1. Introduction
Over the last thirty years, research programs focusing on the chemical study of the metagenomic content of marine sponges have led to a plethora of novel molecules, often showing innovative skeletons, and being used as lead compounds in the search for novel therapeutic approaches [
1,
2,
3,
4].
In the framework of our research program, named BlueGenics (
https://cordis.europa.eu/result/rcn/193365_en.html), devoting to the sustainable exploitation of bioactive marine compounds, the chemical composition of the dichloromethane extract of the marine sponge
Acanthostrongylophora ingens, collected off the coast of South Sulawesi, Indonesia, was analyzed, and chloromethylhalicyclamine B (
5), a novel selective CK1δ/ε kinase inhibitor with an IC
50 value of 6 µM, was described [
5]. An in-depth re-examination of the organic extracts of
A. ingens revealed, in addition to halicyclamine B (
4) and chloromethylhalicyclamine B (
5), small amounts of tetradehydrohalicyclamine B (
1b) and its epimer, a new dehydrohalicyclamine derivative named
epi-tetradehydrohalicyclamine B (
1a) as well as its chloromethyl derivative (
2), acanthocyclamine A (
3), and seven diketopiperazines (DKPs) (
6–
12). Tetradehydrohalicyclamine B (
1b) was recently reported from the same Indonesian
A. ingens by a Japanese group [
6]. They investigated the proteasome inhibitor activity of tetradehydrohalicyclamine B (
1b) and halicyclamine B (
4), showing that both have proteasome inhibitor activity at micromolar concentration,
4 being more potent than
1b, suggesting their possible role as anticancer lead compounds [
7].
Our screening program includes antibacterial, antikinases, and amyloid β-42 assays. The obtained data are reported here, together with structural elucidation of the new alkaloid epi-tetradehydrohalicyclamine B (1a) found in the sponge extract.
3. Discussion and Conclusions
Alkaloids are pharmacologically well characterized and are used in therapy, ranging from chemotherapeutics to analgesics.
This chemical study of the marine sponge
A. ingens allowed the isolation of 13 alkaloids, of which, one,
1a is the epimer of the tetradehydrohalicyclamine B (
1b), just recently published. Compounds
1–
12 were analyzed for their biological activity using our standard panel assays that include antibacterial, antikinases, and amyloid β-42 assays. Acanthocyclamine A (
3) showed a selective antimicrobial activity against
E. coli and an inhibition of the amyloid β-42 production induced by aftin-5 at 26 µM, without cytotoxicity at this dose. These results highlight the potentiality of a bipiperidine scaffold as a promising skeleton to develop products able to prevent or reduce the production of amyloid β-42, a key player in the initiation of Alzheimer’s disease. A previous study revealed that chloromethylhalicyclamine B (
5) appeared to be a selective CK1δ/ε inhibitor at low micromolar concentrations, while halicyclamine B (
4) was inactive. Docking studies showed that chloromethylhalicyclamine B (
5) can efficiently interact with the ATP-binding site of CK1δ in spite of its globular structure, very different from the planar structure of known inhibitors of CK1δ [
5]. Because no CK1δ inhibitory activity was observed for the chloromethyltetradehydrohalicyclamine B (
2), the presence of a tetrahydropyridine appears to be essential for the inhibitory activity towards CK1δ.
Moreover, the diketopiperazine cyclo (d-Pro-l-Phe) (6) revealed a selective antikinase activity against CDK2/cyclin A with an IC50 value of 1 µM. In comparison with the inactive diketopiperazine cyclo (L-Pro-L-Tyr) (12), we can hypothesize that hydroxylation of the phenyl group leads to a loss of CDK2/cyclin A kinase inhibitory activity.
The marine sponge A. ingens is showed to be a rich source of a number of bioactive alkaloids, some of them having an unusual skeleton, and sometimes being halogenated.
This study completes the previous works on A. ingens and points out the growing interest in studying this sponge family with unique and diverse chemical structures. These data highlight the potentiality of these molecules as lead compounds.
4. Materials and Methods
4.1. General Experimental Procedures
Mass spectra were recorded on an API Q-STAR PULSAR I (Applied Biosystem, Concord, ON, Canada). NMR spectra were obtained on either a Bruker Avance 400 or 600 spectrometer (Bruker, Wissenbourg, France) using standard pulse sequences. The acquisition of HMBC spectra were optimized for either 7 or 8.3 Hz. Other NMR spectra were recorded on Varian Unity Inova spectrometers at 700 MHz (Agilent Technology, Cernusco sul Naviglio, Italy); chemical shifts were referenced to the residual solvent signal (CD3OD: δH 3.31, δC 49.00). Flash chromatography was carried out on Buchi C-615 pump system (Rungis, France). Analytical and semi-preparative reversed-phase (Gemini C6-phenyl, Phenomenex, Le Pecq, France) columns were performed with an Alliance HPLC apparatus (model 2695, Waters, Saint-Quentin-en-Yvelines, France), equipped with a photodiode array detector (model 2998, Waters), an evaporative light-scattering detector (model Sedex 80, Sedere, Alfortville, France), and the software Empower (Waters). Chromatography columns (CC) were performed using silica gel (200~400 mesh; Merck, Darmstadt, Germany) and Sephadex LH-20 (Amersham Pharmacia, Uppsala, Sweden).
The Marfey’s experiments were performed using a Thermo LTQ Orbitrap XL mass spectrometer coupled to a Thermo Ultimate 3000 RS system (Thermo Fisher Scientific Spa, Rodano, Italy), which included solvent reservoir, in-line degasser, ternary pump, column thermostat, and refrigerated autosampler. LC–MS data were recorded and analyzed using the software Thermo Xcalibur 2.07 (Thermo Fisher Scientific Spa). The samples (5 μL) were applied on to an analytical reversed-phase column (Phenomenex Kinetex C18, 100 × 2.1 mm, particle size 5 μm), which was eluted at 200 μL/min. The elution procedure consisted of an isocratic profile of acetonitrile–water (5:95, v/v) for 3 min, followed by a linear gradient from 5% to 60% ACN/H2O over 20 min, a linear gradient from 60% to 90% ACN/H2O over 1 min, and an isocratic profile over 5 min.
4.2. Sponge Material
Specimens of Acanthostrongylophora ingens (Thiele, 1900) (class Demospongiae, order Haplosclerida, family Petrosiidae) were collected off the South Sulawesi, Indonesia and Makassar (Ujong Pandang), Spermonde Archipelago, north-west Lankai Island, reef slope at 14 m depth, 5.019° S 119.063° E, 29 April 1998, coll. B.W. Hoeksema, and were identified by one of the authors (R.V.S.) at the University of Amsterdam, The Netherlands, where a voucher specimen was deposited under the registration code ZMA Por. 14471.
4.3. Isolation and Purification
Sponge specimens (500 g) were immediately immersed in MeOH after collection. The MeOH solution was evaporated and the aqueous residue was extracted and partitioned successively with CH2Cl2 (1L), EtOAc (1L), and BuOH (1L) to obtain the corresponding extracts: Extract A (CH2Cl2, 2.5 g), extract B (EtOAc, 1.6 g), and extract C (BuOH, 8.5 g).
The CH2Cl2 extract was subsequently flash chromatographed on silica gel using a gradient elution system from 100% CH2Cl2 to 100% MeOH, obtaining six fractions (A1–A6).
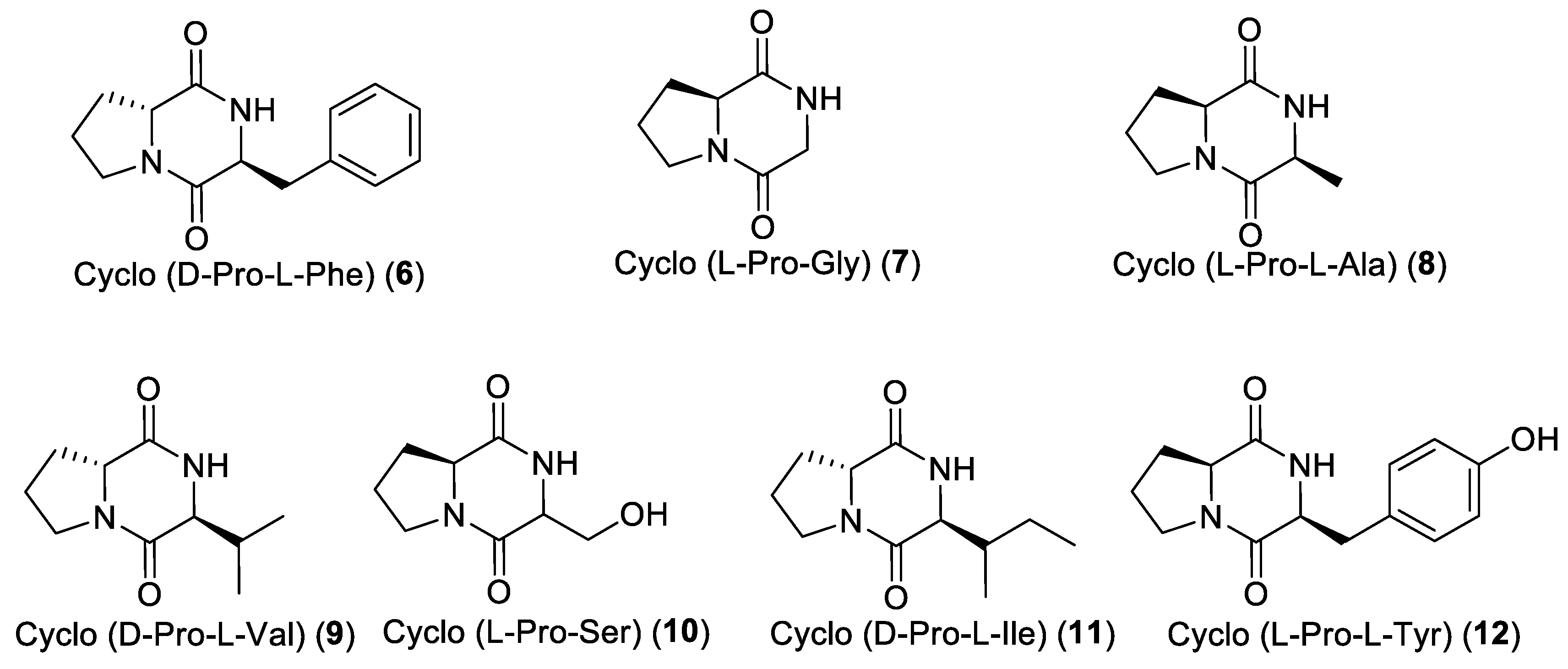
Fraction A3 (313 mg) was further fractionated into 14 sub-fractions on silica gel column chromatography eluted with the CH2Cl2–acetone gradient system. Sub-fraction A3-7 (22 mg) was subjected to C6-phenyl semi-preparative reversed-phase HPLC using, as eluent, the gradient ACN/H2O/HCOOH from 5/95/0.1 to 50/50/0.1 for 35 min (flow rate: 3 mL/min, wavelength: 201 nm) and yielded the diketopiperazine cyclo-(D-Pro-L-Ile) (11, 4 mg). From the sub-fraction A3-8 (17 mg), the three diketopiperazines cyclo-(D-Pro-L-Val) (9, 5 mg), cyclo-(L-Pro-L-Tyr) (12, 3 mg) and cyclo(D-Pro-L-Phe) (6, 4 mg) were purified after C6-phenyl semi-preparative reversed-phase HPLC using the gradient ACN/H2O/HCOOH from 5/95/0.1 to 28/72/0.1 for 27 min (flow rate: 3 mL/min, wavelength: 201 nm).
Fraction A5 (250 mg) was applied on a Sephadex LH-20 column using MeOH as eluent and gave eleven sub-fractions. Sub-fraction A5-3 (8 mg) was subjected to C6-phenyl semi-preparative reversed-phase HPLC using the gradient ACN/H2O/HCOOH from 5/95/0.1 to 40/60/0.1 for 11 min (flow rate: 3 mL/min, wavelength: 201 nm) to yield 3 mg of a mixture of 1a and 1b with a ratio of 65:35 by 1H NMR integration.
Fraction A6 (150 mg) was applied on a Sephadex LH-20 column and eluting with MeOH to give nine sub-fractions. Sub-fraction A6-2 was purified by C6-phenyl analytical reversed-phase HPLC using the gradient ACN/H2O/HCOOH from 5/95/0.1 to 13/87/0.1 for 25 min (flow rate: 1 mL/min, wavelength: 201 nm) to yield chloromethyltetradehydrohalicyclamine B (2, 1.2 mg). Using a gradient system ACN/H2O/HCOOH (5/95/0.1 to 20/80/0.1 for 25 min, flow rate 1 mL/min, wavelength 254 nm), sub-fraction A6-9 was purified by C6-phenyl analytical reversed-phase HPLC yielding chloromethylhalicyclamine B (5, 2.5 mg) and halicyclamine B (4, 1.8 mg).
An aliquot of the BuOH extract (2 g) was subsequently flash chromatographed on a silica gel column using the elution gradient system from 100% CH2Cl2 to 100% MeOH, to yield eighteen fractions (C1-C18). Fraction C6 was subjected to C6-phenyl semi-preparative reversed-phase HPLC using ACN/H2O/HCOOH from 5/95/0.1 to 35/65/0.1 as elution gradient for 25 min (flow rate: 3 mL/min, wavelength: 201 nm) and yielded cyclo-(L-Pro-Gly) (7, 2 mg), cyclo-(L-Pro-L-Ala) (8, 2.5 mg), and cyclo-(L-Pro-Ser) (10, 2.7 mg).
Fraction C13 was applied on a Sephadex LH-20 column using MeOH as eluent and yielded three sub-fractions. Sub-fraction C13-2 was subjected to C6-phenyl semi-preparative reversed-phase HPLC using the gradient ACN/H2O/HCOOH from 5/95/0.1 to 25/75/0.1 for 28 min (flow rate: 3 mL/min, wavelength: 201 nm) to yield acanthocyclamine A (3, 5 mg).
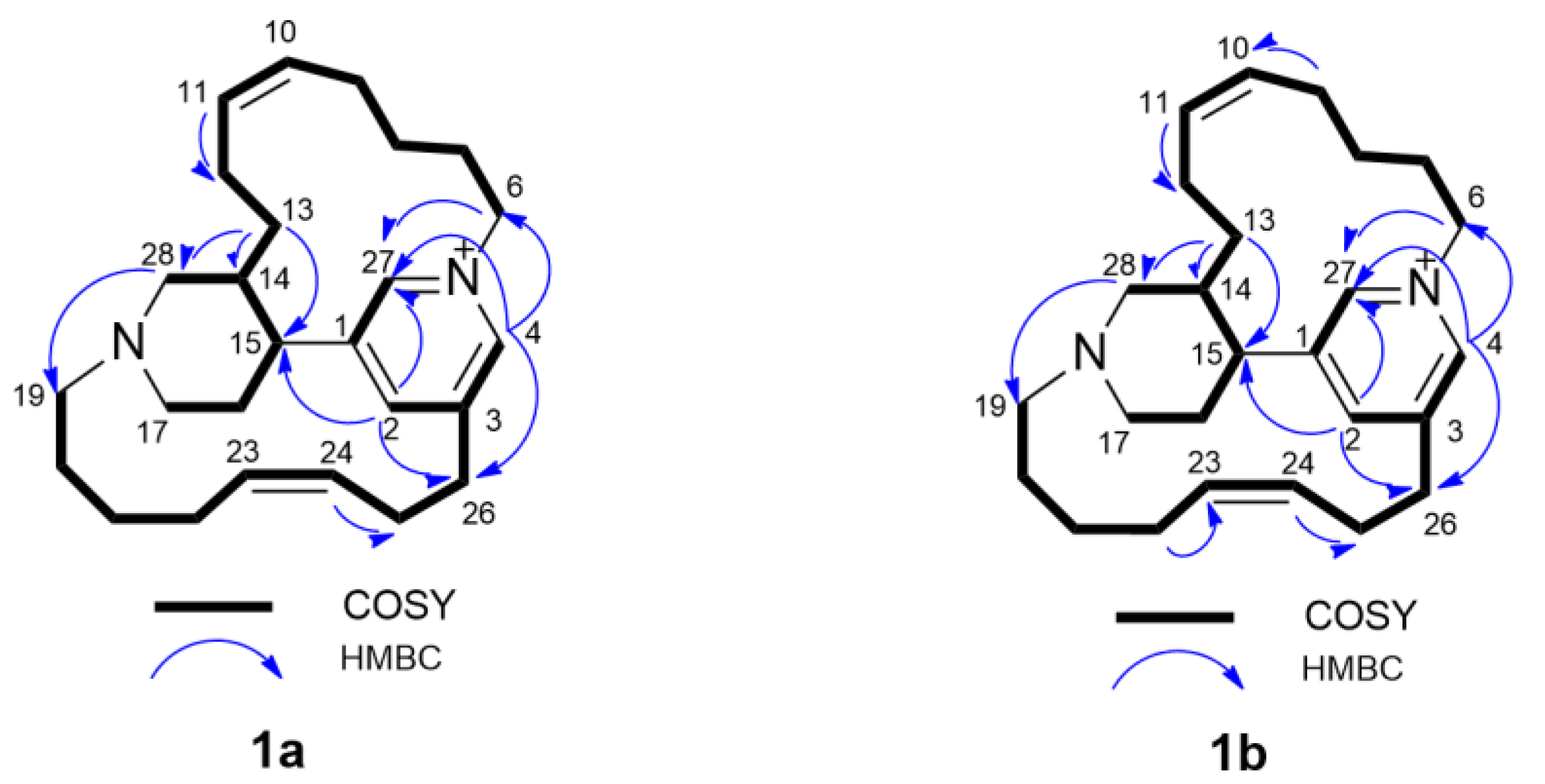
Epi-tetradehydrohalicyclamine B (
1a):
1H and
13C NMR data, see
Table 1, (+)- HR-ESIMS
m/z 379.3132 [M]
+ (calcd for C
26H
39N
2+, 379.3107).
4.4. Marfey’s Analysis
Marfey’s analysis was used to determine the configuration of the amino acids of DKPs cyclo(Pro-Phe), cyclo(Pro-Gly), cyclo(Pro-Ala), cyclo(Pro-Val), cyclo(Pro-Ser), cyclo(Pro-Ile), and cyclo(Pro-Tyr). The compounds were subjected to hydrolysis and derivatization with the L-enantiomer of Marfey’s reagent (FDAA, or 1-fluoro-2,4-dinitrophenyl-5-alanine amide) [
12]. Only 10 μg of compounds were degraded, and the obtained derivatives were analyzed by HR-ESIMS-HPLC.
The samples were treated with 6 N HCl and heated in a flame-sealed glass tube at 180 °C for 2 h. The residual HCl fumes were removed in vacuo. The hydrolysate of the diketopiperazines were dissolved in triethylamine/ACN (2:3) (100 µL), and these solutions were then treated with 1-fluoro-2,4-dinitrophenyl-5-l-alaninamide (L-FDAA) in ACN/acetone (1:2) (100 μL). The vials were heated at 50 °C for 1 h. The mixture was dried and re-suspended in ACN/H2O (5:95) (500 μL) for subsequent LC-MS analysis. Authentic L-Phe, L-Pro, L-Tyr, L-Val, L-Ile, L-Ala standards were treated with L-FDAA and D-FDAA.
In spite of the low amounts used, the extracted-ion chromatograms of the diketopiperazines at m/z 418.1357 (FDAA-Phe), 368.1201 (FDAA-Pro), 434.1306 (FDAA-Tyr), 370.1357 (FDAA-Val), 384.1514 (FDAA-Ile), and 342.1044 (FDAA-Ala) were almost devoid of noise.
In DKP cyclo(Pro-Phe) (
6), the phenylalanine residue was found to have L configuration while the proline residue was found to have D configuration on the basis of the retention times of their respective Marfey’s derivatives (
Figure 5).
In DKP cyclo(Pro-Gly) (
7), L configuration was found for proline residue (
Figure S1).
In DKP cyclo(Pro-Ala) (
8), both alanine and proline residues were found to have L configuration (
Figure S2).
In DKP cyclo(Pro-Val) (
9), the valine residue was found to have L configuration while the proline residue was found to have D configuration (
Figure S3).
In DKP cyclo(Pro-Ser) (
10), L configuration was found for proline residue while it was not possible to establish the configuration for the serine residue, probably because of the low amount of diketopiperazine (
Figure S4).
In DKP cyclo(Pro-Ile) (
11), the isoleucine residue was found to have L configuration while the proline residue was found to have the D configuration (
Figure S5).
In DKP cyclo(Pro-Tyr) (
12), the tyrosine and the proline residues were found to have the L configuration (
Figure S6).
4.5. Quantum Mechanical Prediction of 1H-1H Coupling Constants of epi-Tetradehydrohalicyclamine B (1a)
Initial conformational search was performed using molecular dynamics simulations at different temperatures, performed in vacuo in the CFF91 force field using the Insight II/Discover package (BIOVIA: San Diego, CA, USA). The resulting conformers (42 conformers within 4 kcal/mol from the lowest-energy conformer) were used as starting structure for quantum mechanical calculations with the Gaussian 09 program [
13]. Geometries were optimized at the B3LYP/6-31G(d) level of theory and the continuum-solvent (PCM) model for MeOH, and
1H-
1H coupling constants were calculated for each conformer at the B3LYP/6-31G(d,p) level of theory according to the suggestion by Bally and Rablen [
11], i.e., considering only the Fermi contact contribution to
J and scaling the calculated value by 0.9117. The averaged coupling constants shown in
Table 2 were obtained using populations calculated from the CFF91 energies, and including in the calculations only the conformers populated by more than 1%. It is interesting to note that using quantum mechanical calculations to evaluate energies and populations of conformers produced a remarkably worse fit with the experimental values. The inaccuracy of relative energies of conformers is a known problem of DFT calculations [
10], which in this particular study could not be avoided even when a higher level of theory (e.g., B3LYP-D3/TZVP) was used.
4.6. Antimicrobial Assays
Assays were performed using the disk diffusion assay method. Pure compounds (100 µg) were solubilized in DMSO and deposited on a 6 mm paper disk and put on agar plated seeded with reference strains Staphylococcus aureus (ATCC 6538) and Escherichia coli (ATCC 8739). Antimicrobial activity was determined by measuring the diameter of the inhibition zone after 24 h of incubation at 37 °C. Cefotaxime (30 µg) and amoxicillin (25 µg) were used as positive controls against S. aureus and E. coli, giving 30 and 21 mm of inhibition zones, respectively.
4.7. Antikinases Assays
Evaluation of the protein kinase activity was performed in vitro as previously described [
14]. Briefly, the buffers used in the experiments were prepared as follows: Homogenization buffer: 60 mM β-glycerophosphate, 15 mM
p-nitrophenylphosphate, 25 mM Mops (pH 7.2), 15 mM EGTA, 15 mM MgCl
2, 1 mM dithiothreitol, 1 mM sodium vanadate, 1mM NaF, 1 mM phenylphosphate, 10 µg leupeptin/mL, 10 µg aprotinin/mL, 10 µg soybean trypsin inhibitor/mL, and 100 µg benzamidine; buffer A: 10 mM MgCl
2, 1mM EGTA, 1 mM dithiothreitol, 25 mM Tris-HCl pH 7.5, 50 µg heparin.mL
−1; buffer C: Homogenization buffer but 5 mM EGTA, no NaF and no protease inhibitors. Kinase activities were assayed in duplicates in buffer A or C at 30 °C, at a final ATP concentration of 15 µM. The order of mixing the reagents was: Buffers, substrate, enzyme, and inhibitor. Isolated compounds were tested against a panel of eight kinases; namely, dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A), cyclin-dependent kinase 5 (CDK5/p25), glycogen synthase kinase-3 (GSK-3 α/β), CDC-like kinase 1 (CLK-1), casein kinase 1 (CK1δ/ε), cyclin-dependent kinase 1 (CDK1/cyclin B), cyclin-dependent kinase 2 (CDK2/cyclin A), and cyclin-dependent kinase 9 (CDK9/cyclin T).
4.8. Amyloid β42 Induction Assay
This assay, described in detail in reference [
15] allows the detection of molecules able to induce the production of extracellular amyloid β-42 peptide.
N2a cells stably transfected with human APP695 were maintained in Dulbecco’s modified Eagle’s media (DMEM/optiMEM, Gibco, InVitrogen, St. Aubin, France), supplemented with 5% fetal bovine serum (Gibco, InVitrogen, St. Aubin, France), 1% penicillin-streptomycin solution (Sigma Aldrich, Saint- Quentin Fallavier, France), and G418 (0.1 mg/mL) in a humidified atmosphere at 37 °C with 5% CO2. N2a-APP695 cells were plated at a density of approximately 10,000 cells per well in 96-well plates in modified media (DMEM/optiMEM) with 0.5% FBS. After 18 h incubation, the conditioned media were replaced by new media containing compounds at the final concentrations of 0.1, 1.0, 10 μg/mL. After 18 h incubation, the cultured media were harvested for amyloid β-42 determination by ELISA assay.
Amyloid β-42 levels were measured in a double antibody sandwich ELISA using a combination of monoclonal antibody (mAb) 6E10 (SIG-39320, Covance, Eurogentec, Seraing, Belgium) and biotinylated polyclonal amyloid β-42 antibody (provided by Dr. P.D. Mehta, Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA). Briefly, wells of microtiter plates (Maxisorp, Nunc, ThermoFisher Scientific, Illkirch, France) were coated 100 μL mAb 6E10 diluted in carbonate-bicarbonate buffer (buffer (0.015 M Na2CO3 + 0.035 M NaHCO3) pH 9.6) at a 1.5 μg/mL final concentration, and plates were incubated overnight at 4 °C. The plates were then washed with PBST (PBS containing 0.05% Tween-20) and blocked for 1 h with 1% BSA in PBST to avoid non-specific binding. Following a washing step, 100 μL of cell supernatant was added and incubated for 2 h at room temperature (RT) on a shaking device. Plates were then washed with PBST and 100 μL of biotinylated antibodies (diluted to 1 μL/mL in PBST containing 0.5% BSA) were added and incubation was carried out for 75 min at RT under constant shaking. After a washing step, streptavidin-Poly-HRP (horseradish peroxidase) conjugate (Pierce, ThermoFisher Scientific, Illkirch, France), diluted in PBS + 1% BSA, was added and incubation was carried out for 45 min at RT under continuous shaking. After washing, 100 μL of OPD (o-Phenylenediamine dihydrochloride, Pierce, ThermoFisher Scientific, Illkirch, France) in pH 5.0 citrate buffer (0.049M citric acid monohydrate + 0.1M Na2HPO4·2H2O + 1 mL H2O2 30%/L) were added as a substrate and after 15 min incubation at room temperature, the reaction was stopped by addition of 100 μL 1 N sulfuric acid. Optical density (OD) was measured at 490 nm using a plate reader (BioTek Instruments, El 800, Gen 5 software, Winooski, VT, USA).
Amyloidβ–42 quantification was calculated using standard curves, which were prepared with synthetic Aβ-42 HFIP treated (JPT Peptide Technologies, Berlin, Germany) and Aβ-42 specific polyclonal antibody. Curve fitting was performed using a 4 parameters sigmoid equation (SigmaPlot, Systat, Sigma). Results are expressed as fold change ± s.d. All experiments were performed in triplicate.
4.9. Inhibition of Amyloid β-42 Production Induced by Aftin-5 Assay
This assay allows the detection of molecules able to inhibit the production of extracellular Amyloid β peptides induced by a pre-treatment with 100 μM of aftin-5. Aftin-5 is available from Adipogen International, San Diego, CA, USA.
N2a cells stably transfected with human APP695 were maintained in Dulbecco’s modified Eagle’s media (DMEM/optiMEM), supplemented with 5% fetal bovine serum, 1% penicillin-streptomycin solution (Sigma), and G418 (0.1 mg/mL) in a humidified atmosphere at 37 °C with 5% CO2. N2a-APP695 cells were plated at a density of approximately 10,000 cells per well in 96-well plates in modified media (DMEM/optiMEM) with 0.5% FBS. After 18 h incubation, the conditioned media were replaced by new media containing compounds at the final concentrations of 0.1, 1.0, 10 μg/mL. After 1 h incubation, aftin-5 was added (100 μM 1% DMSO final). After 18 h incubation, the cultured media were harvested for amyloid β-42 determination by ELISA assay.
4.10. Cytotoxic Assay: Effects on N2a-APP695 Viability (MTS Survival Assay)
This assay allows evaluating the survival rate of cultured mammalian cells exposed to extracts or pure compounds. It allows the detection of cell death-inducing molecules.
N2a cells stably transfected with human APP695 were maintained in Dulbecco’s modified Eagle’s medium (DMEM/optiMEM), supplemented with 5% fetal bovine serum, 1% penicillin-streptomycin solution, and G418 (Sigma, St. Louis, MO, USA) (0.1 mg/mL) in a humidified atmosphere at 37 °C with 5% CO2.
N2a-APP695 cells were plated at a density of approximately 10,000 cells per well on 96-well plates in Dulbecco’s modified Eagle’s medium (DMEM/optiMEM), supplemented with 0.5% fetal bovine serum. After 24 h incubation, the conditioned media were replaced by new media containing compounds at the final concentrations of 0.1, 1.0, 10 µg/mL. Viability of cells was measured by MTS-formazan reduction using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) at 18 h post treatment. Incubation was pursued for 1.5 h (37 °C, 5% CO2, and 95% humidity). Optical density (OD) was measured at 490 and 630 nm using a microELISA reader (BioTek Instruments, Winooski, VT, USA.).

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}