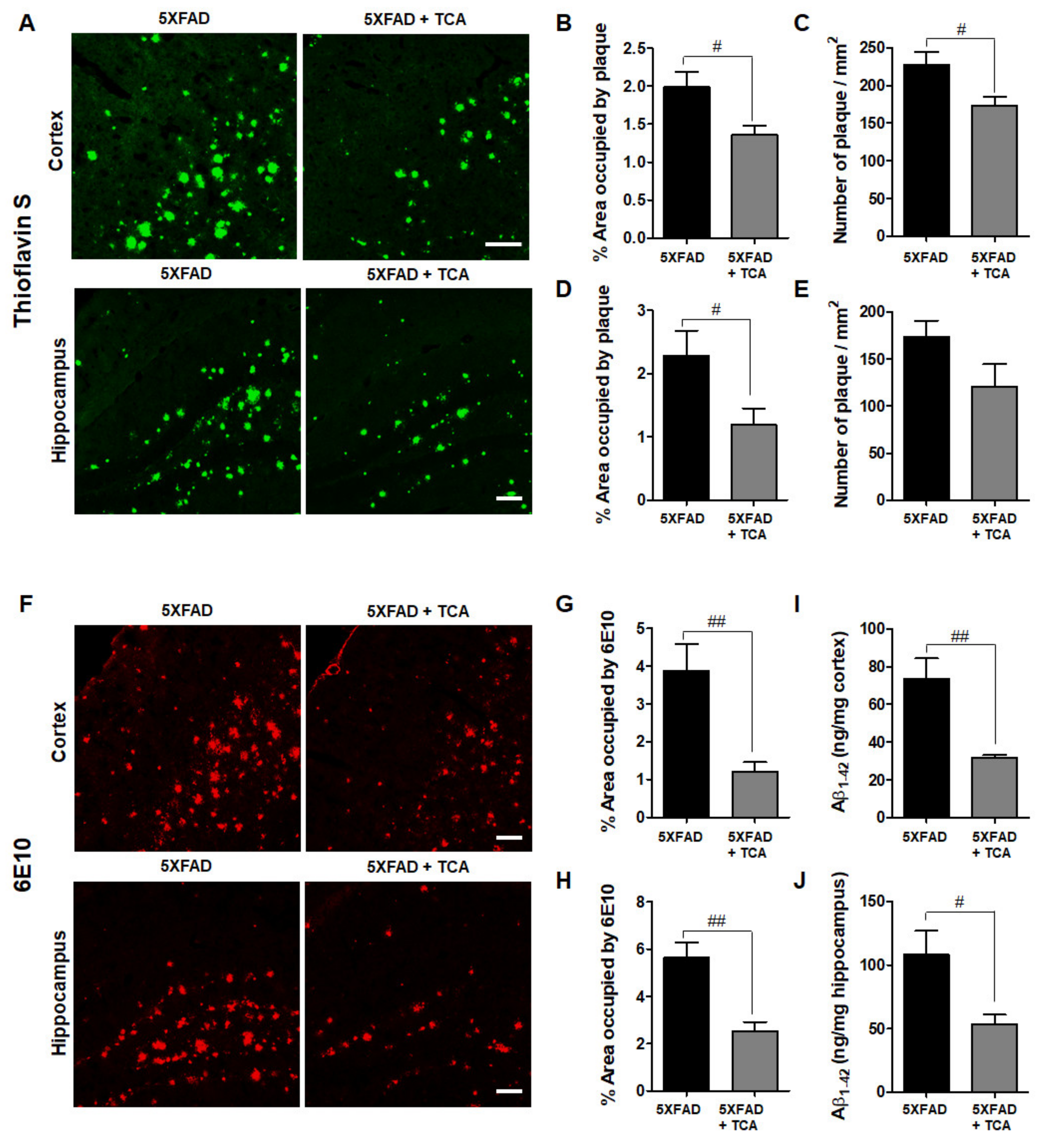
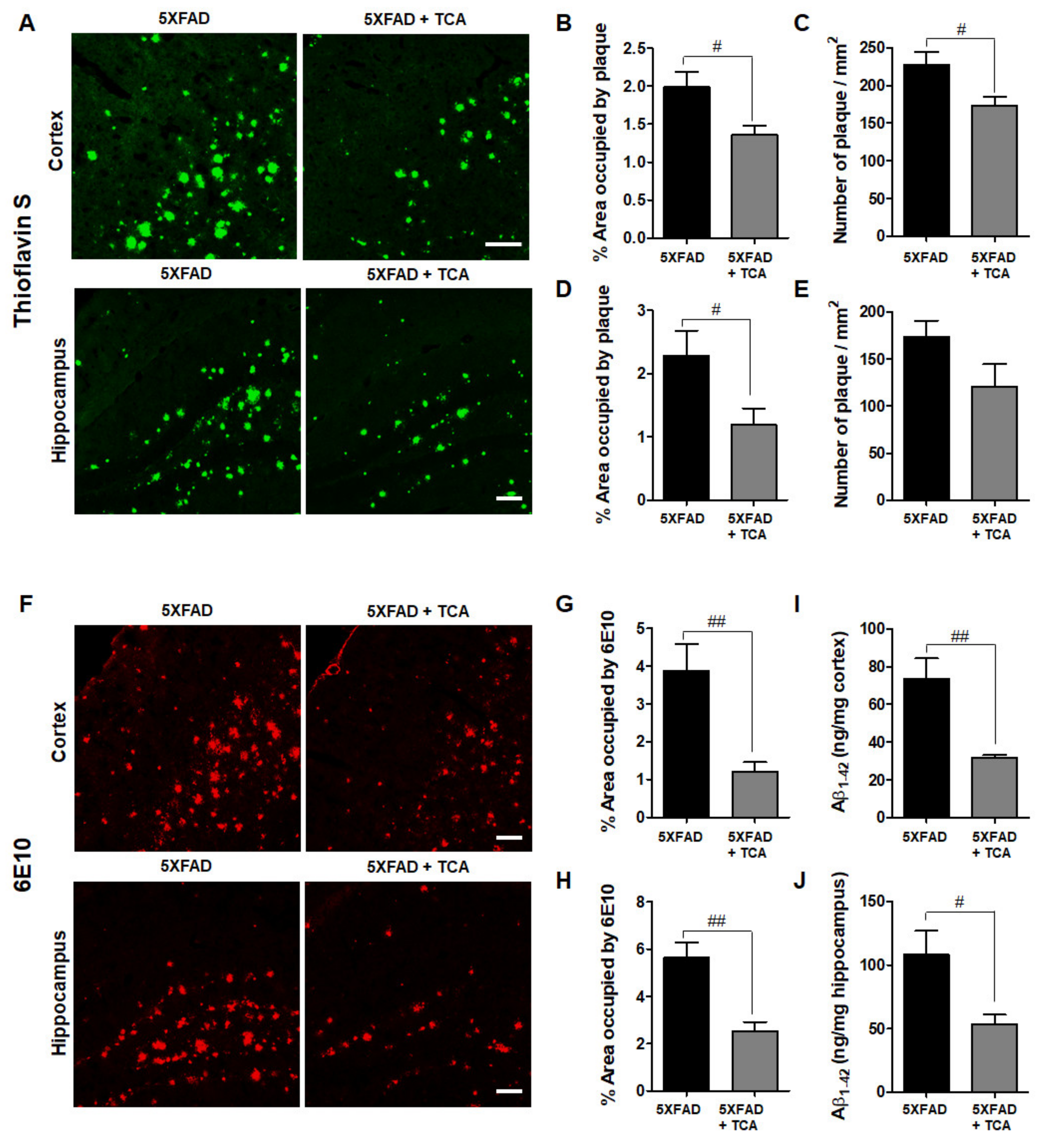
2.2. TCA Reduces Aβ Deposition in the Brains of 5XFAD Mice
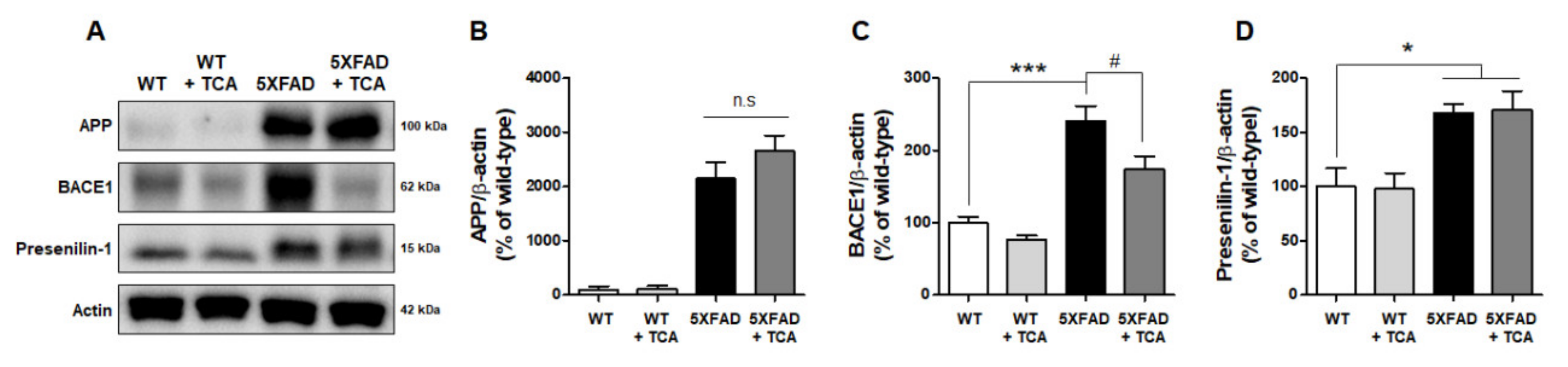
2.3. TCA Decreases BACE1 Levels in the Brains of 5XFAD Mice
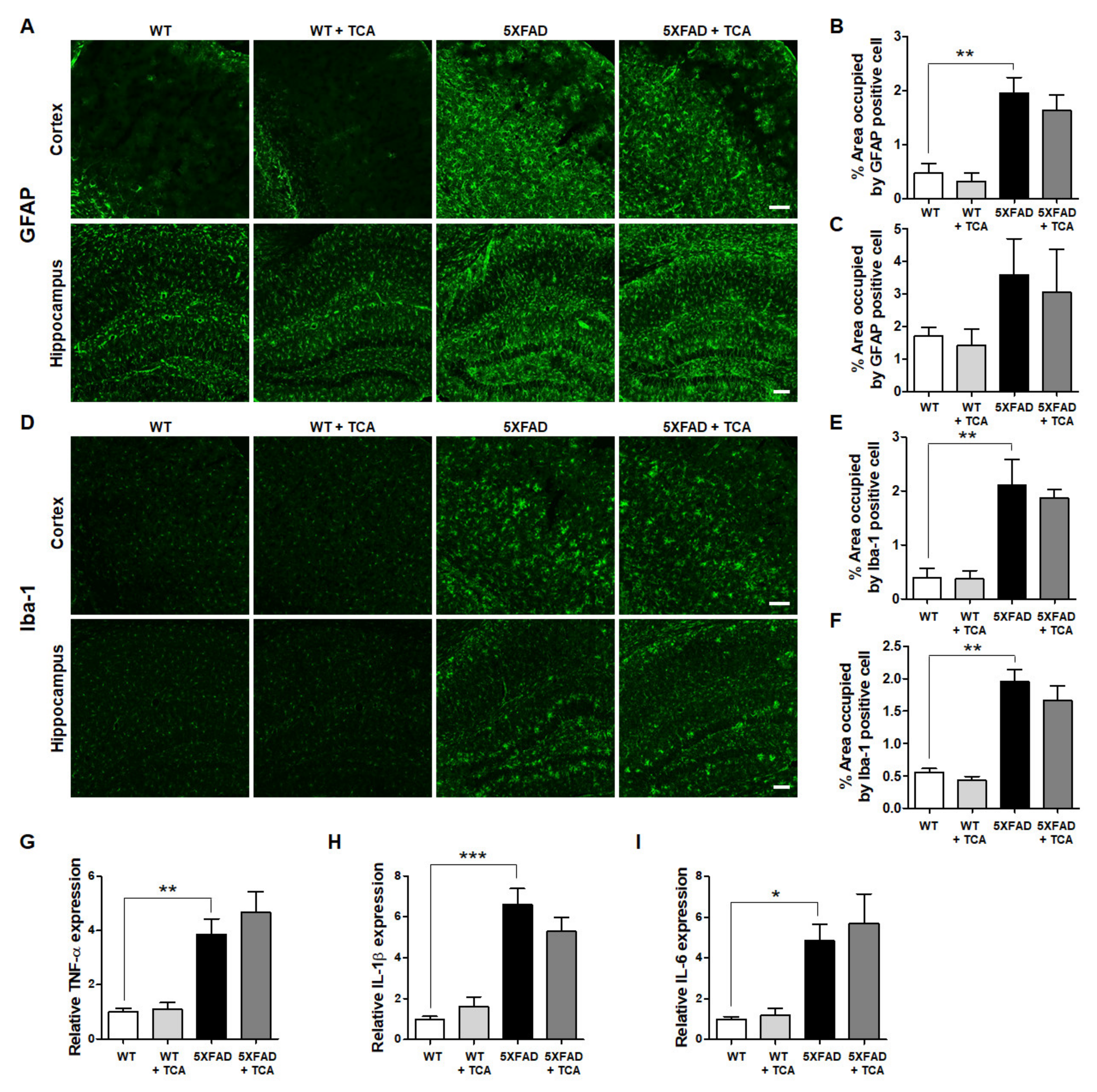
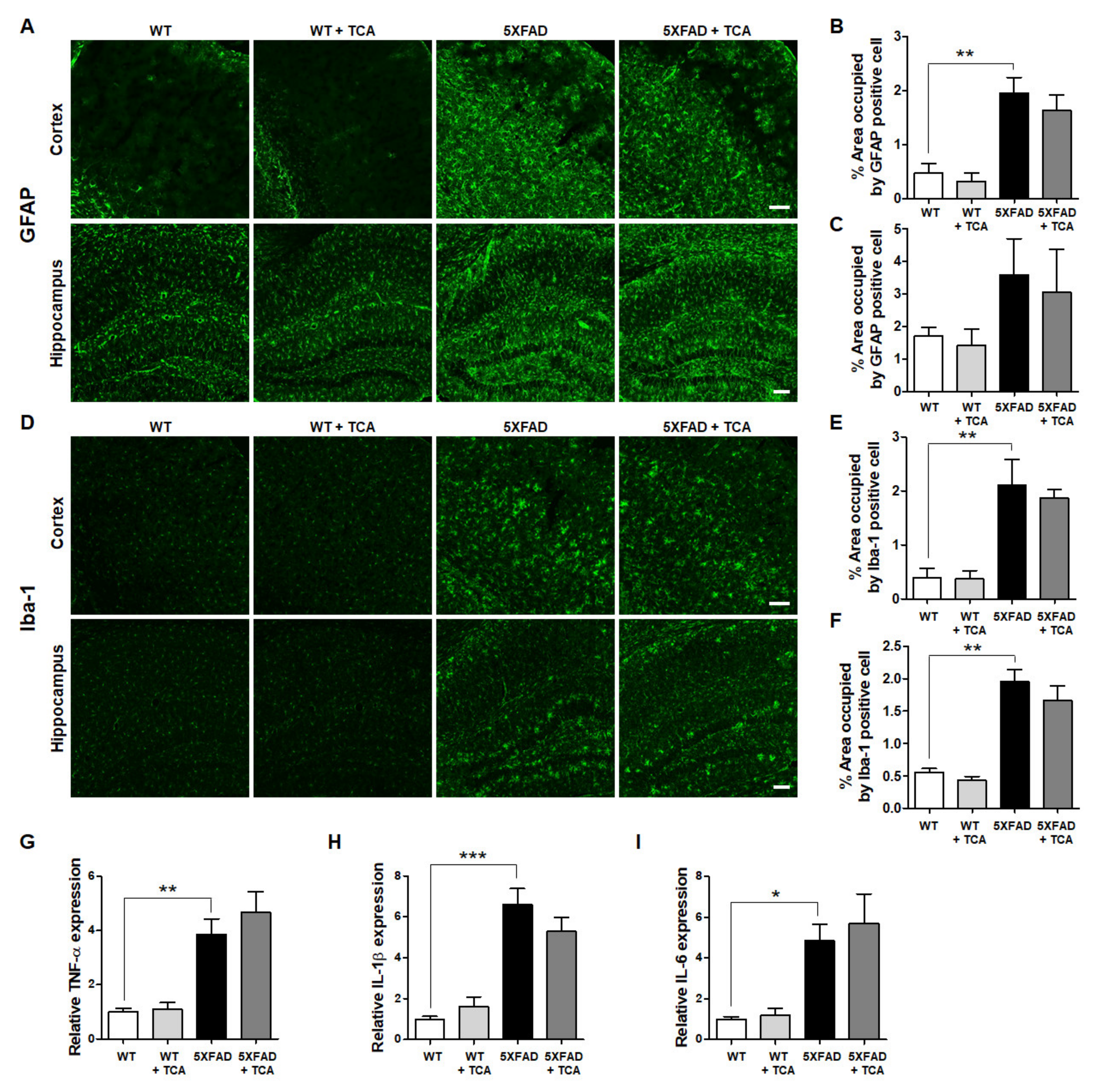
2.4. TCA Does Not Affect the Levels of Inflammatory Factors in the Brains of 5XFAD Mice
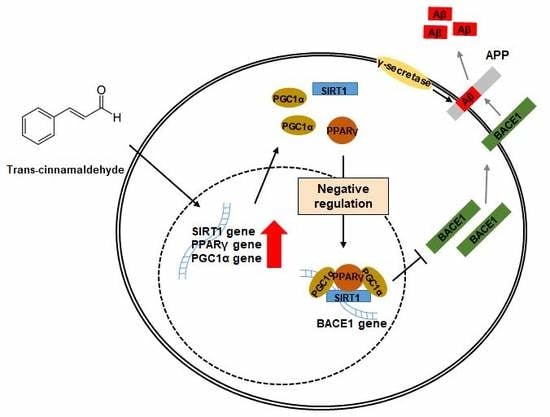
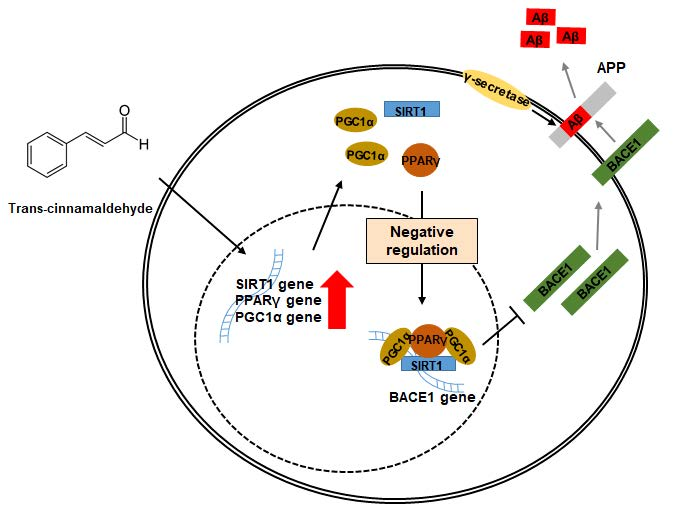
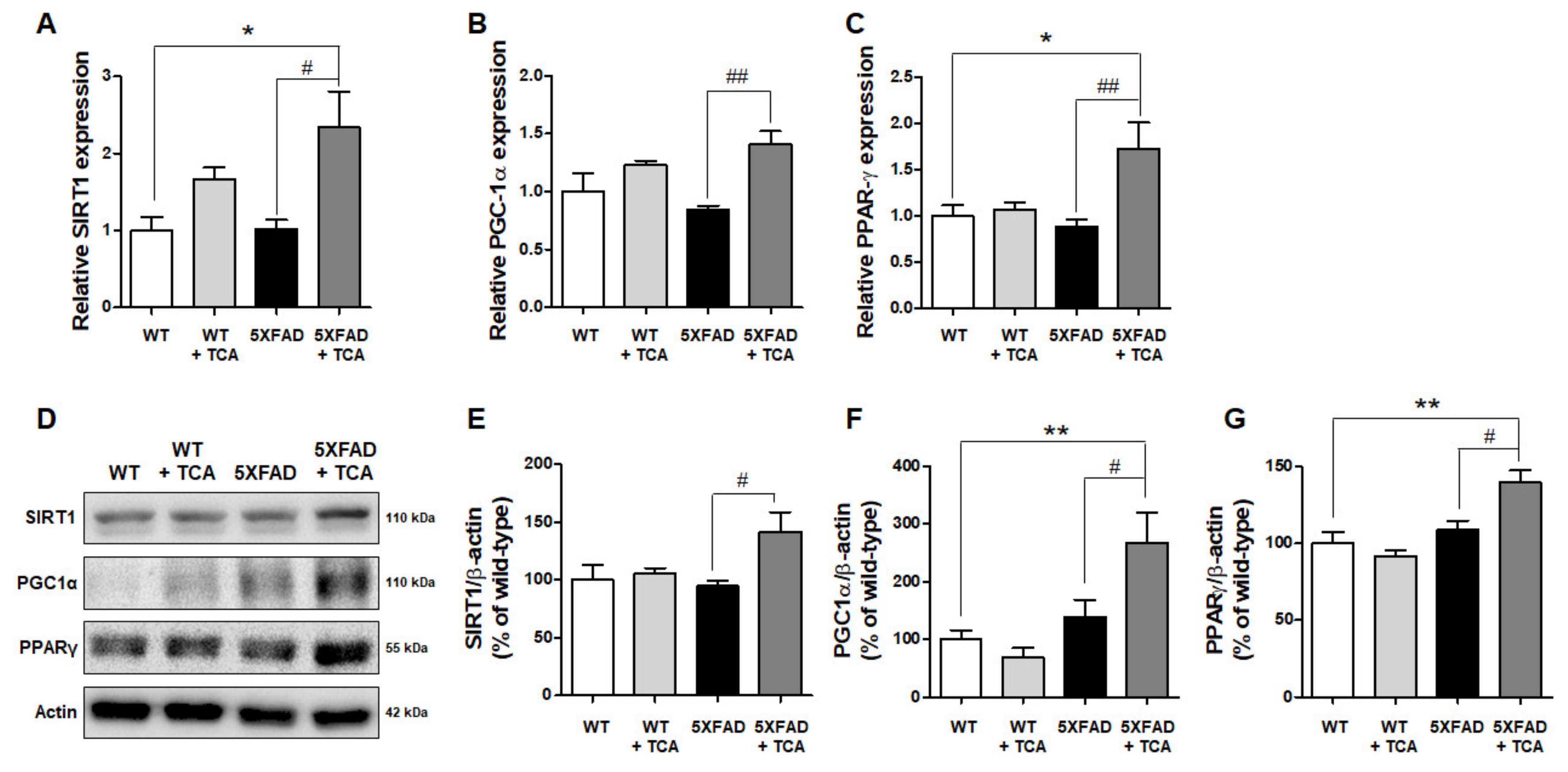
2.5. TCA Activates the SIRT1-PGC1α-PPARγ Pathway That Regulates the Expression of BACE1
3. Discussion
4. Materials and Methods
4.1. Materials
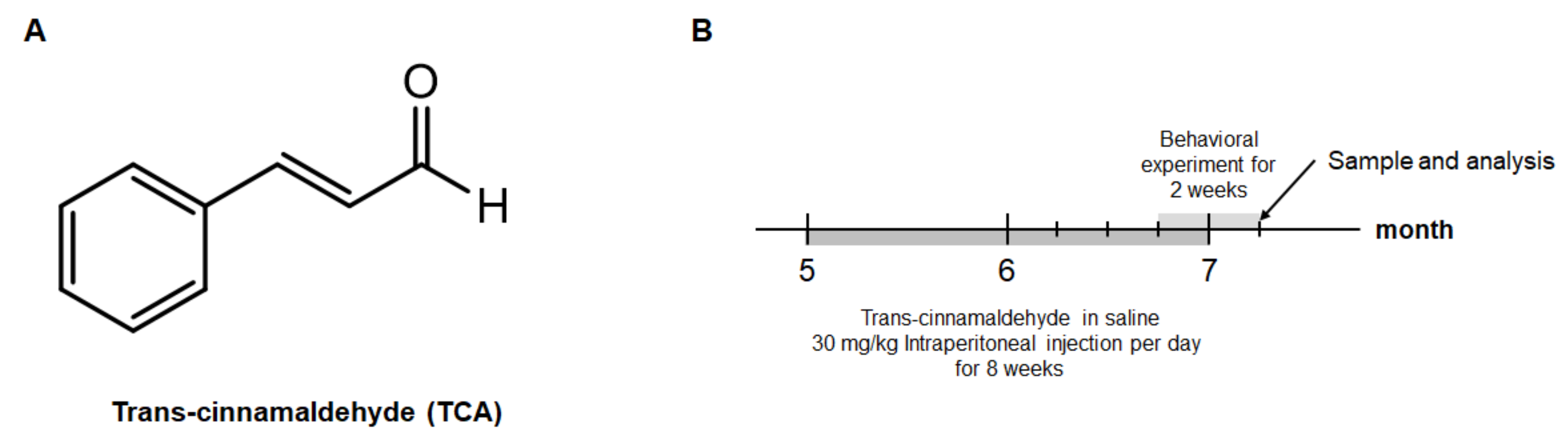
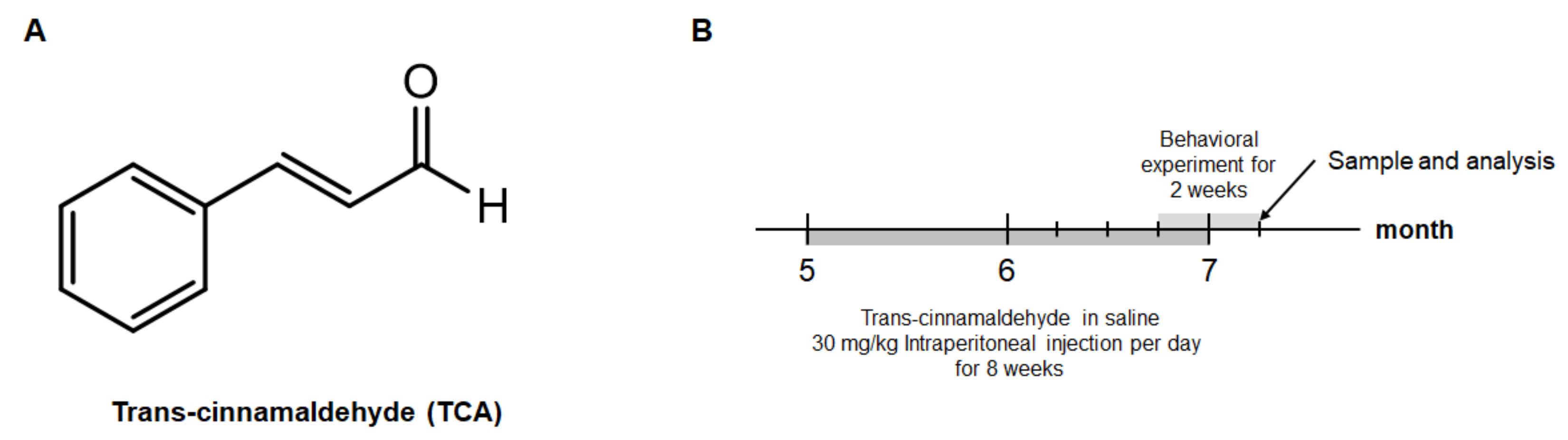
4.2. Animals and Treatment
4.3. Behavioral Studies
4.4. Brain Tissue Preparation
4.5. Thioflavin S Staining
4.6. Immunofluorescence
4.7. Aβ 1–42 ELISA
4.8. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| BACE1 | β-secretase |
| APP | amyloid precursor protein |
| TCA | trans-cinnamaldehyde |
| SIRT1 | silent information regulator 1 |
| PGC1α | peroxisome proliferator-activated receptor γ coactivator 1α |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PS1 | presenilin-1 |
| TNF-α | tumor necrosis factor-α |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| Iba-1 | ionized calcium-binding adapter molecule 1 |
| GFAP | glial fibrillary acidic protein |
| RXR | retinoid X receptor |
| PPRE | peroxisome proliferator response elements |
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci 2017, 24, 47. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Roussel, A.M.; Hininger, I.; Benaraba, R.; Ziegenfuss, T.N.; Anderson, R.A. Antioxidant effects of a cinnamon extract in people with impaired fasting glucose that are overweight or obese. J. Am. Coll. Nutr. 2009, 28, 16–21. [Google Scholar] [CrossRef]
- Kim, S.H.; Hyun, S.H.; Choung, S.Y. Anti-diabetic effect of cinnamon extract on blood glucose in db/db mice. J. Ethnopharmacol. 2006, 104, 119–123. [Google Scholar] [CrossRef]
- Schink, A.; Naumoska, K.; Kitanovski, Z.; Kampf, C.J.; Frohlich-Nowoisky, J.; Thines, E.; Poschl, U.; Schuppan, D.; Lucas, K. Anti-inflammatory effects of cinnamon extract and identification of active compounds influencing the TLR2 and TLR4 signaling pathways. Food Funct. 2018, 9, 5950–5964. [Google Scholar] [CrossRef] [Green Version]
- Frydman-Marom, A.; Levin, A.; Farfara, D.; Benromano, T.; Scherzer-Attali, R.; Peled, S.; Vassar, R.; Segal, D.; Gazit, E.; Frenkel, D.; et al. Orally administrated cinnamon extract reduces beta-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS ONE 2011, 6, e16564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.V.; Gan, S.H. Cinnamon: A multifaceted medicinal plant. Evid. Based Complement. Alternat. Med. 2014, 2014, 642942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, W.Y.; Choi, J.S.; Jeong, J.W. The Neuroprotective Effects of Cinnamic Aldehyde in an MPTP Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.H.; Jeong, Y.K.; Yeo, S.; Lee, J.H.; Jeong, M.Y.; Kim, S.H.; Choi, Y.G.; Lim, S. Neuroprotective effect of trans-cinnamaldehyde on the 6-hydroxydopamine-induced dopaminergic injury. Biol. Pharm. Bull. 2013, 36, 1928–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Bueler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s beta-secretase gene transcription via SIRT1-PPARgamma-PGC-1 in neurons. Cell. Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [Green Version]
- Blasko, I.; Beer, R.; Bigl, M.; Apelt, J.; Franz, G.; Rudzki, D.; Ransmayr, G.; Kampfl, A.; Schliebs, R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J. Neural. Transm. (Vienna) 2004, 111, 523–536. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [Green Version]
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 2004, 41, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Chami, L.; Checler, F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Lange-Dohna, C.; Zeitschel, U.; Gaunitz, F.; Perez-Polo, J.R.; Bigl, V.; Rossner, S. Cloning and expression of the rat BACE1 promoter. J. Neurosci. Res. 2003, 73, 73–80. [Google Scholar] [CrossRef]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, X.; Zhang, Y.W.; Rockenstein, E.; Bu, G.; Golde, T.E.; Masliah, E.; Xu, H. Alzheimer’s beta-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of beta-amyloid. J. Neurosci. 2012, 32, 11390–11395. [Google Scholar] [CrossRef] [Green Version]
- Bourne, K.Z.; Ferrari, D.C.; Lange-Dohna, C.; Rossner, S.; Wood, T.G.; Perez-Polo, J.R. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J. Neurosci. Res. 2007, 85, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.E.; Futawaka, K.; Yamamoto, H.; Kasahara, M.; Tagami, T.; Liu, T.H.; Moriyama, K. Cinnamaldehyde Contributes to Insulin Sensitivity by Activating PPARdelta, PPARgamma, and RXR. Am. J. Chin. Med. 2015, 43, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, A.A.; Ghaznavi, S.A.; Sande Mathias, I.; Ellard, K.K.; Janos, J.A.; Sylvia, L.G. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 761–769. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Katsouri, L.; Parr, C.; Bogdanovic, N.; Willem, M.; Sastre, M. PPARgamma co-activator-1alpha (PGC-1alpha) reduces amyloid-beta generation through a PPARgamma-dependent mechanism. J. Alzheimers Dis. 2011, 25, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Schuchman, E.H.; Jin, H.K.; Bae, J.S. Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid beta ameliorates Alzheimer’s disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells 2012, 30, 1544–1555. [Google Scholar] [CrossRef]
- Kim, N.; Martinez, C.C.; Jang, D.S.; Lee, J.K.; Oh, M.S. Anti-neuroinflammatory effect of Iresine celosia on lipopolysaccharide-stimulated microglial cells and mouse. Biomed. Pharmacother. 2019, 111, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Do, J.; Bae, J.S.; Jin, H.K.; Kim, J.H.; Inn, K.S.; Oh, M.S.; Lee, J.K. Piperlongumine inhibits neuroinflammation via regulating NF-kappaB signaling pathways in lipopolysaccharide-stimulated BV2 microglia cells. J. Pharmacol. Sci. 2018, 137, 195–201. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, J.; Kim, N.; Jeon, S.H.; Gee, M.S.; Ju, Y.-J.; Kim, J.-H.; Oh, M.S.; Lee, J.K. Trans-Cinnamaldehyde Alleviates Amyloid-Beta Pathogenesis via the SIRT1-PGC1α-PPARγ Pathway in 5XFAD Transgenic Mice. Int. J. Mol. Sci. 2020, 21, 4492. https://doi.org/10.3390/ijms21124492
Do J, Kim N, Jeon SH, Gee MS, Ju Y-J, Kim J-H, Oh MS, Lee JK. Trans-Cinnamaldehyde Alleviates Amyloid-Beta Pathogenesis via the SIRT1-PGC1α-PPARγ Pathway in 5XFAD Transgenic Mice. International Journal of Molecular Sciences. 2020; 21(12):4492. https://doi.org/10.3390/ijms21124492
Chicago/Turabian StyleDo, Jimin, Namkwon Kim, Seung Ho Jeon, Min Sung Gee, Yeon-Joo Ju, Jong-Ho Kim, Myung Sook Oh, and Jong Kil Lee. 2020. "Trans-Cinnamaldehyde Alleviates Amyloid-Beta Pathogenesis via the SIRT1-PGC1α-PPARγ Pathway in 5XFAD Transgenic Mice" International Journal of Molecular Sciences 21, no. 12: 4492. https://doi.org/10.3390/ijms21124492