Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-hydroxy-1-naphthaldehyde Complexes


Abstract
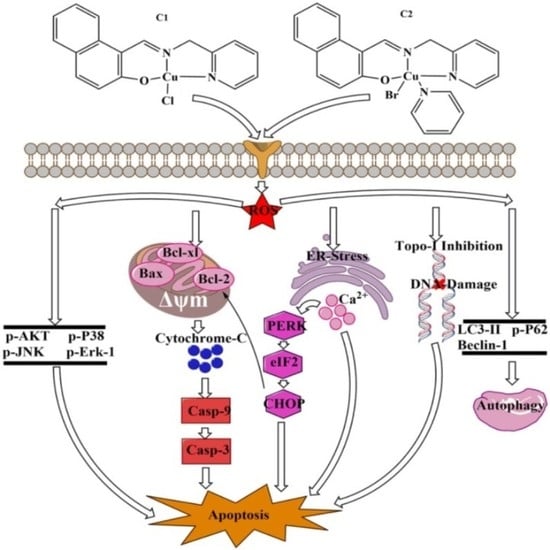
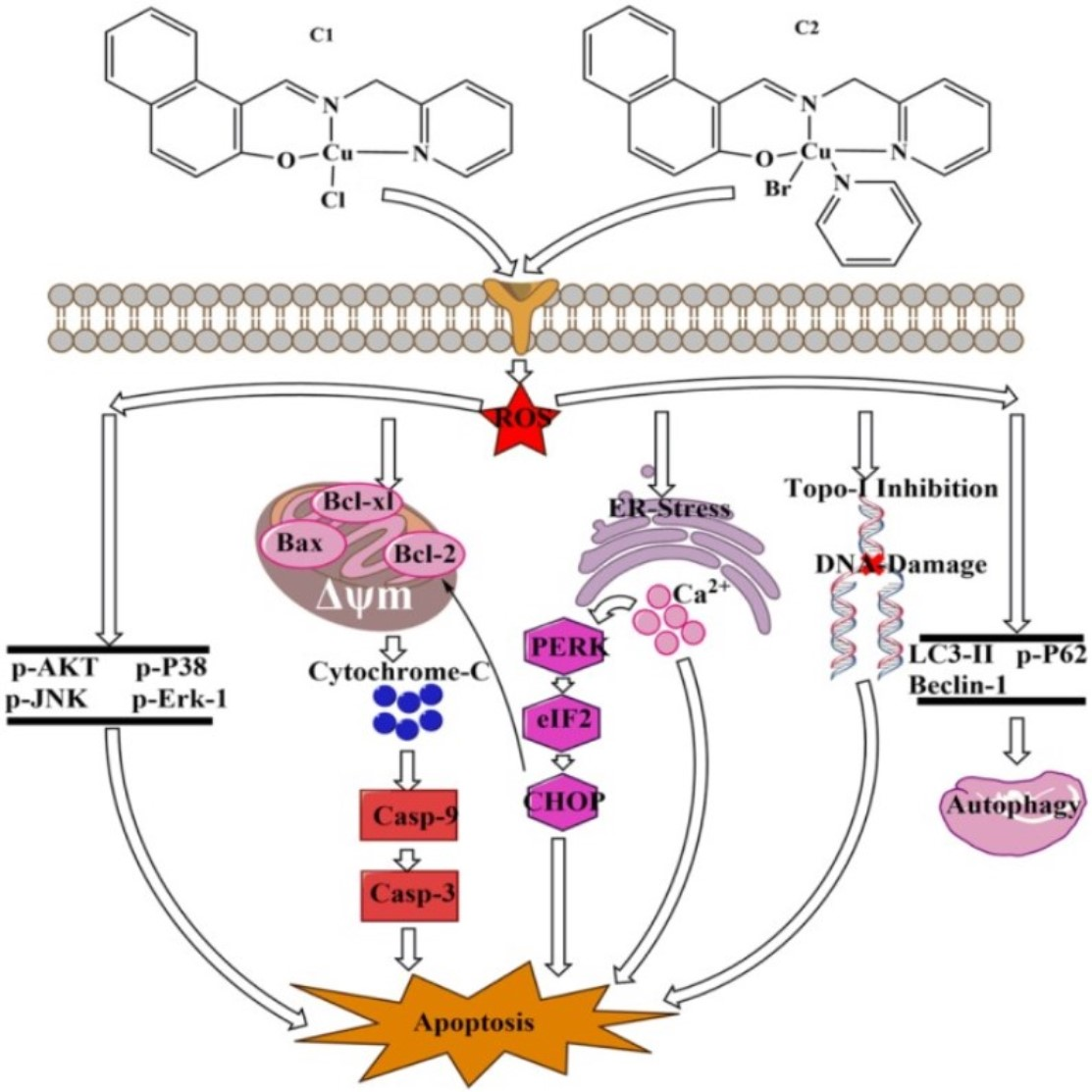
1. Introduction
2. Results
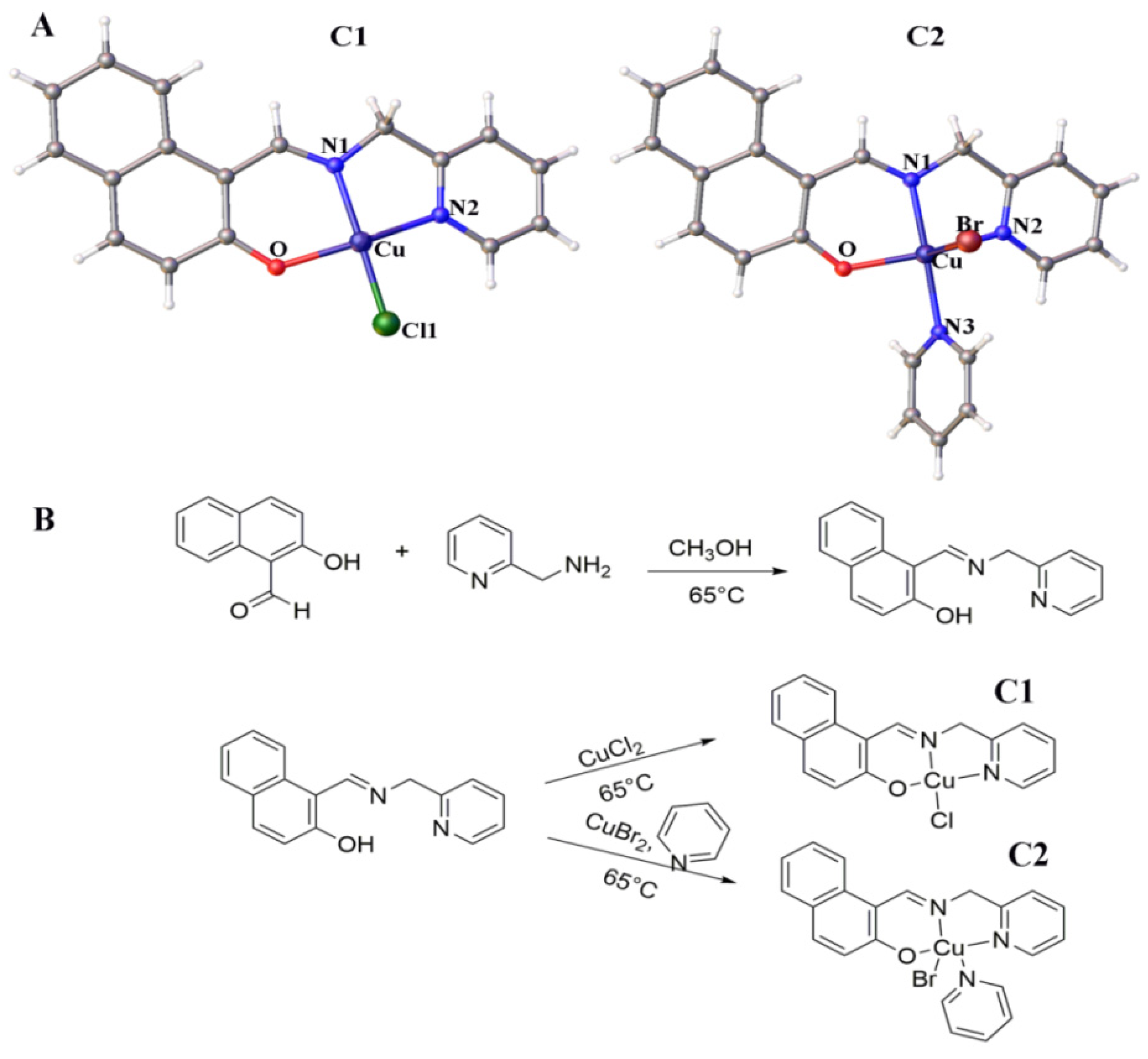
2.1. Development and Structure of Cu2+ Complexes
2.2. Integrality of Cu2+ Complexes in Solution
2.3. Anticancer Properties of C1 and C2
2.3.1. Comparative Cytotoxicity of C1 and C2 In Vitro
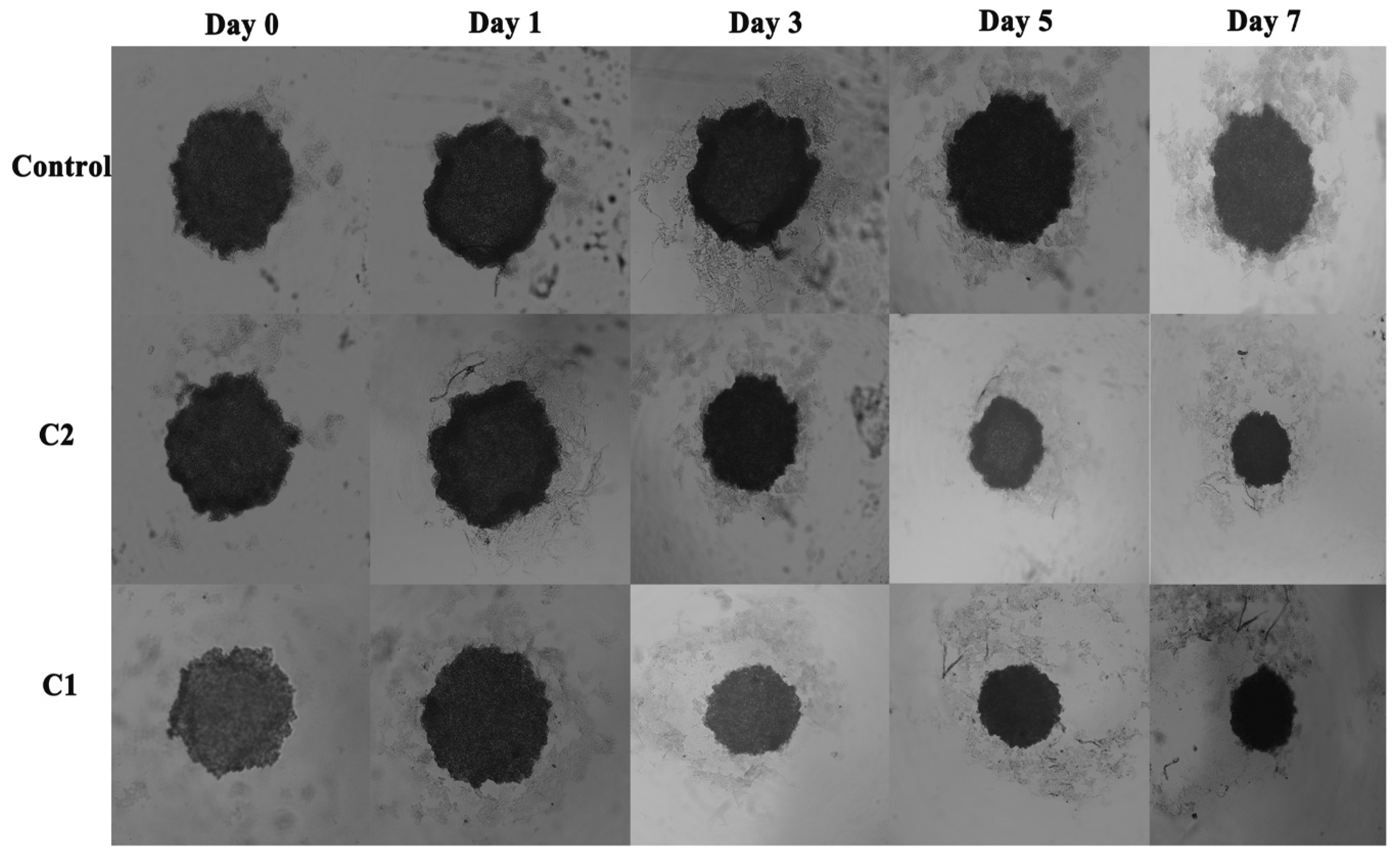
2.3.2. Comparative Influence of C1 and C2 on Spheroid Growth
2.4. ROS-Mediated Multi-Targeted Anticancer Mechanisms of Cu2+ Compounds
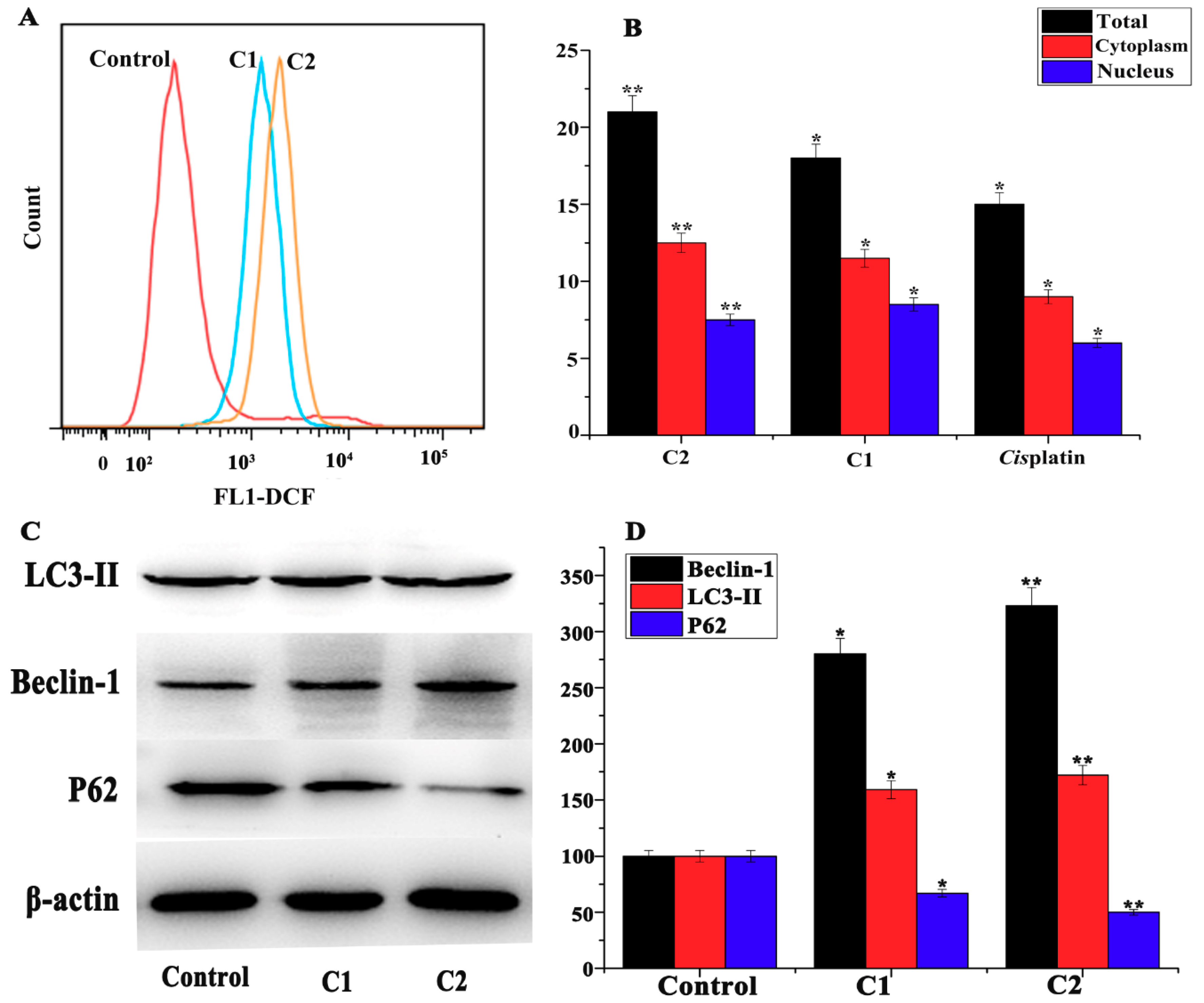
2.4.1. Analysis of the Stimulation of ROS in A-549 Cells
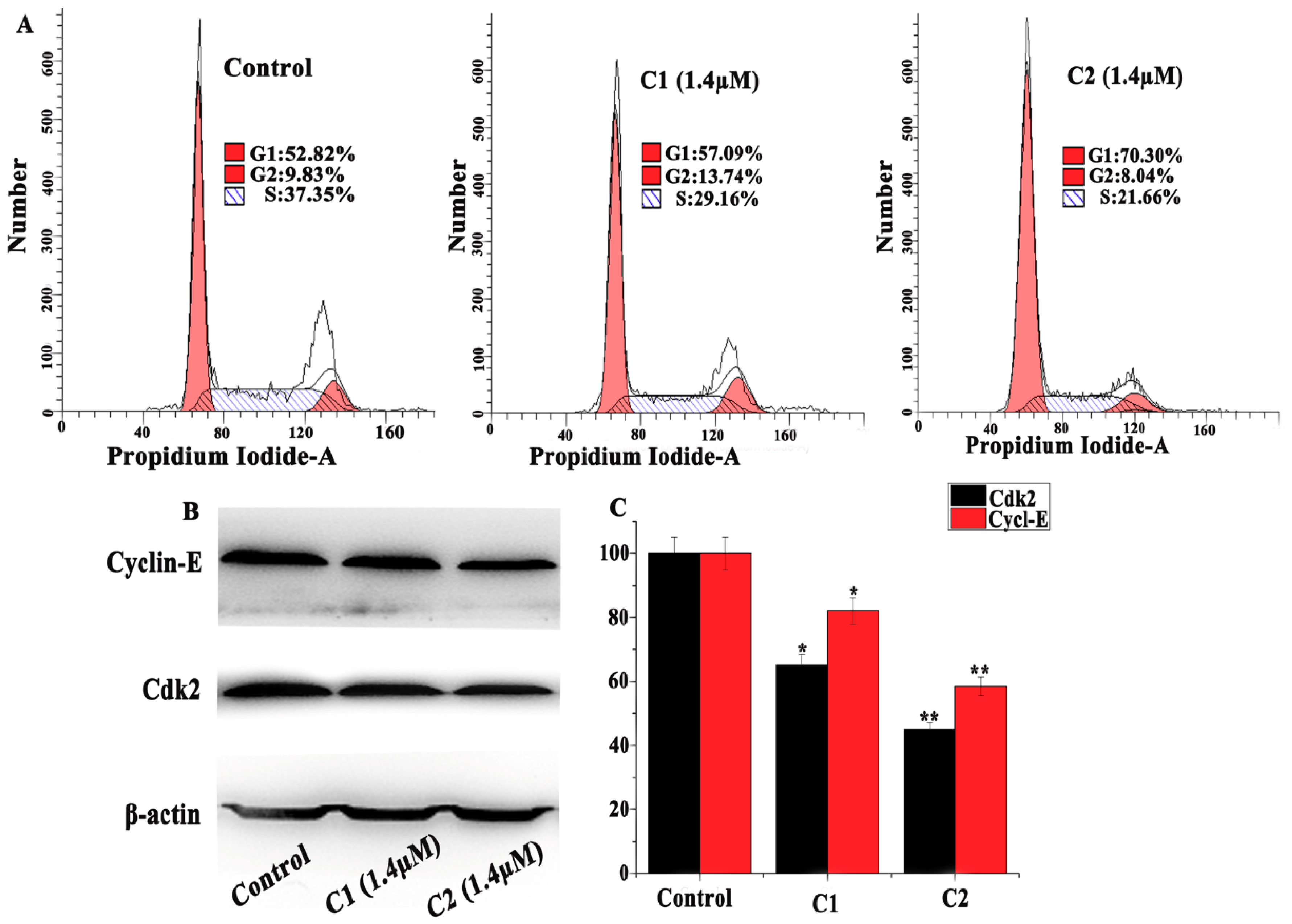
2.4.2. Regulation of the Cell Cycle
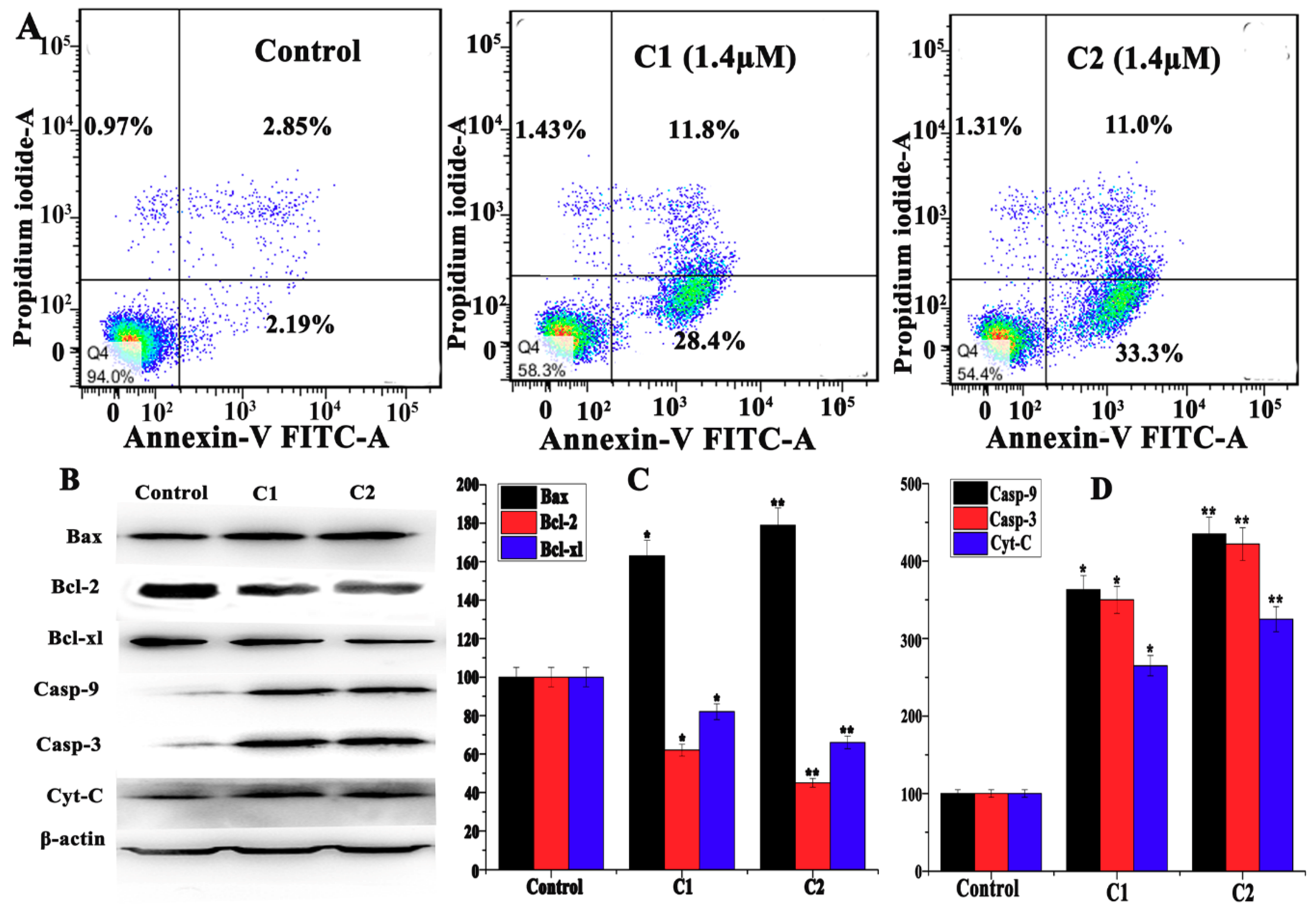
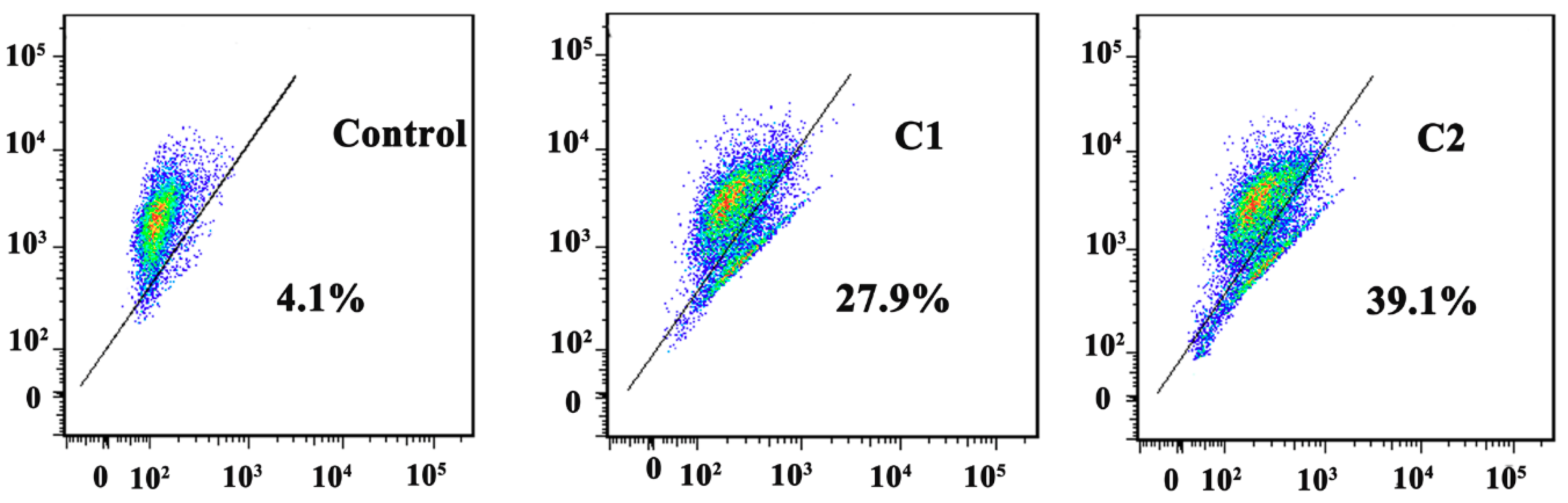
2.4.3. Study of Mitochondrial apoptosis in A-549 induced by Cu2+ Complexes
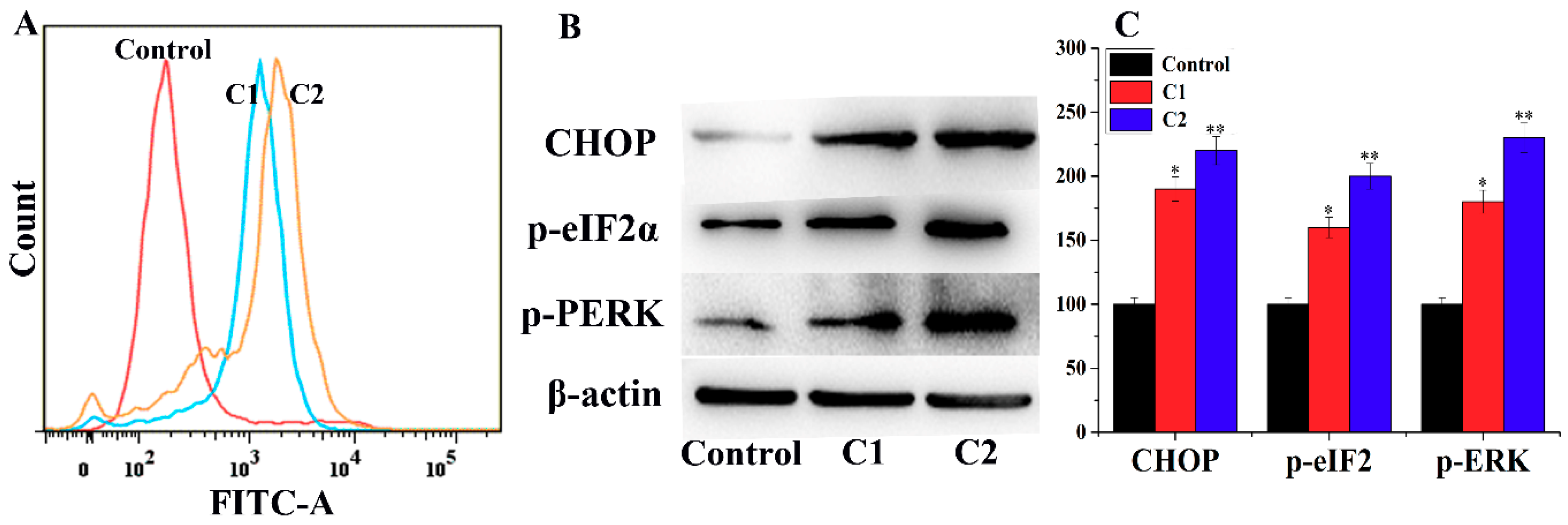
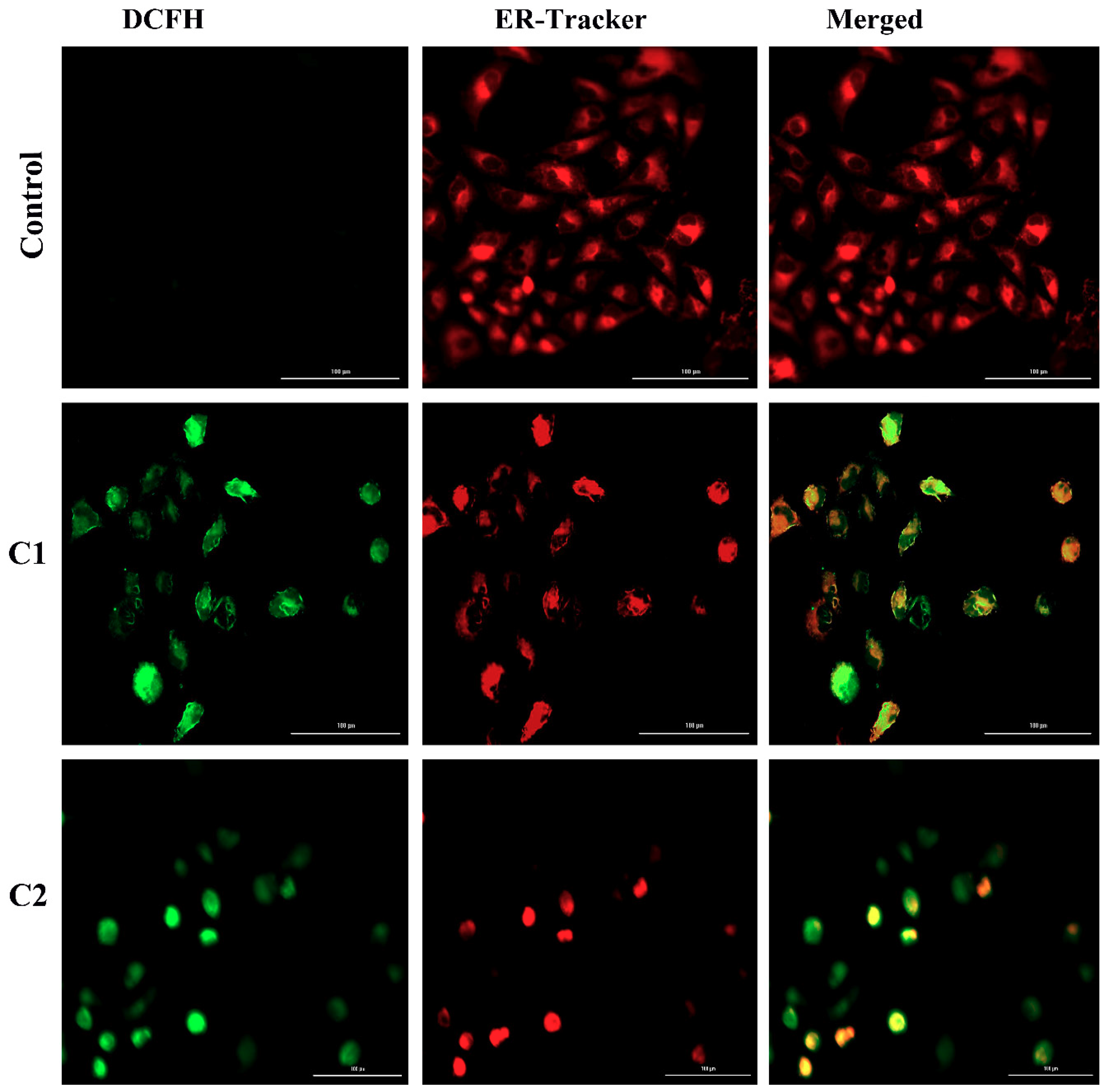
2.4.4. ROS-Mediated Endoplasmic Reticulum Stress Pathway
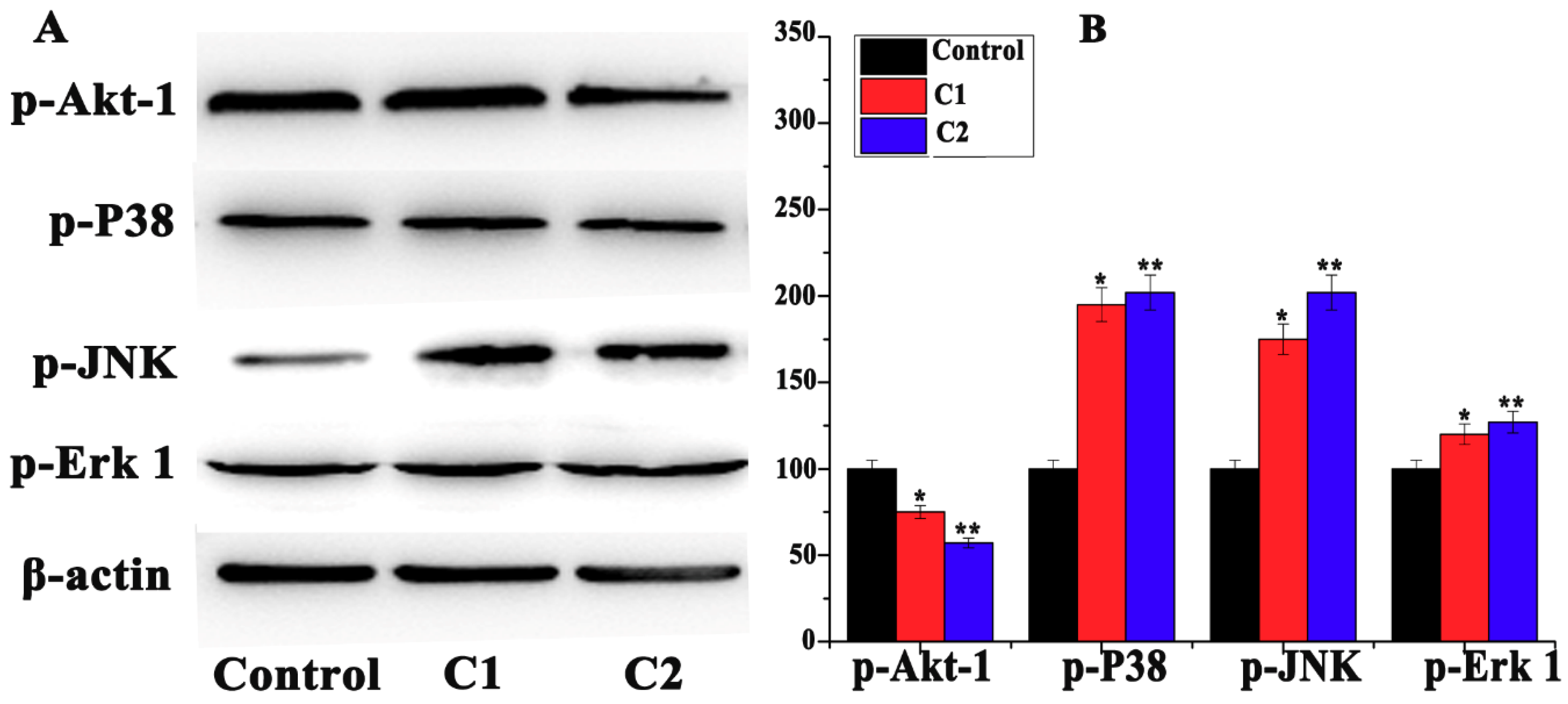
2.4.5. Mitogen-Activated Protein Kinase (MAPK) Pathway Study
2.4.6. Study of Autophagy in A-549 Cells
2.4.7. Topoisomerase I Inhibition
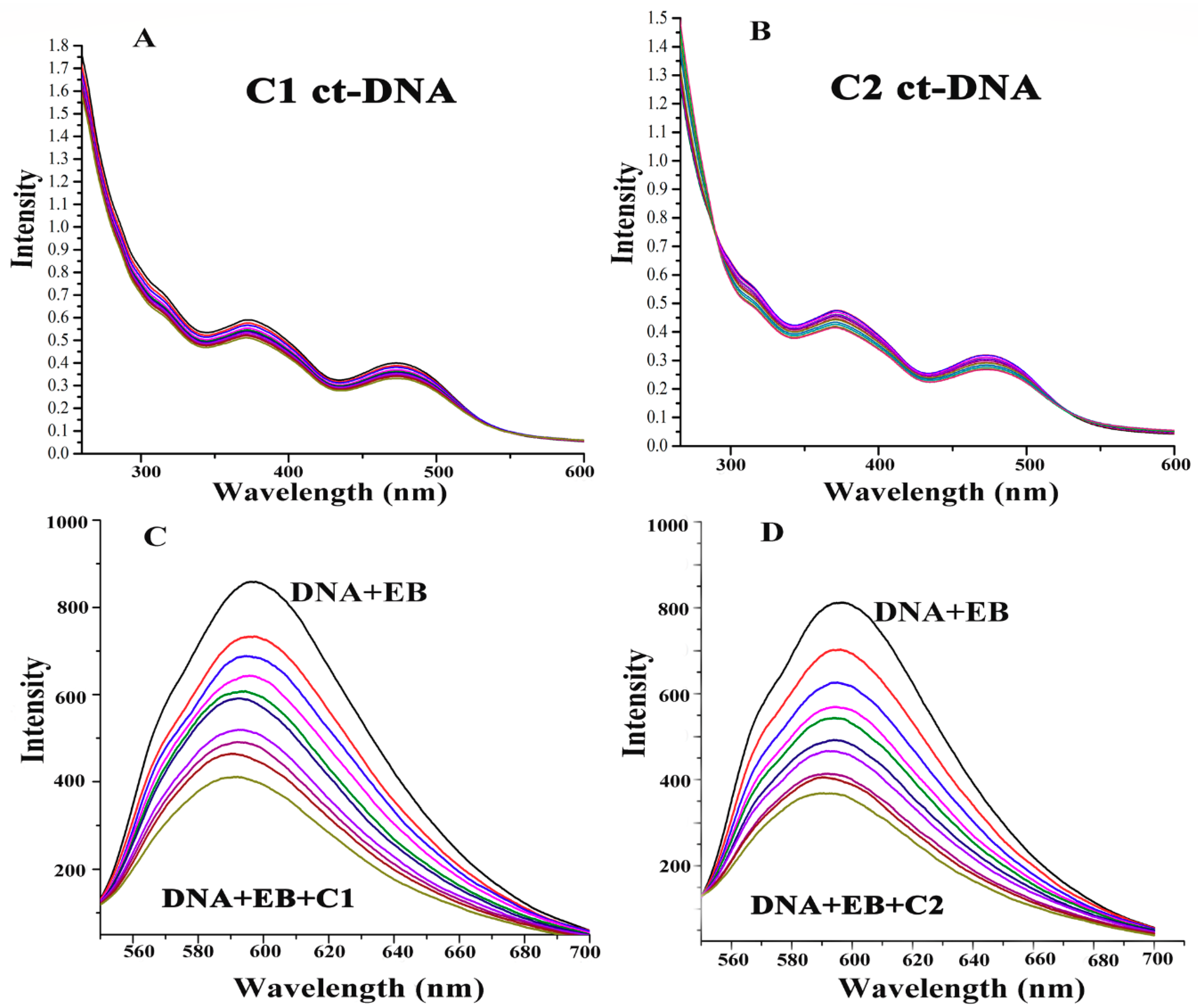
2.4.8. ROS-Mediated DNA Binding and Cleavage Study
3. Discussion
4. Experiments
4.1. Material and Methods
4.2. Development of Cu2+ Complexes
4.2.1. Synthesis of Ligand (L)
4.2.2. Synthesis of C1
4.2.3. Synthesis of C2
4.2.4. X-Ray Crystallography of C1 and C2
4.3. Anticancer Activity of the C1 and C2 Complexes
4.3.1. Cell Culture
4.3.2. Cytotoxicity Assay (MTT)
4.3.3. Tumor Spheroid Growth Inhibition
4.4. Determination of Possible Anticancer Mechanisms of Cu2+ Compounds
4.4.1. Cell Cycle Distribution Analysis
4.4.2. Apoptosis by Flow Cytometry
4.4.3. Mitochondrial Membrane Potential Assay
4.4.4. Intracellular ROS Assay
4.4.5. Visualization of ROS in the Endoplasmic Reticulum
4.4.6. Detection of Intracellular Ca2+ Ions
4.4.7. Topoisomerase I Inhibition Assay
4.4.8. DNA Binding and Cleavage Study
4.4.9. Western Blot Analysis
4.4.10. NE-PER Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R. Preclinical Perspectives on Platinum Resistance. Drugs 2000, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Grant, M.P.; Harper, B.W.; Krause-Heuer, A.M.; Manohar, M.; Orkey, N.; Aldrich-Wright, J.R. Transition Metal-Based Anticancer Drugs. Curr. Top. Med. Chem. 2011, 11, 521–542. [Google Scholar] [CrossRef] [PubMed]
- Shokohi-Pour, Z.; Chiniforoshan, H.; Momtazi-Borojeni, A.A.; Notash, B. A novel Schiff base derived from the gabapentin drug and copper (II) complex: Synthesis, characterization, interaction with DNA/protein and cytotoxic activity. J. Photochem. Photobiol. B Biol. 2016, 162, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, Z.; Salehi, M.; Khaleghian, A.; Kubicki, M. In vitro cytotoxic activity of a novel Schiff base ligand derived from 2-hydroxy-1-naphthaldehyde and its mononuclear metal complexes. J. Mol. Struct. 2018, 1173, 213–220. [Google Scholar] [CrossRef]
- Raman, N.; Jeyamurugan, R.; Senthilkumar, R.; Rajkapoor, B.; Franzblau, S.G. In vivo and in vitro evaluation of highly specific thiolate carrier group copper(II) and zinc(II) complexes on Ehrlich ascites carcinoma tumor model. Eur. J. Med. Chem. 2010, 45, 5438–5451. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Kong, S.M.; Tiong, Y.L.; Maah, M.J.; Sukram, N.; Ahmad, M.; Khoo, A.S.B. Selective anticancer copper(II)-mixed ligand complexes: Targeting of ROS and proteasomes. Metallomics 2014, 6, 892–906. [Google Scholar] [CrossRef]
- DeNoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Cantini, F.; Baffoni, S.C. Cellular copper distribution: A mechanistic systems biology approach. Cell. Mol. Life Sci. 2010, 67, 2563–2589. [Google Scholar] [CrossRef]
- Delgado, M.; Pérez-Miguelsanz, J.; Garrido, F.; Rodríguez-Tarduchy, G.; Pérez-Sala, D.; Pajares, M. Ángeles Early effects of copper accumulation on methionine metabolism. Cell. Mol. Life Sci. 2008, 65, 2080–2090. [Google Scholar] [CrossRef]
- MacPherson, I.S.; Murphy, M.E.P. Type-2 copper-containing enzymes. Cell. Mol. Life Sci. 2007, 64, 2887–2899. [Google Scholar] [CrossRef] [PubMed]
- Kowol, C.R.; Heffeter, P.; Miklos, W.; Gille, L.; Trondl, R.; Cappellacci, L.; Berger, W.; Keppler, B.K. Mechanisms underlying reductant-induced reactive oxygen species formation by anticancer copper (II) compounds. J. Biol. Inorg. Chem. 2012, 17, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.; Potapov, A.; Khlebnikov, A.; Korotkova, E.; Lukina, A.; Malovichko, G.; Kirpotina, L.; Quinn, M.T. Decomposition of reactive oxygen species by copper(II) bis(1-pyrazolyl)methane complexes. JBIC J. Boil. Inorg. Chem. 2006, 11, 499–513. [Google Scholar] [CrossRef]
- Potapov, A.S.; Nudnova, E.A.; Domina, G.A.; Kirpotina, L.N.; Quinn, M.T.; Khlebnikov, A.I.; Schepetkin, I.A. Synthesis, characterization and potent superoxide dismutase like activity of novel bis(pyrazole)—2,2’-bipyridyl mixed ligand copper(II) complexes. Dalton Trans. 2009, 23, 4488–4498. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, 1005–1028. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Donovan, M.; Cotter, T.G. Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 2002, 265, 49–72. [Google Scholar] [CrossRef]
- You, Z.-L.; Zhu, H.-L. (Nitrato-κ O)[1-(pyridin-2-ylmethyliminomethyl)naphthalen-2-olato-κ O ]copper(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, m1079–m1080. [Google Scholar] [CrossRef]
- Effect of cooperative non-covalent interactions on the solid statenheterochiral self-assembly: The concepts of isotactic and syndiotactic arrangements in coordination complex. Inorg. Chim. Acta 2014, 410, 156–170. [CrossRef]
- You, Z.-L.; Zhu, H.-L. Syntheses, Crystal Structures, and Antibacterial Activities of Four Schiff Base Complexes of Copper and Zinc. Z. Anorg. Allg. Chem. 2004, 630, 2754–2760. [Google Scholar] [CrossRef]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A review of the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11. [Google Scholar]
- Ali, K.A.; Abd-Elzaher, M.M.; Mahmoud, K. Synthesis and anticancer properties of silver (I) complexes containing 2, 6-Bis (substituted) pyridine derivatives. Int. J. Med. Chem. 2013, 2013, 256836. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, J.; Khan, M.; Yu, P.; Yang, F.; Liang, H. Structure and biological properties of five Pt(II) complexes as potential anticancer agents. J. Inorg. Biochem. 2018, 185, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Sant, S.; Johnston, P.A. The production of 3D tumor spheroids for cancer drug discovery. Drug Discov. Today Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive Oxygen Species, Cellular Redox Systems and Apoptosis. Free Radic. Boil. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD (P)+ oxidation–reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.-J.; Ye, S.-S.; Cao, N.; Huang, J.; Gao, J.; Chen, Q.-Y. ROS-mediated autophagy was involved in cancer cell death induced by novel copper(II) complex. Exp. Toxicol. Pathol. 2010, 62, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Gladden, A.B.; Diehl, J.A. Cell cycle progression without cyclin E/CDK2: Breaking down the walls of dogma. Cancer Cell 2003, 4, 160–162. [Google Scholar] [CrossRef]
- Ivanchuk, S.M.; Rutka, J.T. Regulation of the Cell Cycle and Interventional Developmental Therapeutics. In Handbook of Brain Tumor Chemotherapy; Elsevier BV: Amsterdam, The Netherlands, 2006; pp. 123–140. [Google Scholar]
- Wong, W.W.-L.; Puthalakath, H. Bcl-2 family proteins: The sentinels of the mitochondrial apoptosis pathway. IUBMB Life 2008, 60, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Créancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.-M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to p53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Wu, C.-C.; Bratton, S.B. Regulation of the Intrinsic Apoptosis Pathway by Reactive Oxygen Species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.-Y.; Wei, M.C.; Weiler, S.; A Flavell, R.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-XL Sequester BH3 Domain-Only Molecules Preventing BAX- and BAK-Mediated Mitochondrial Apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Inayat-Hussain, S.; Annuar, B.; Din, L.; Ali, A.; Ross, D. Loss of mitochondrial transmembrane potential and caspase-9 activation during apoptosis induced by the novel styryl-lactone goniothalamin in HL-60 leukemia cells. Toxicol. Vitr. 2003, 17, 433–439. [Google Scholar] [CrossRef]
- Diehl, J.A.; Haines, D.S.; Fuchs, S.Y.; Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and Implications of Reactive Oxygen Species Generation During the Unfolded Protein Response: Roles of Endoplasmic Reticulum Oxidoreductases, Mitochondrial Electron Transport, and NADPH Oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ye, J.; Alonso-Basanta, M.; Hahn, S.M.; Koumenis, C.; Dorsey, J.F. Modulation of CCAAT/Enhancer Binding Protein Homologous Protein (CHOP)-dependent DR5 Expression by Nelfinavir Sensitizes Glioblastoma Multiforme Cells to Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) *. J. Boil. Chem. 2011, 286, 29408–29416. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Naguro, I.; Runchel, C.; Ichijo, H. The roles of ASK family proteins in stress responses and diseases. Cell Commun. Signal. 2009, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Orton, R.J.; Sturm, O.E.; Vyshemirsky, V.; Calder, M.; Gilbert, D.R.; Kolch, W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem. J. 2005, 392, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.; Bibbs, L.; Ulevitch, R. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Boil. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, X.; Li, M.; Liu, J. P44/42 MAPK signal pathway-mediated hyperphosphorylation of paxillin and redistribution of E-cadherin was involved in microcystin-LR-reduced cellular adhesion in a human liver cell line. Chemosphere 2018, 200, 594–602. [Google Scholar] [CrossRef]
- Pearson, G. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar] [PubMed]
- Park, S.; Choi, S.-G.; Yoo, S.-M.; Son, J.H.; Jung, Y.-K. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014, 10, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, J.; Geng, J.; Nair, U.; Klionsky, D.J. Atg22 Recycles Amino Acids to Link the Degradative and Recycling Functions of Autophagy. Mol. Boil. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kawiak, A.; Piosik, J.; Stasilojc, G.; Gwizdek-Wiśniewska, A.; Marczak, L.; Stobiecki, M.; Bigda, J.; Lojkowska, E. Induction of apoptosis by plumbagin through reactive oxygen species-mediated inhibition of topoisomerase II. Toxicol. Appl. Pharmacol. 2007, 223, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. DNA Topoisomerases and Cancer; Springer: Berlin, Germany, 2012. [Google Scholar]
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Future Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yao, Q.; Tian, L.; Wang, Y. Piperidylthiosemicarbazones Cu(II) complexes with a high anticancer activity by catalyzing hydrogen peroxide to degrade DNA and promote apoptosis. Eur. J. Med. Chem. 2018, 158, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Bozas, G.; Kafantari, E.; Tsiatas, M.; Nikitas, N.; Dimopoulos, M.-A.; Papadimitriou, C.A. Endometrial Cancer: What Is New in Adjuvant and Molecularly Targeted Therapy? Obstet. Gynecol. Int. 2010, 2010, 749579. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Pevarello, P.; Barbacid, M.; Bischoff, J.R. CDK inhibitors in cancer therapy: What is next? Trends Pharmacol. Sci. 2008, 29, 16–21. [Google Scholar] [CrossRef]
- Krieger, M.L.; Eckstein, N.; Schneider, V.; Koch, M.; Royer, H.-D.; Jaehde, U.; Bendas, G. Overcoming cisplatin resistance of ovarian cancer cells by targeted liposomes in vitro. Int. J. Pharm. 2010, 389, 10–17. [Google Scholar] [CrossRef]
- He, F.; Wang, Q.; Zheng, X.-L.; Yan, J.-Q.; Yang, L.; Sun, H.; Hu, L.-N.; Lin, Y.; Wang, X. Wogonin potentiates cisplatin-induced cancer cell apoptosis through accumulation of intracellular reactive oxygen species. Oncol. Rep. 2012, 28, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qian, H.; Yiu, S.-M.; Sun, J.; Zhu, G. Multi-targeted organometallic ruthenium(II)–arene anticancer complexes bearing inhibitors of poly(ADP-ribose) polymerase-1: A strategy to improve cytotoxicity. J. Inorg. Biochem. 2014, 131, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Petrelli, A. From Single- to Multi-Target Drugs in Cancer Therapy: When Aspecificity Becomes an Advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef]
- Hong, M.; Yin, H.; Zhang, X.; Li, C.; Yue, C.; Cheng, S. Di- and tri-organotin(IV) complexes with 2-hydroxy-1-naphthaldehyde 5-chloro-2-hydroxybenzoylhydrazone: Synthesis, characterization and in vitro antitumor activities. J. Organomet. Chem. 2013, 724, 23–31. [Google Scholar] [CrossRef]
- Sugumar, P.; Butcher, R.J.; Vijayan, P.; Ponnuswamy, M.N.; Balakumaran, M.D.; Kalaichelvan, P.T.; Velmurugan, K.; Nandhakumar, R.; Viswanathamurthi, P. Unprecedented formation of organo-ruthenium(II) complexes containing 2-hydroxy-1-naphthaldehyde S-benzyldithiocarbazate: Synthesis, X-ray crystal structure, DFT study and their biological activities in vitro. Inorg. Chem. Front. 2015, 2, 620–639. [Google Scholar]
- Reddy, P.R.; Shilpa, A. 2-Hydroxynaphthalene-1-carbaldehyde-and 2-(Aminomethyl) pyridine-Based Schiff Base CuII Complexes for DNA Binding and Cleavage. Chem. Biodivers. 2012, 9, 2262–2281. [Google Scholar] [CrossRef]
- Puschmann, H.; Dolomanov, O.; Bourhis, L.; Gildea, R.; Howard, J. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | C1 | C2 |
|---|---|---|
| Empirical formula | C17H13ClCuN2O | C22H18BrCuN3O |
| Formula weight | 360.30 | 483.86 |
| Temperature/K | 296.15 | 296.15 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/n | P21/n |
| a/Å | 8.100(6) | 8.6992(9) |
| b/Å | 8.632(7) | 13.6084(14) |
| c/Å | 21.270(16) | 16.4250(18) |
| α/° | 90 | 90 |
| β/° | 100.245(12) | 90.070(2) |
| γ/° | 90 | 90 |
| Volume/Å3 | 1463.4(19) | 1944.4(4) |
| Z | 4 | 4 |
| ρcalcg/cm3 | 1.6353 | 1.6527 |
| μ(Mo-Kα) mm−1 | 1.677 | 3.196 |
| F(000) | 734.2 | 972.6 |
| Data/restraints/parameters | 3007/0/199 | 4211/0/253 |
| Goodness of fit on F2 | 1.054 | 1.027 |
| Final R indexes (I ≥ 2σ (I)) | R1 = 0.0421 | R1 = 0.0375 |
| wR2 = 0.1358 | wR2 = 0.1023 | |
| Largest diff. peak/hole /e Å-3 | 0.63/−0.69 | 0.86/−0.46 |
| Compounds IC50 ± SD (µM) | ||||
|---|---|---|---|---|
| A-549 | MGC-803 | T24 | HL-7702 | |
| L | >50 | >50 | >50 | >50 |
| C1 | 1.06 ± 0.01 | 1.98 ± 0.14 | 2.21 ± 0.21 | 11.02 ± 0.15 |
| C2 | 0.7 ± 0.01 | 1.17 ± 0.09 | 2.09 ± 0.26 | 10.05 ± 0.12 |
| Cisplatin | 21.74 ± 1.52 | 17.87 ± 0.35 | 22.21 ± 0.54 | 10.21 ± 0.24 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.H.; Cai, M.; Deng, J.; Yu, P.; Liang, H.; Yang, F. Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-hydroxy-1-naphthaldehyde Complexes. Molecules 2019, 24, 2544. https://doi.org/10.3390/molecules24142544
Khan MH, Cai M, Deng J, Yu P, Liang H, Yang F. Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-hydroxy-1-naphthaldehyde Complexes. Molecules. 2019; 24(14):2544. https://doi.org/10.3390/molecules24142544
Chicago/Turabian StyleKhan, Muhammad Hamid, Meiling Cai, Jungang Deng, Ping Yu, Hong Liang, and Feng Yang. 2019. "Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-hydroxy-1-naphthaldehyde Complexes" Molecules 24, no. 14: 2544. https://doi.org/10.3390/molecules24142544
APA StyleKhan, M. H., Cai, M., Deng, J., Yu, P., Liang, H., & Yang, F. (2019). Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-hydroxy-1-naphthaldehyde Complexes. Molecules, 24(14), 2544. https://doi.org/10.3390/molecules24142544