
The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease


Abstract
1. Introduction
2. Role of the CaSR for the Developing NS: Differentiation of Neural Cells
2.1. CaSR in Neuronal Differentiation
2.2. CaSR Relevance in Oligodendrocyte Differentiation
2.3. CaSR in Astrocyte Differentiation
3. Role of CaSR for the Adult NS
3.1. Neurotransmission and Excitability
3.2. Heterodimerization with Other GPCRs
4. CaSR as a Potential Target for Disorders of Nervous System
4.1. Ischemia and Hypoxia
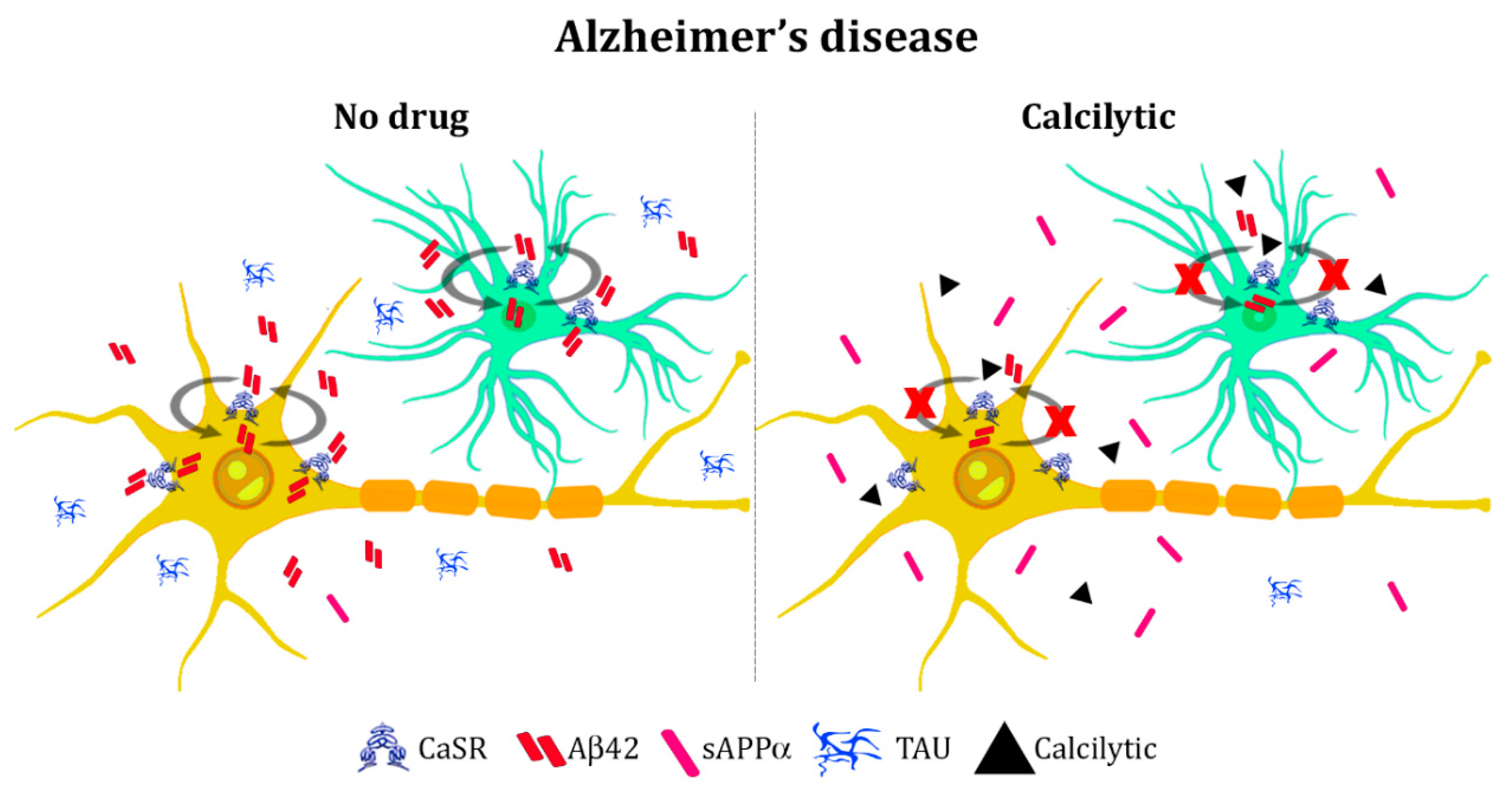
4.2. Alzheimer’s Disease
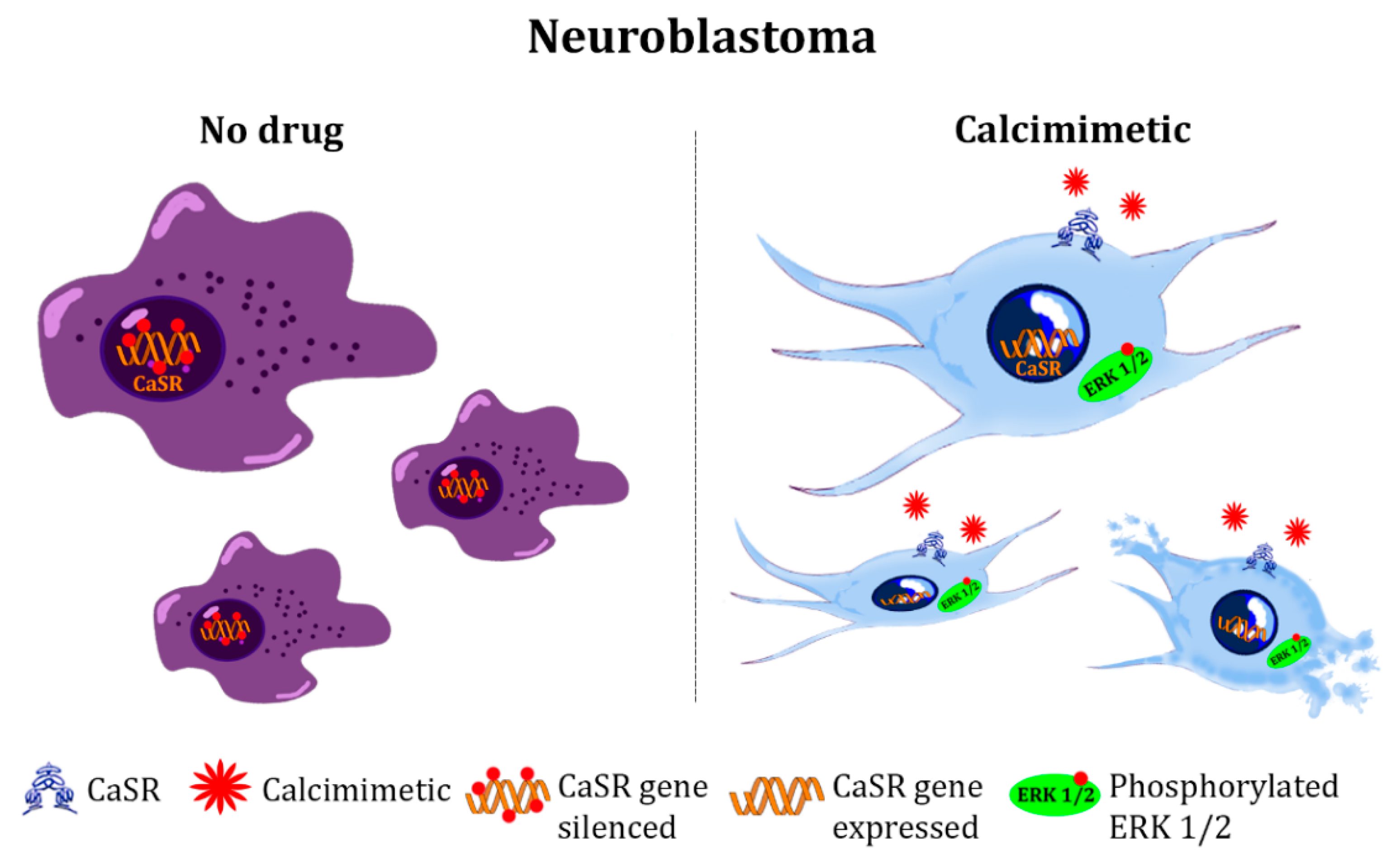
4.3. Tumors of Nervous System
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Hendy, G.N.; Canaff, L. Calcium-Sensing Receptor Gene: Regulation of Expression. Front. Physiol. 2016, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Hendy, G.N. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J. Biol. Chem. 2002, 277, 30337–30350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, T.; Zou, J.; Miller, C.L.; Gorkhali, R.; Yang, J.-Y.; Schilmiller, A.; Wang, S.; Huang, K.; Brown, E.M.; et al. Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci. Adv. 2016, 2, e1600241. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.; et al. Structural mechanism of ligand activation in human calcium-sensing receptor. elife 2016, 5, e13662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Miller, C.L.; Brown, E.M.; Yang, J.J. The calcium sensing receptor: From calcium sensing to signaling. Sci. China Life Sci. 2015, 58, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; McLarnon, S.J.; Mora, S.; Jiang, J.; Thomas, C.; Jacobson, K.A.; Spiegel, A.M. A Region in the Seven-transmembrane Domain of the Human Ca2+ Receptor Critical for Response to Ca2+. J. Biol. Chem. 2005, 280, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.F. Allosteric modulators of the extracellular calcium receptor. Drug Discov. Today Technol. 2013, 10, e277–e284. [Google Scholar] [CrossRef]
- Gama, L.; Breitwieser, G.E. A carboxyl-terminal domain controls the cooperativity for extracellular Ca2+ activation of the human calcium sensing receptor. A study with receptor-green fluorescent protein fusions. J. Biol. Chem. 1998, 273, 29712–29718. [Google Scholar] [CrossRef]
- Bai, M.; Trivedi, S.; Lane, C.R.; Yang, Y.; Quinn, S.J.; Brown, E.M. Protein kinase C phosphorylation of threonine at position 888 in Ca2+ o-sensing receptor (CaR) inhibits coupling to Ca2+ store release. J. Biol. Chem. 1998, 273, 21267–21275. [Google Scholar] [CrossRef]
- Jiang, Y.-F.; Zhang, Z.; Kifor, O.; Lane, C.R.; Quinn, S.J.; Bai, M. Protein kinase C (PKC) phosphorylation of the Ca2+ o-sensing receptor (CaR) modulates functional interaction of G proteins with the CaR cytoplasmic tail. J. Biol. Chem. 2002, 277, 50543–50549. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Singh, G.; Ghosh, E. Emerging structural insights into biased GPCR signaling. Trends Biochem. Sci. 2014, 39, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Towards tissue-specific pharmacology: Insights from the calcium-sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol. Sci. 2015, 36, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Saidak, Z.; Brazier, M.; Kamel, S.; Mentaverri, R. Agonists and Allosteric Modulators of the Calcium-Sensing Receptor and Their Therapeutic Applications. Mol. Pharmacol. 2009, 76, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538. [Google Scholar] [CrossRef]
- Conigrave, A.D.; Quinn, S.J.; Brown, E.M. L-Amino acid sensing by the extracellular Ca2+-sensing receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4814–4819. [Google Scholar] [CrossRef]
- Nemeth, E.F.; Goodman, W.G. Calcimimetic and Calcilytic Drugs: Feats, Flops, and Futures. Calcif. Tissue Int. 2016, 98, 341–358. [Google Scholar] [CrossRef]
- Kifor, O.; Diaz, R.; Butters, R.; Brown, E.M. The Ca2+-sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR-transfected, human embryonic kidney (HEK293) cells. J. Bone Miner. Res. 1997, 12, 715–725. [Google Scholar] [CrossRef]
- Kifor, O.; MacLeod, R.J.; Diaz, R.; Bai, M.; Yamaguchi, T.; Yao, T.; Kifor, I.; Brown, E.M. Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells. Am. J. Physiol. Ren. Physiol. 2001, 280, F291–F302. [Google Scholar] [CrossRef]
- Thomsen, A.R.B.; Hvidtfeldt, M.; Bräuner-Osborne, H. Biased agonism of the calcium-sensing receptor. Cell Calcium 2012, 51, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.L.; Gibbons, C.E.; Vizard, T.; Ward, D.T. Ca2+-sensing receptor induces Rho kinase-mediated actin stress fiber assembly and altered cell morphology, but not in response to aromatic amino acids. Am. J. Physiol. Cell Physiol. 2006, 290, C1543–C1551. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Oakley, R.H.; Gesty-Palmer, D.; Cruickshank, R.D.; Spurney, R.F.; Luttrell, L.M.; Quarles, L.D. Beta-arrestin- and G protein receptor kinase-mediated calcium-sensing receptor desensitization. Mol. Endocrinol. 2005, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Kemp, P.J. The calcium-sensing receptor beyond extracellular calcium homeostasis: Conception, development, adult physiology, and disease. Annu. Rev. Physiol. 2012, 74, 271–297. [Google Scholar] [CrossRef] [PubMed]
- Conigrave, A.D.; Ward, D.T. Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Conigrave, A.D. The Calcium-Sensing Receptor and the Parathyroid: Past, Present, Future. Front. Physiol. 2016, 7, 563. [Google Scholar] [CrossRef]
- Brown, E.M.; Gardner, D.G.; Windeck, R.A.; Aurbach, G.D. Relationship of Intracellular 3′,5′-Adenosine Monophosphate Accumulation to Parathyroid Hormone Release from Dispersed Bovine Parathyroid Cells. Endocrinology 1978, 103, 2323–2333. [Google Scholar] [CrossRef]
- Brown, E.M.; Fuleihan, G.H.; Chen, C.J.; Kifor, O. A comparison of the effects of divalent and trivalent cations on parathyroid hormone release, 3′,5′-cyclic-adenosine monophosphate accumulation, and the levels of inositol phosphates in bovine parathyroid cells. Endocrinology 1990, 127, 1064–1071. [Google Scholar] [CrossRef]
- Nemeth, E.F.; Scarpa, A. Rapid mobilization of cellular Ca2+ in bovine parathyroid cells evoked by extracellular divalent cations. Evidence for a cell surface calcium receptor. J. Biol. Chem. 1987, 262, 5188–5196. [Google Scholar]
- Riccardi, D.; Brown, E.M. Physiology and pathophysiology of the calcium-sensing receptor in the kidney. Am. J. Physiol. Ren. Physiol. 2010, 298, F485–F499. [Google Scholar] [CrossRef]
- Dufner, M.M.; Kirchhoff, P.; Remy, C.; Hafner, P.; Müller, M.K.; Cheng, S.X.; Tang, L.-Q.; Hebert, S.C.; Geibel, J.P.; Wagner, C.A. The calcium-sensing receptor acts as a modulator of gastric acid secretion in freshly isolated human gastric glands. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1084–G1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, Y.-H.; He, Y.; Liao, X.-L.; Niu, Y.-C.; Wang, G.; Zhao, C.; Wang, L.; Tian, M.-J.; Li, Y.; Sun, C.-H. The calcium-sensing receptor promotes adipocyte differentiation and adipogenesis through PPARγ pathway. Mol. Cell. Biochem. 2012, 361, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.-L.; Oda, Y.; Komuves, L.; Bikle, D.D. The role of the calcium-sensing receptor in epidermal differentiation. Cell Calcium 2004, 35, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Theman, T.A.; Collins, M.T. The role of the calcium-sensing receptor in bone biology and pathophysiology. Curr. Pharm. Biotechnol. 2009, 10, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Kim, J.M.; Choi, Y.; Park, K. MTA promotes chemotaxis and chemokinesis of immune cells through distinct calcium-sensing receptor signaling pathways. Biomaterials 2018, 150, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Ruat, M.; Molliver, M.E.; Snowman, A.M.; Snyder, S.H. Calcium sensing receptor: Molecular cloning in rat and localization to nerve terminals. Proc. Natl. Acad. Sci. USA 1995, 92, 3161–3165. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Calcium Control of Neurotransmitter Release. Cold Spring Harb. Perspect. Biol. 2012, 4, a011353. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef]
- Leclerc, C.; Néant, I.; Webb, S.E.; Miller, A.L.; Moreau, M. Calcium transients and calcium signalling during early neurogenesis in the amphibian embryo Xenopus laevis. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.; Néant, I.; Webb, S.E.; Miller, A.L.; Leclerc, C. Calcium signalling during neural induction in Xenopus laevis embryos. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, C.; Néant, I.; Moreau, M. The calcium: An early signal that initiates the formation of the nervous system during embryogenesis. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Conner, D.A.; Pollak, M.R.; Ladd, D.J.; Kifor, O.; Warren, H.B.; Brown, E.M.; Seidman, J.G.; Seidman, C.E. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat. Genet. 1995, 11, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Kos, C.H.; Karaplis, A.C.; Peng, J.-B.; Hediger, M.A.; Goltzman, D.; Mohammad, K.S.; Guise, T.A.; Pollak, M.R. The calcium-sensing receptor is required for normal calcium homeostasis independent of parathyroid hormone. J. Clin. Investig. 2003, 111, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lv, F.; Sun, W.; Tao, C.; Ding, G.; Karaplis, A.; Brown, E.; Goltzman, D.; Miao, D. The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and PTH. PLoS Genet. 2011, 7, e1002294. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Lu, Y.-S.; Gao, J.-Y.; Marshall, C.; Xiao, M.; Miao, D.-S.; Karaplis, A.; Goltzman, D.; Ding, J. Calcium Sensing Receptor Absence Delays Postnatal Brain Development via Direct and Indirect Mechanisms. Mol. Neurobiol. 2013, 48, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Glebova, N.O.; Ginty, D.D. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J. Neurosci. 2004, 24, 743–751. [Google Scholar] [CrossRef]
- Vizard, T.N.; O’Keeffe, G.W.; Gutierrez, H.; Kos, C.H.; Riccardi, D.; Davies, A.M. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat. Neurosci. 2008, 11, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Vizard, T.N.; Newton, M.; Howard, L.; Wyatt, S.; Davies, A.M. ERK signaling mediates CaSR-promoted axon growth. Neurosci. Lett. 2015, 603, 77–83. [Google Scholar] [CrossRef][Green Version]
- Chattopadhyay, N.; Jeong, K.-H.; Yano, S.; Huang, S.; Pang, J.L.; Ren, X.; Terwilliger, E.; Kaiser, U.B.; Vassilev, P.M.; Pollak, M.R.; et al. Calcium receptor stimulates chemotaxis and secretion of MCP-1 in GnRH neurons in vitro: Potential impact on reduced GnRH neuron population in CaR-null mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E523–E532. [Google Scholar] [CrossRef][Green Version]
- Widera, D.; Holtkamp, W.; Entschladen, F.; Niggemann, B.; Zänker, K.; Kaltschmidt, B.; Kaltschmidt, C. MCP-1 induces migration of adult neural stem cells. Eur. J. Cell Biol. 2004, 83, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, S.; Wu, C.; Hampson, D.R. The calcium-sensing receptor and integrins modulate cerebellar granule cell precursor differentiation and migration. Dev. Neurobiol. 2016, 76, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.K.; Magno, A.L.; Davis, E.A.; Hanyaloglu, A.C.; Stuckey, B.G.A.; Burrows, M.; Eidne, K.A.; Charles, A.K.; Ratajczak, T. Functional deletion of the calcium-sensing receptor in a case of neonatal severe hyperparathyroidism. J. Clin. Endocrinol. Metab. 2004, 89, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Espinosa-Jeffrey, A.; Tfelt-Hansen, J.; Yano, S.; Bandyopadhyay, S.; Brown, E.M.; de Vellis, J. Calcium receptor expression and function in oligodendrocyte commitment and lineage progression: Potential impact on reduced myelin basic protein in CaR-null mice. J. Neurosci. Res. 2008, 86, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Ye, C.P.; Yamaguchi, T.; Kifor, O.; Vassilev, P.M.; Nishimura, R.; Brown, E.M. Extracellular calcium-sensing receptor in rat oligodendrocytes: Expression and potential role in regulation of cellular proliferation and an outward K+ channel. Glia 1998, 24, 449–458. [Google Scholar] [CrossRef]
- Ferry, S.; Traiffort, E.; Stinnakre, J.; Ruat, M. Developmental and adult expression of rat calcium-sensing receptor transcripts in neurons and oligodendrocytes. Eur. J. Neurosci. 2000, 12, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Ye, C.P.; Yamaguchi, T.; Kerner, R.; Vassilev, P.M.; Brown, E.M. Extracellular calcium-sensing receptor induces cellular proliferation and activation of a nonselective cation channel in U373 human astrocytoma cells. Brain Res. 1999, 851, 116–124. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Ye, C.P.; Yamaguchi, T.; Vassilev, P.M.; Brown, E.M. Evidence for extracellular calcium-sensing receptor mediated opening of an outward K+ channel in a human astrocytoma cell line (U87). Glia 1999, 26, 64–72. [Google Scholar] [CrossRef]
- Gleichmann, M.; Mattson, M.P. Neuronal Calcium Homeostasis and Dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef]
- Hendy, G.N.; D’Souza-Li, L.; Yang, B.; Canaff, L.; Cole, D.E.C. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum. Mutat. 2000, 16, 281–296. [Google Scholar] [CrossRef]
- Kapoor, A.; Satishchandra, P.; Ratnapriya, R.; Reddy, R.; Kadandale, J.; Shankar, S.K.; Anand, A. An idiopathic epilepsy syndrome linked to 3q13.3-q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann. Neurol. 2008, 64, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Stepanchick, A.; McKenna, J.; McGovern, O.; Huang, Y.; Breitwieser, G.E. Calcium sensing receptor mutations implicated in pancreatitis and idiopathic epilepsy syndrome disrupt an arginine-rich retention motif. Cell. Physiol. Biochem. 2010, 26, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Kanazirska, M.; Quinn, S.; Brown, E.M.; Vassilev, P.M. Modulation by polycationic Ca(2+)-sensing receptor agonists of nonselective cation channels in rat hippocampal neurons. Biochem. Biophys. Res. Commun. 1996, 224, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Ho-Pao, C.L.; Kanazirska, M.; Quinn, S.; Seidman, C.E.; Seidman, J.G.; Brown, E.M.; Vassilev, P.M. Deficient cation channel regulation in neurons from mice with targeted disruption of the extracellular Ca2+-sensing receptor gene. Brain Res. Bull. 1997, 44, 75–84. [Google Scholar] [CrossRef]
- Ye, C.; Rogers, K.; Bai, M.; Quinn, S.J.; Brown, E.M.; Vassilev, P.M. Agonists of the Ca(2+)-sensing receptor (CaR) activate nonselective cation channels in HEK293 cells stably transfected with the human CaR. Biochem. Biophys. Res. Commun. 1996, 226, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, P.M.; Ho-Pao, C.L.; Kanazirska, M.P.; Ye, C.; Hong, K.; Seidman, C.E.; Seidman, J.G.; Brown, E.M. Cao-sensing receptor (CaR)-mediated activation of K+ channels is blunted in CaR gene-deficient mouse neurons. Neuroreport 1997, 8, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Bergsman, J.B.; Harata, N.C.; Scheller, R.H.; Tsien, R.W. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron 2004, 41, 243–256. [Google Scholar] [CrossRef]
- Phillips, C.G.; Harnett, M.T.; Chen, W.; Smith, S.M. Calcium-sensing receptor activation depresses synaptic transmission. J. Neurosci. 2008, 28, 12062–12070. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bergsman, J.B.; Wang, X.; Gilkey, G.; Pierpoint, C.-R.; Daniel, E.A.; Awumey, E.M.; Dauban, P.; Dodd, R.H.; Ruat, M.; et al. Presynaptic external calcium signaling involves the calcium-sensing receptor in neocortical nerve terminals. PLoS ONE 2010, 5, e8563. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, Q.; Wang, H.; Wang, Y.; Nakayama, M.; Ren, D. Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron 2010, 68, 488–499. [Google Scholar] [CrossRef]
- Jones, B.L.; Smith, S.M. Calcium-Sensing Receptor: A Key Target for Extracellular Calcium Signaling in Neurons. Front. Physiol. 2016, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Vyleta, N.P.; Smith, S.M. Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium-sensing receptor. J. Neurosci. 2011, 31, 4593–4606. [Google Scholar] [CrossRef] [PubMed]
- Babiec, W.E.; O’Dell, T.J. Novel Ca2+-dependent mechanisms regulate spontaneous release at excitatory synapses onto CA1 pyramidal cells. J. Neurophysiol. 2018, 119, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Miyashita, T.; Murata, Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 1998, 279, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.; Green, A.; Main, M.J.; Wilson, R.; Fraser, N.; Marshall, F.H. Calcium sensing properties of the GABA(B) receptor. Neuropharmacology 1999, 38, 1647–1656. [Google Scholar] [CrossRef]
- Gama, L.; Wilt, S.G.; Breitwieser, G.E. Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. J. Biol. Chem. 2001, 276, 39053–39059. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Tu, C.; Cheng, Z.; Rodriguez, L.; Chen, T.-H.; Gassmann, M.; Bettler, B.; Margeta, M.; Jan, L.Y.; Shoback, D. Complex formation with the Type B gamma-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor. Studies with HEK-293 cells and neurons. J. Biol. Chem. 2007, 282, 25030–25040. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated with Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Lipton, S.A. Pathologically activated therapeutics for neuroprotection. Nat. Rev. Neurosci. 2007, 8, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.T. Calcium homeostasis following traumatic neuronal injury. Curr. Neurovasc. Res. 2004, 1, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.S.; Pak, H.-J.; Shin, Y.-J.; Riew, T.-R.; Park, J.-H.; Moon, Y.W.; Lee, M.-Y. Differential expression of the calcium-sensing receptor in the ischemic and border zones after transient focal cerebral ischemia in rats. J. Chem. Neuroanat. 2015, 66–67, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Pak, H.-J.; Riew, T.-R.; Shin, Y.-J.; Choi, J.-H.; Jin, X.; Lee, M.-Y. Enhanced expression of the calcium-sensing receptor in reactive astrocytes following ischemic injury in vivo and in vitro. J. Neurol. Sci. 2016, 366, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, N.; Yenari, M.A.; Chang, W. Mild Hypothermia Suppresses Calcium-Sensing Receptor (CaSR) Induction Following Forebrain Ischemia While Increasing GABA-B Receptor 1 (GABA-B-R1) Expression. Transl. Stroke Res. 2011, 2, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ho, H.; Kim, N.; Liu, J.; Tu, C.-L.; Yenari, M.A.; Chang, W. Calcium-sensing receptor (CaSR) as a novel target for ischemic neuroprotection. Ann. Clin. Transl. Neurol. 2014, 1, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, L.; Wang, S.; Li, S.; Li, Y.; Zhang, L. Effects of calcium-sensing receptors on apoptosis in rat hippocampus during hypoxia/reoxygenation through the ERK1/2 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10808–10815. [Google Scholar] [PubMed]
- Xue, Z.; Song, Z.; Wan, Y.; Wang, K.; Mo, L.; Wang, Y. Calcium-sensing receptor antagonist NPS2390 attenuates neuronal apoptosis though intrinsic pathway following traumatic brain injury in rats. Biochem. Biophys. Res. Commun. 2017, 486, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Mao, M.; Tian, L.; Yu, Y.; Zeng, J.; Ouyang, K.; Yu, L.; Li, L.; Wang, D.; Deng, X.; et al. Calcium sensing receptor mediated the excessive generation of β-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem. Biophys. Res. Commun. 2015, 459, 568–573. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Ho-Pao, C.L.; Kanazirska, M.; Quinn, S.; Rogers, K.; Seidman, C.E.; Seidman, J.G.; Brown, E.M.; Vassilev, P.M. Amyloid-beta proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J. Neurosci. Res. 1997, 47, 547–554. [Google Scholar] [CrossRef]
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-Sensing Receptors of Human Astrocyte-Neuron Teams: Amyloid-β-Driven Mediators and Therapeutic Targets of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 353–364. [Google Scholar] [PubMed]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential Astrocytic Receptors and Transporters in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons-therapeutic relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [PubMed]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased Calcium-Sensing Receptor Immunoreactivity in the Hippocampus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Conley, Y.P.; Mukherjee, A.; Kammerer, C.; DeKosky, S.T.; Kamboh, M.I.; Finegold, D.N.; Ferrell, R.E. Evidence supporting a role for the calcium-sensing receptor in Alzheimer disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150, 703–709. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Spalletta, G.; Souza, R.P.; Graff, A.; Bastos-Rodrigues, L.; Camargos Bicalho, M.A. Definition of Late Onset Alzheimer’s Disease and Anticipation Effect of Genome-Wide Significant Risk Variants: Pilot Study of the APOE e4 Allele. Neuropsychobiology 2019, 77, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflügers Arch. Eur. J. Physiol. 2010, 459, 441–449. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. ER calcium and Alzheimer’s disease: In a state of flux. Sci. Signal. 2010, 3, pe10. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; LaFerla, F.M. Linking Calcium to Aβ and Alzheimer’s Disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Luksch, R.; Castellani, M.R.; Collini, P.; De Bernardi, B.; Conte, M.; Gambini, C.; Gandola, L.; Garaventa, A.; Biasoni, D.; Podda, M.; et al. Neuroblastoma (Peripheral neuroblastic tumours). Crit. Rev. Oncol. Hematol. 2016, 107, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.-D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef]
- Mateo-Lozano, S.; García, M.; Rodríguez-Hernández, C.J.; de Torres, C. Regulation of Differentiation by Calcium-Sensing Receptor in Normal and Tumoral Developing Nervous System. Front. Physiol. 2016, 7, 169. [Google Scholar] [CrossRef] [PubMed]
- De Torres, C.; Beleta, H.; Díaz, R.; Toran, N.; Rodríguez, E.; Lavarino, C.; García, I.; Acosta, S.; Suñol, M.; Mora, J. The calcium-sensing receptor and parathyroid hormone-related protein are expressed in differentiated, favorable neuroblastic tumors. Cancer 2009, 115, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Casalà, C.; Gil-Guiñón, E.; Ordóñez, J.L.; Miguel-Queralt, S.; Rodríguez, E.; Galván, P.; Lavarino, C.; Munell, F.; de Alava, E.; Mora, J.; et al. The calcium-sensing receptor is silenced by genetic and epigenetic mechanisms in unfavorable neuroblastomas and its reactivation induces ERK1/2-dependent apoptosis. Carcinogenesis 2013, 34, 268–276. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, C.J.; Mateo-Lozano, S.; García, M.; Casalà, C.; Briansó, F.; Castrejón, N.; Rodríguez, E.; Suñol, M.; Carcaboso, A.M.; Lavarino, C.; et al. Cinacalcet inhibits neuroblastoma tumor growth and upregulates cancer-testis antigens. Oncotarget 2016, 7, 16112–16129. [Google Scholar] [CrossRef]
- Haven, C.J.; van Puijenbroek, M.; Karperien, M.; Fleuren, G.-J.; Morreau, H. Differential expression of the calcium sensing receptor and combined loss of chromosomes 1q and 11q in parathyroid carcinoma. J. Pathol. 2004, 202, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Grigorieva, I.V.; Thakker, R. V Transcription factors in parathyroid development: Lessons from hypoparathyroid disorders. Ann. N. Y. Acad. Sci. 2011, 1237, 24–38. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Lee, C.-T.; Bendriem, R.M.; Wu, W.W.; Shen, R.-F. 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef]
- Broccoli, V.; Giannelli, S.G.; Mazzara, P.G. Modeling physiological and pathological human neurogenesis in the dish. Front. Neurosci. 2014, 8, 183. [Google Scholar] [CrossRef]
- Xie, Y.Z.; Zhang, R.X. Neurodegenerative diseases in a dish: The promise of iPSC technology in disease modeling and therapeutic discovery. Neurol. Sci. 2015, 36, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Espuny-Camacho, I.; Michelsen, K.A.; Gall, D.; Linaro, D.; Hasche, A.; Bonnefont, J.; Bali, C.; Orduz, D.; Bilheu, A.; Herpoel, A.; et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 2013, 77, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.K. GPCR drug discovery: Novel ligands for CNS receptors. Recent Pat. CNS Drug Discov. 2007, 2, 107–112. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell Types | Model | Role of CaSR | References |
|---|---|---|---|
| Neurons | Superior cervical ganglion (SCG) sympathetic neurons | Promotes axonal and dendritic growth and extension through ERK1/2 activation. | [48,49] |
| GnRH neuronal cell lines GN11 and GT1-7 | Induces neuronal migration and chemotaxis by the secretion of monocyte chemoattractant protein-1, MCP-1; supports the neuronal survival of GnRH neuronal population. | [50] | |
| Cerebellar granule-cell precursor (GCP) neurons | Stimulates GCPs migration through the activation of MAPK signaling. | [52] | |
| Neurons differentiated from NSCs of newborn CaSR-/- mice | Serves for neurite growth. | [46] | |
| Oligodendrocytes | Oligodendrocytes differentiated from NSCs of newborn CaSR-/- mice | Serves for oligodendrocytes development. | [46] |
| Oligodendrocytes differentiated from rat NSCs | Favors oligodendrocyte commitment and lineage progression; stimulates oligodendrocyte proliferation; induces the opening of a Ca-activated K+ Channel. | [54,55] | |
| 20 days post-natal oligodendrocytes from rat brain | Mediates an intracellular calcium mobilization and inositol phosphate accumulation—PLC mediated. | [56] | |
| Astrocytes | U373 astrocytoma cell line | Increases cell proliferation; activates a nonselective cation channel (NCC). | [57] |
| U87 astrocytoma cell line | Stimulates the opening of an outward K+ channel. | [58] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giudice, M.L.; Mihalik, B.; Dinnyés, A.; Kobolák, J. The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease. Molecules 2019, 24, 2546. https://doi.org/10.3390/molecules24142546
Giudice ML, Mihalik B, Dinnyés A, Kobolák J. The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease. Molecules. 2019; 24(14):2546. https://doi.org/10.3390/molecules24142546
Chicago/Turabian StyleGiudice, Maria Lo, Balázs Mihalik, András Dinnyés, and Julianna Kobolák. 2019. "The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease" Molecules 24, no. 14: 2546. https://doi.org/10.3390/molecules24142546
APA StyleGiudice, M. L., Mihalik, B., Dinnyés, A., & Kobolák, J. (2019). The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease. Molecules, 24(14), 2546. https://doi.org/10.3390/molecules24142546