PPARs as Nuclear Receptors for Nutrient and Energy Metabolism


{kind=link}
{kind=link}
Abstract
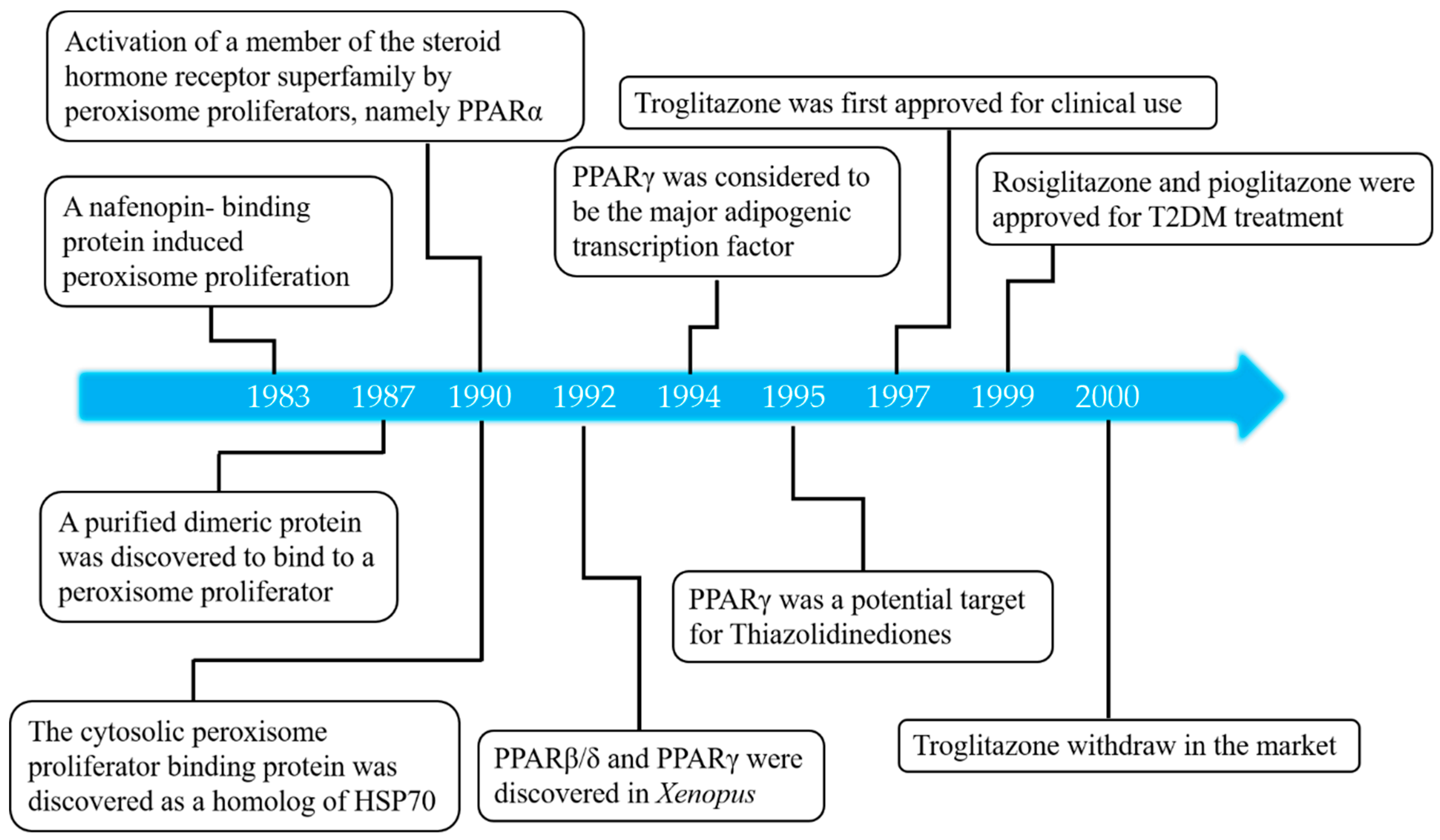
1. Introduction
2. Action of PPARs in Nutrient Metabolism
2.1. PPARs in Lipid Metabolism
2.2. PPARs in Glucose Homeostasis
2.3. PPARs in Cholesterol Metabolism
2.4. PPARs in Animals Fed a High-Fat Diet
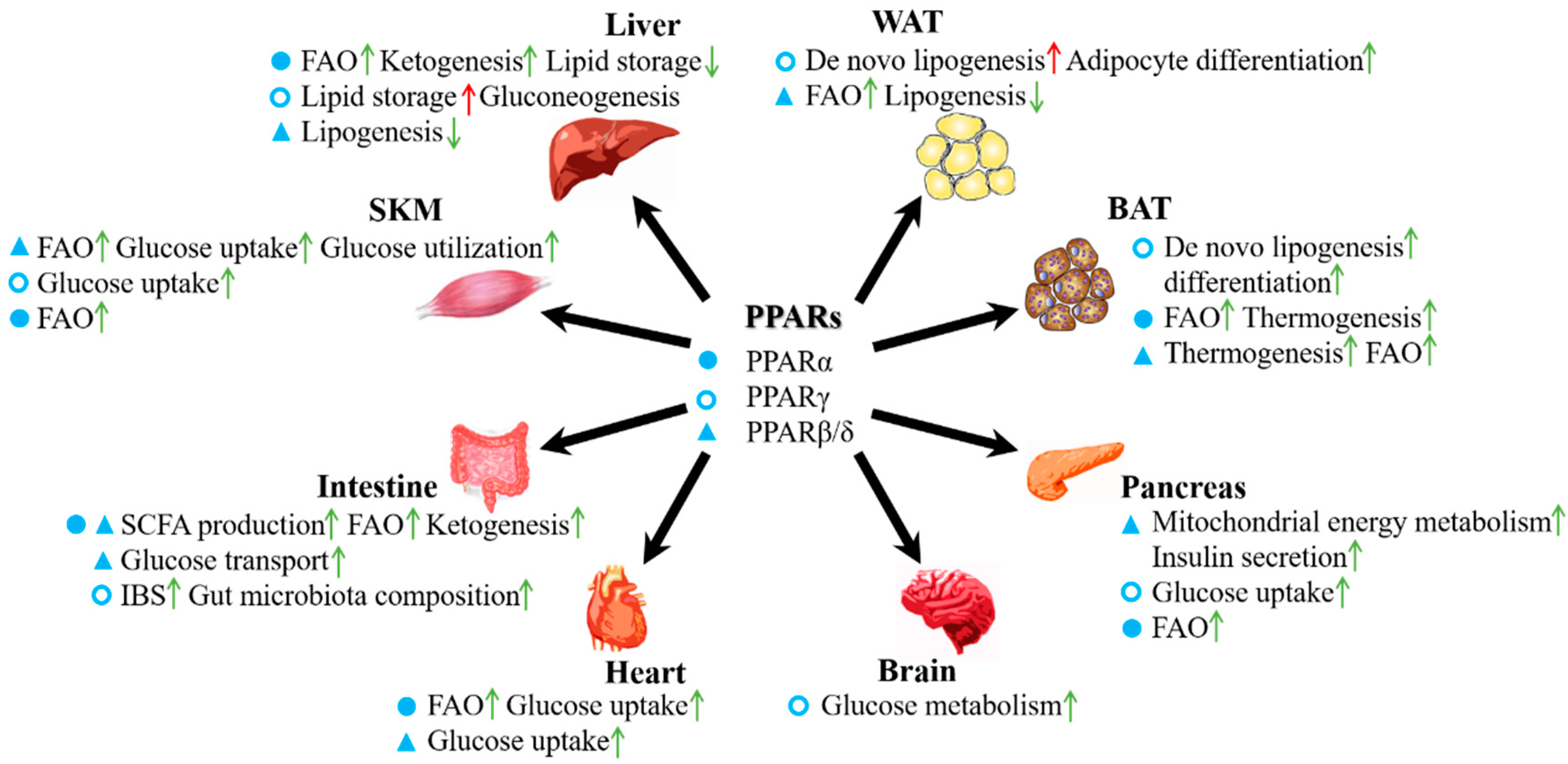
3. Roles of PPARs in the Energy Metabolism of Various Organs
3.1. PPARs in Adipose Tissue
3.2. PPARs in the Liver
3.3. PPARs in the Intestine
3.4. PPARs in Skeletal Muscle
3.5. PPARs in the Pancreas
3.6. PPARs in the Heart
4. Functions of PPARs beyond Being “Nutrient and Energy Metabolite Receptors”
5. Conclusions
Funding
Conflicts of Interest
References
- Hess, R.; Staubli, W.; Riess, W. Nature of the hepatomegalic effect produced by ethyl-chlorophenoxy-isobutyrate in the rat. Nature 1965, 208, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Krishnakantha, T.P. Hepatic peroxisome proliferation: Induction by two novel compounds structurally unrelated to clofibrate. Science 1975, 190, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B.; De Duve, C. A fatty acyl-coa oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhai, Y.; Wang, J. The role of PPAR and its cross-talk with car and lxr in obesity and atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, N.D.; Fahl, W.E.; Reddy, J.K. Detection of a nafenopin-binding protein in rat liver cytosol associated with the induction of peroxisome proliferation by hypolipidemic compounds. Biochem. Biophys. Res. Commun. 1983, 116, 388–393. [Google Scholar] [CrossRef]
- Lalwani, N.D.; Alvares, K.; Reddy, M.K.; Reddy, M.N.; Parikh, I.; Reddy, J.K. Peroxisome proliferator-binding protein: Identification and partial characterization of nafenopin-, clofibric acid-, and ciprofibrate-binding proteins from rat liver. Proc. Natl. Acad. Sci. USA 1987, 84, 5242–5246. [Google Scholar] [CrossRef] [PubMed]
- Alvares, K.; Carrillo, A.; Yuan, P.M.; Kawano, H.; Morimoto, R.I.; Reddy, J.K. Identification of cytosolic peroxisome proliferator binding protein as a member of the heat shock protein hsp70 family. Proc. Natl. Acad. Sci. USA 1990, 87, 5293–5297. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Gottlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. Cdna cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Davis, L.; Breyer, M.D. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am. J. Physiol. 1997, 273, F1013–F1022. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Endo, N.; Rutledge, S.J.; Vogel, R.; Shinar, D.; Rodan, G.A. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 1992, 6, 1634–1641. [Google Scholar]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Jow, L.; Noonan, D.; McDonnell, D.P. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J. Steroid Biochem. Mol. Biol. 1994, 51, 157–166. [Google Scholar] [CrossRef]
- Zhu, Y.; Alvares, K.; Huang, Q.; Rao, M.S.; Reddy, J.K. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 1993, 268, 26817–26820. [Google Scholar] [PubMed]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cdna: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and metabolic disorders associated with challenged adipose tissue plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Lazar, M.A. Endogenous ligands for nuclear receptors: Digging deeper. J. Biol. Chem. 2010, 285, 40409–40415. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced t2dm, dyslipidaemia and nafld. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Woller, A.; Duez, H.; Staels, B.; Lefranc, M. A mathematical model of the liver circadian clock linking feeding and fasting cycles to clock function. Cell Rep. 2016, 17, 1087–1097. [Google Scholar] [CrossRef]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. Pparalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a ppre in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [CrossRef]
- Berthou, L.; Duverger, N.; Emmanuel, F.; Langouet, S.; Auwerx, J.; Guillouzo, A.; Fruchart, J.C.; Rubin, E.; Denefle, P.; Staels, B.; et al. Opposite regulation of human versus mouse apolipoprotein a-i by fibrates in human apolipoprotein a-i transgenic mice. J. Clin. Investig. 1996, 97, 2408–2416. [Google Scholar] [CrossRef]
- Shah, A.; Rader, D.J.; Millar, J.S. The effect of PPAR-alpha agonism on apolipoprotein metabolism in humans. Atherosclerosis 2010, 210, 35–40. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Gervois, P.; Jakel, H.; Nowak, M.; Bauge, E.; Dehondt, H.; Staels, B.; Pennacchio, L.A.; Rubin, E.M.; Fruchart-Najib, J.; et al. Apolipoprotein a5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J. Biol. Chem. 2003, 278, 17982–17985. [Google Scholar] [CrossRef]
- Hiukka, A.; Leinonen, E.; Jauhiainen, M.; Sundvall, J.; Ehnholm, C.; Keech, A.C.; Taskinen, M.R. Long-term effects of fenofibrate on vldl and hdl subspecies in participants with type 2 diabetes mellitus. Diabetologia 2007, 50, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T.; Group, K.S. Effects of k-877, a novel selective PPARα modulator (spparmalpha), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Kajita, K.; Mune, T.; Ikeda, T.; Matsumoto, M.; Uno, Y.; Sugiyama, C.; Matsubara, K.; Morita, H.; Takemura, M.; Seishima, M.; et al. Effect of fasting on PPARgamma and ampk activity in adipocytes. Diabetes Res. Clin. Pract. 2008, 81, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. Ppardelta activation attenuates hepatic steatosis in ldlr-/- mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xie, X.; Fan, Y.; Tian, J.; Guan, Y.; Wang, X.; Zhu, Y.; Wang, N. Peroxisome proliferator-activated receptor-delta induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008, 48, 432–441. [Google Scholar] [CrossRef]
- Peeters, A.; Baes, M. Role of PPARα in hepatic carbohydrate metabolism. PPAR Res. 2010, 2010, 572405. [Google Scholar] [CrossRef]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. Ppargamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. Ppardelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor δ/β in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. Pgc1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.H.; Pedersen, A.J.; Birk, J.B.; Kristensen, D.E.; Vind, B.F.; Baba, O.; Nohr, J.; Hojlund, K.; Wojtaszewski, J.F. Human muscle fiber type-specific insulin signaling: Impact of obesity and type 2 diabetes. Diabetes 2015, 64, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Manninen, V.; Tenkanen, L.; Koskinen, P.; Huttunen, J.K.; Manttari, M.; Heinonen, O.P.; Frick, M.H. Joint effects of serum triglyceride and ldl cholesterol and hdl cholesterol concentrations on coronary heart disease risk in the helsinki heart study. Implications for treatment. Circulation 1992, 85, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans affairs high-density lipoprotein cholesterol intervention trial study group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Bezafibrate Infarction Prevention. Secondary prevention by raising hdl cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000, 102, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the field study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Group, A.S.; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., 3rd; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar]
- Marrapodi, M.; Chiang, J.Y. Peroxisome proliferator-activated receptor alpha (PPARα) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J. Lipid Res. 2000, 41, 514–520. [Google Scholar] [PubMed]
- Kok, T.; Bloks, V.W.; Wolters, H.; Havinga, R.; Jansen, P.L.; Staels, B.; Kuipers, F. Peroxisome proliferator-activated receptor alpha (PPARα)-mediated regulation of multidrug resistance 2 (mdr2) expression and function in mice. Biochem. J. 2003, 369, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Charbonnel, B.; Staels, B. Thiazolidinediones and PPARgamma agonists: Time for a reassessment. Trends Endocrinol. Metab. 2012, 23, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hong, F.; Wang, J.; Wang, J.; Zhao, X.; Wang, S.; Xue, T.; Xu, J.; Zheng, X.; Zhai, Y. Dbz is a putative PPARgamma agonist that prevents high fat diet-induced obesity, insulin resistance and gut dysbiosis. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wang, J.; Hong, F.; Wang, S.; Jin, X.; Xue, T.; Jia, L.; Zhai, Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J. Pineal Res. 2017, 62, e12399. [Google Scholar] [CrossRef]
- Wang, J.; Xu, P.; Xie, X.; Li, J.; Zhang, J.; Wang, J.; Hong, F.; Li, J.; Zhang, Y.; Song, Y.; et al. Dbz (danshensu bingpian zhi), a novel natural compound derivative, attenuates atherosclerosis in apolipoprotein e-deficient mice. J. Am. Heart Assoc. 2017, 6, e006297. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. Mbx-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef]
- Leibowitz, M.D.; Fievet, C.; Hennuyer, N.; Peinado-Onsurbe, J.; Duez, H.; Bergera, J.; Cullinan, C.A.; Sparrow, C.P.; Baffic, J.; Berger, G.D.; et al. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000, 473, 333–336. [Google Scholar] [CrossRef]
- Olson, E.J.; Pearce, G.L.; Jones, N.P.; Sprecher, D.L. Lipid effects of peroxisome proliferator-activated receptor-delta agonist gw501516 in subjects with low high-density lipoprotein cholesterol: Characteristics of metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2289–2294. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Vrins, C.L.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.; Huet, S.; Oude Elferink, R.P.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Lee, J.Y.; Teraminami, A.; Kim, Y.I.; Hirai, S.; Uemura, T.; Inoue, H.; Takahashi, N.; Kawada, T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J. Lipid Res. 2011, 52, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Guerre-Millo, M.; Rouault, C.; Poulain, P.; Andre, J.; Poitout, V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Reach, G.; Staels, B. Ppar-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 2001, 50, 2809–2814. [Google Scholar] [CrossRef] [PubMed]
- Rachid, T.L.; Penna-de-Carvalho, A.; Bringhenti, I.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Fenofibrate (PPARα agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Mol. Cell Endocrinol. 2015, 402, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by sirt1-dependent deacetylation of PPARgamma. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An erk/cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef]
- Eckel-Mahan, K.L.; Patel, V.R.; de Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.A.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the circadian clock by nutritional challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Ma, Y.; Alsaggar, M.; Liu, D. Dual outcomes of rosiglitazone treatment on fatty liver. AAPS J. 2016, 18, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.; Zhang, Q.; Yu, M.; Xu, J.; Wang, Z. Maternal high-fat diet modulates hepatic glucose, lipid homeostasis and gene expression in the PPAR pathway in the early life of offspring. Int. J. Mol. Sci. 2014, 15, 14967–14983. [Google Scholar] [CrossRef] [PubMed]
- Barroso, E.; Rodriguez-Calvo, R.; Serrano-Marco, L.; Astudillo, A.M.; Balsinde, J.; Palomer, X.; Vazquez-Carrera, M. The PPARbeta/delta activator gw501516 prevents the down-regulation of ampk caused by a high-fat diet in liver and amplifies the pgc-1alpha-lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology 2011, 152, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Heudobler, D.; Rechenmacher, M.; Luke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome proliferator-activated receptors (PPAR)gamma agonists as master modulators of tumor tissue. Int. J. Mol. Sci. 2018, 19, 3540. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. (Lausanne) 2016, 7, 30. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. MPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/ebpalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef]
- Pu, Y.; Veiga-Lopez, A. Ppargamma agonist through the terminal differentiation phase is essential for adipogenic differentiation of fetal ovine preadipocytes. Cell. Mol. Biol. Lett. 2017, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, J.; Li, H.; Liu, J.; He, L.; Zhang, J.; Zhai, Y. Inhibition of adipocyte differentiation and adipogenesis by the traditional chinese herb sibiraea angustata. Exp. Biol. Med. (Maywood) 2010, 235, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Li, Y.; Stockton, P.; Foley, J. Fasting induces the expression of pgc-1alpha and err isoforms in the outer stripe of the outer medulla (osom) of the mouse kidney. PLoS ONE 2011, 6, e26961. [Google Scholar] [CrossRef] [PubMed]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E. The peroxisome proliferator-activated receptor beta/delta agonist, gw501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Fan, W.; Evans, R. PPARs and errs: Molecular mediators of mitochondrial metabolism. Curr. Opin Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Preidis, G.A.; Kim, K.H.; Moore, D.D. Nutrient-sensing nuclear receptors PPARα and fxr control liver energy balance. J. Clin. Investig. 2017, 127, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Korecka, A.; Polizzi, A.; Lippi, Y.; Blum, Y.; Canlet, C.; Tremblay-Franco, M.; Gautier-Stein, A.; Burcelin, R.; Yen, Y.C.; et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci. Rep. 2016, 6, 20127. [Google Scholar] [CrossRef]
- Regnier, M.; Polizzi, A.; Lippi, Y.; Fouche, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Costet, P.; Legendre, C.; More, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against nafld. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Memon, R.A.; Tecott, L.H.; Nonogaki, K.; Beigneux, A.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141, 4021–4031. [Google Scholar]
- Moran-Salvador, E.; Lopez-Parra, M.; Garcia-Alonso, V.; Titos, E.; Martinez-Clemente, M.; Gonzalez-Periz, A.; Lopez-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter cd36 is a common target of lxr, pxr, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef]
- Hoekstra, M.; Kruijt, J.K.; Van Eck, M.; Van Berkel, T.J. Specific gene expression of atp-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and kupffer cells. J. Biol. Chem. 2003, 278, 25448–25453. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Muller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARα and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol. Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef]
- Hasan, A.U.; Rahman, A.; Kobori, H. Interactions between host PPARs and gut microbiota in health and disease. Int. J. Mol. Sci. 2019, 20, 387. [Google Scholar] [CrossRef]
- Karimian Azari, E.; Leitner, C.; Jaggi, T.; Langhans, W.; Mansouri, A. Possible role of intestinal fatty acid oxidation in the eating-inhibitory effect of the PPAR-alpha agonist wy-14643 in high-fat diet fed rats. PLoS ONE 2013, 8, e74869. [Google Scholar] [CrossRef]
- Van den Bosch, H.M.; Bunger, M.; de Groot, P.J.; van der Meijde, J.; Hooiveld, G.J.; Muller, M. Gene expression of transporters and phase i/ii metabolic enzymes in murine small intestine during fasting. BMC Genom. 2007, 8, 267. [Google Scholar] [CrossRef]
- Nozu, T.; Miyagishi, S.; Nozu, R.; Takakusaki, K.; Okumura, T. Pioglitazone improves visceral sensation and colonic permeability in a rat model of irritable bowel syndrome. J. Pharm. Sci. 2019, 139, 46–49. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Parajuli, D.; McClain, C.J. Microbiome in nafld and ald. Clin. Liver Dis. 2015, 6, 55–58. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Knights, D.; Kostic, A.D.; Knight, R.; Xavier, R.J. A microbiome foundation for the study of crohn’s disease. Cell Host Microbe 2017, 21, 301–304. [Google Scholar] [CrossRef]
- Schnuck, J.K.; Sunderland, K.L.; Gannon, N.P.; Kuennen, M.R.; Vaughan, R.A. Leucine stimulates PPARbeta/delta-dependent mitochondrial biogenesis and oxidative metabolism with enhanced glut4 content and glucose uptake in myotubes. Biochimie 2016, 128–129, 1–7. [Google Scholar] [CrossRef]
- Koh, J.H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.H. Pparbeta is essential for maintaining normal levels of pgc-1alpha and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef]
- Kinouchi, K.; Magnan, C.; Ceglia, N.; Liu, Y.; Cervantes, M.; Pastore, N.; Huynh, T.; Ballabio, A.; Baldi, P.; Masri, S.; et al. Fasting imparts a switch to alternative daily pathways in liver and muscle. Cell Rep. 2018, 25, 3299–3314.e6. [Google Scholar] [CrossRef]
- Wan, J.; Jiang, L.; Lu, Q.; Ke, L.; Li, X.; Tong, N. Activation of PPARdelta up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2010, 391, 1567–1572. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Zhang, Y.; Pan, Z.; Wu, B.; Huang, X.; Zhang, Y.; Mei, Y.; Ge, L.; Shen, G.; et al. Peroxisome proliferator-activated receptorbeta/delta activation is essential for modulating p-foxo1/foxo1 status in functional insulin-positive cell differentiation. Cell Death Dis. 2015, 6, e1715. [Google Scholar] [CrossRef]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef]
- Jin, X.; Jia, T.; Liu, R.; Xu, S. The antagonistic effect of selenium on cadmium-induced apoptosis via PPAR-gamma/pi3k/akt pathway in chicken pancreas. J. Hazard. Mater. 2018, 357, 355–362. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Lee, T.W.; Bai, K.J.; Lee, T.I.; Chao, T.F.; Kao, Y.H.; Chen, Y.J. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 2017, 24, 5. [Google Scholar] [CrossRef]
- Vazquez-Carrera, M. Unraveling the effects of PPARbeta/delta on insulin resistance and cardiovascular disease. Trends Endocrinol. Metab. 2016, 27, 319–334. [Google Scholar] [CrossRef]
- Duan, S.Z.; Ivashchenko, C.Y.; Russell, M.W.; Milstone, D.S.; Mortensen, R.M. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ. Res. 2005, 97, 372–379. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef]
- Poynter, M.E.; Daynes, R.A. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappab signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar] [CrossRef]
- Chung, S.W.; Kang, B.Y.; Kim, S.H.; Pak, Y.K.; Cho, D.; Trinchieri, G.; Kim, T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa b. J. Biol. Chem. 2000, 275, 32681–32687. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, J.; Liu, D.; Tuo, X.; Yu, Y.; Yang, H.; Wang, N. Nuciferine inhibits proinflammatory cytokines via the PPARs in lps-induced raw264.7 cells. Molecules 2018, 23, 2723. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Liu, M.; Bachstetter, A.D.; Cass, W.A.; Lifshitz, J.; Bing, G. Pioglitazone attenuates neuroinflammation and promotes dopaminergic neuronal survival in the nigrostriatal system of rats after diffuse brain injury. J. Neurotrauma 2017, 34, 414–422. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Fox, S.H.; Brotchie, J.M. Pioglitazone may impair l-dopa anti-parkinsonian efficacy in the mptp-lesioned macaque: Results of a pilot study. Synapse 2015, 69, 99–102. [Google Scholar] [CrossRef]
- Li, X.; Song, D.; Leng, S.X. Link between type 2 diabetes and Alzheimer’s disease: From epidemiology to mechanism and treatment. Clin. Interv. Aging 2015, 10, 549–560. [Google Scholar] [CrossRef]
- Deardorff, W.J.; Grossberg, G.T. Targeting neuroinflammation in Alzheimer’s disease: Evidence for nsaids and novel therapeutics. Expert Rev. Neurother. 2017, 17, 17–32. [Google Scholar] [CrossRef]
- Skerrett, R.; Pellegrino, M.P.; Casali, B.T.; Taraboanta, L.; Landreth, G.E. Combined liver x receptor/peroxisome proliferator-activated receptor gamma agonist treatment reduces amyloid beta levels and improves behavior in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem. 2015, 290, 21591–21602. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, H.; Zhao, H.; Qi, B.; Li, F.; An, L. Ppargamma activation ameliorates postoperative cognitive decline probably through suppressing hippocampal neuroinflammation in aged mice. Int. Immunopharmacol. 2017, 43, 53–61. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef]
- Hanyu, H.; Sato, T.; Kiuchi, A.; Sakurai, H.; Iwamoto, T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 2009, 57, 177–179. [Google Scholar] [CrossRef]
- Azuma, Y.T.; Nishiyama, K.; Matsuo, Y.; Kuwamura, M.; Morioka, A.; Nakajima, H.; Takeuchi, T. Pparalpha contributes to colonic protection in mice with dss-induced colitis. Int. Immunopharmacol. 2010, 10, 1261–1267. [Google Scholar] [CrossRef]
- Drori, S.; Girnun, G.D.; Tou, L.; Szwaya, J.D.; Mueller, E.; Xia, K.; Shivdasani, R.A.; Spiegelman, B.M. Hic-5 regulates an epithelial program mediated by PPARgamma. Genes Dev. 2005, 19, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kim, E.; Wang, C.C.; Harrison, L.E. Ciglitazone-induced p27 gene transcriptional activity is mediated through sp1 and is negatively regulated by the mapk signaling pathway. Cell Signal. 2005, 17, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Yao, P.L.; Gonzalez, F.J. Targeting peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) for cancer chemoprevention. Curr. Pharm. Rep. 2015, 1, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The role of PPAR-delta in metabolism, inflammation, and cancer: Many characters of a critical transcription factor. Int. J. Mol. Sci. 2018, 19, 3339. [Google Scholar] [CrossRef] [PubMed]
- Foreman, J.E.; Sorg, J.M.; McGinnis, K.S.; Rigas, B.; Williams, J.L.; Clapper, M.L.; Gonzalez, F.J.; Peters, J.M. Regulation of peroxisome proliferator-activated receptor-beta/delta by the apc/beta-catenin pathway and nonsteroidal antiinflammatory drugs. Mol. Carcinog. 2009, 48, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Wang, Y.; Williams, C.R.; Risingsong, R.; Gilmer, T.; Willson, T.M.; Sporn, M.B. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), gw7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999, 59, 5671–5673. [Google Scholar]
- Mehta, R.G.; Williamson, E.; Patel, M.K.; Koeffler, H.P. A ligand of peroxisome proliferator-activated receptor gamma, retinoids, and prevention of preneoplastic mammary lesions. J. Natl. Cancer Inst. 2000, 92, 418–423. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. Ppar-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef]
- Ham, S.A.; Yoo, T.; Lee, W.J.; Hwang, J.S.; Hur, J.; Paek, K.S.; Lim, D.S.; Han, S.G.; Lee, C.H.; Seo, H.G. Adamts1-mediated targeting of tsp-1 by PPARdelta suppresses migration and invasion of breast cancer cells. Oncotarget 2017, 8, 94091–94103. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. Ppargamma and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Scott, R.; O’Brien, R.; Fulcher, G.; Pardy, C.; D’Emden, M.; Tse, D.; Taskinen, M.R.; Ehnholm, C.; Keech, A.; Fenofibrate, I.; et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: The fenofibrate intervention and event lowering in diabetes (field) study. Diabetes Care 2009, 32, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Xu, P.; Zhai, Y. The opportunities and challenges of peroxisome proliferator-activated receptors ligands in clinical drug discovery and development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; Bell, S.; Sund, R.; Hartikainen, S.A.; Tuomilehto, J.; Pukkala, E.; Keskimaki, I.; Badrick, E.; Renehan, A.G.; Buchan, I.E.; et al. Pioglitazone and bladder cancer risk: A multipopulation pooled, cumulative exposure analysis. Diabetologia 2015, 58, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, J.P. Diabetes: New conductors for the peroxisome proliferator-activated receptor gamma (PPARgamma) orchestra. Int. J. Biochem. Cell Biol. 2011, 43, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Wang, D.; Katkuri, S.; Wang, H.; Dey, S.K.; DuBois, R.N. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat. Med. 2004, 10, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Cymabay Therapeutics. Corporate Presentation. 2016. Available online: https://content.equisolve.net/cymabay/media/9af11b3859e3b4fa04eff0f43424ffb5.pdf (accessed on 12 July 2019).
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Raval, P.; Jain, M.; Goswami, A.; Basu, S.; Gite, A.; Godha, A.; Pingali, H.; Raval, S.; Giri, S.; Suthar, D.; et al. Revisiting glitazars: Thiophene substituted oxazole containing alpha-ethoxy phenylpropanoic acid derivatives as highly potent PPARα/gamma dual agonists devoid of adverse effects in rodents. Bioorganic Med. Chem. Lett. 2011, 21, 3103–3109. [Google Scholar] [CrossRef]
- Joshi, S.R. Saroglitazar for the treatment of dyslipidemia in diabetic patients. Expert Opin. Pharmacother. 2015, 16, 597–606. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist gft505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist gft505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, gft505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Deochand, C.; Didsbury, J.; de la Monte, S.M. T3d-959: A multi-faceted disease remedial drug candidate for the treatment of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2016, 51, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Dominguez, C.; Didsbury, J.; de la Monte, S.M. Targeting Alzheimer’s disease neuro-metabolic dysfunction with a small molecule nuclear receptor agonist (t3d-959) reverses disease pathologies. J. Alzheimer’s Dis. Parkinsonism 2016, 6. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef]
- Ericsson, C.G.; Hamsten, A.; Nilsson, J.; Grip, L.; Svane, B.; de Faire, U. Angiographic assessment of effects of bezafibrate on progression of coronary artery disease in young male postinfarction patients. Lancet 1996, 347, 849–853. [Google Scholar] [CrossRef]
- He, B.K.; Ning, Z.Q.; Li, Z.B.; Shan, S.; Pan, D.S.; Ko, B.C.; Li, P.P.; Shen, Z.F.; Dou, G.F.; Zhang, B.L.; et al. In vitro and in vivo characterizations of chiglitazar, a newly identified PPAR pan-agonist. PPAR Res. 2012, 2012, 546548. [Google Scholar] [CrossRef]
- Ruzehaji, N.; Frantz, C.; Ponsoye, M.; Avouac, J.; Pezet, S.; Guilbert, T.; Luccarini, J.M.; Broqua, P.; Junien, J.L.; Allanore, Y. Pan PPAR agonist iva337 is effective in prevention and treatment of experimental skin fibrosis. Ann. Rheum. Dis. 2016, 75, 2175–2183. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorg. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARα modulator (SPPARMalpha) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C. Selective peroxisome proliferator-activated receptor alpha modulators (sPPARmalpha): The next generation of peroxisome proliferator-activated receptor alpha-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S.; K-Study Group. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-alpha modulator (sPPARmalpha), in dyslipidemic patients with renal impairment. Int. J. Mol. Sci. 2019, 20, 706. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. https://doi.org/10.3390/molecules24142545
Hong F, Pan S, Guo Y, Xu P, Zhai Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules. 2019; 24(14):2545. https://doi.org/10.3390/molecules24142545
Chicago/Turabian StyleHong, Fan, Shijia Pan, Yuan Guo, Pengfei Xu, and Yonggong Zhai. 2019. "PPARs as Nuclear Receptors for Nutrient and Energy Metabolism" Molecules 24, no. 14: 2545. https://doi.org/10.3390/molecules24142545
APA StyleHong, F., Pan, S., Guo, Y., Xu, P., & Zhai, Y. (2019). PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules, 24(14), 2545. https://doi.org/10.3390/molecules24142545