

The CK2/ECE1c Partnership: An Unveiled Pathway to Aggressiveness in Cancer

 , , and
, , and
Abstract
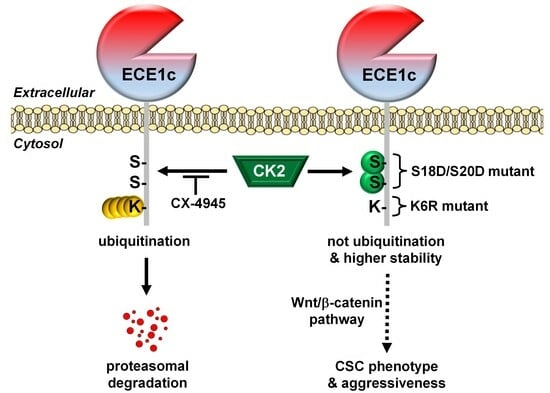
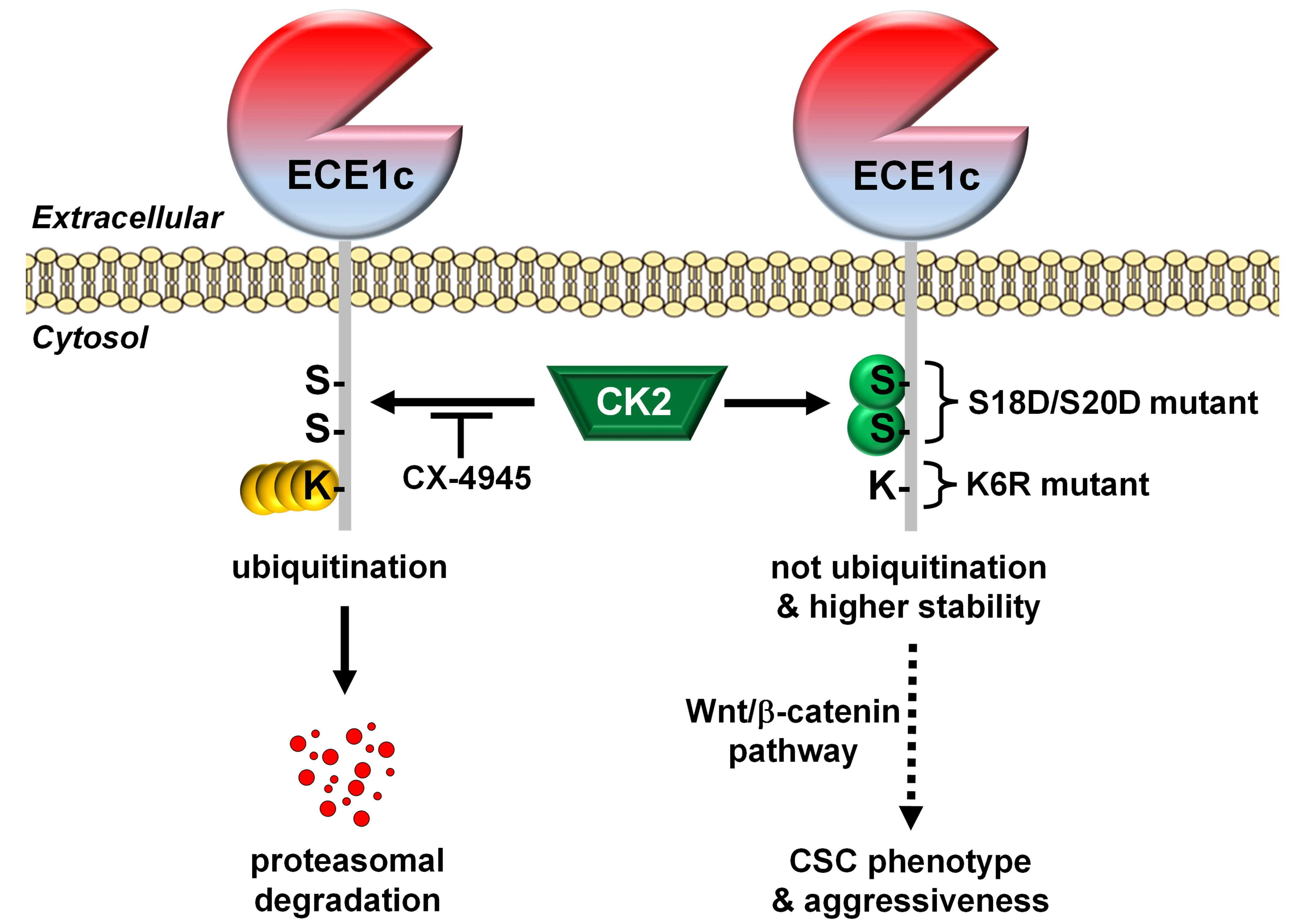
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
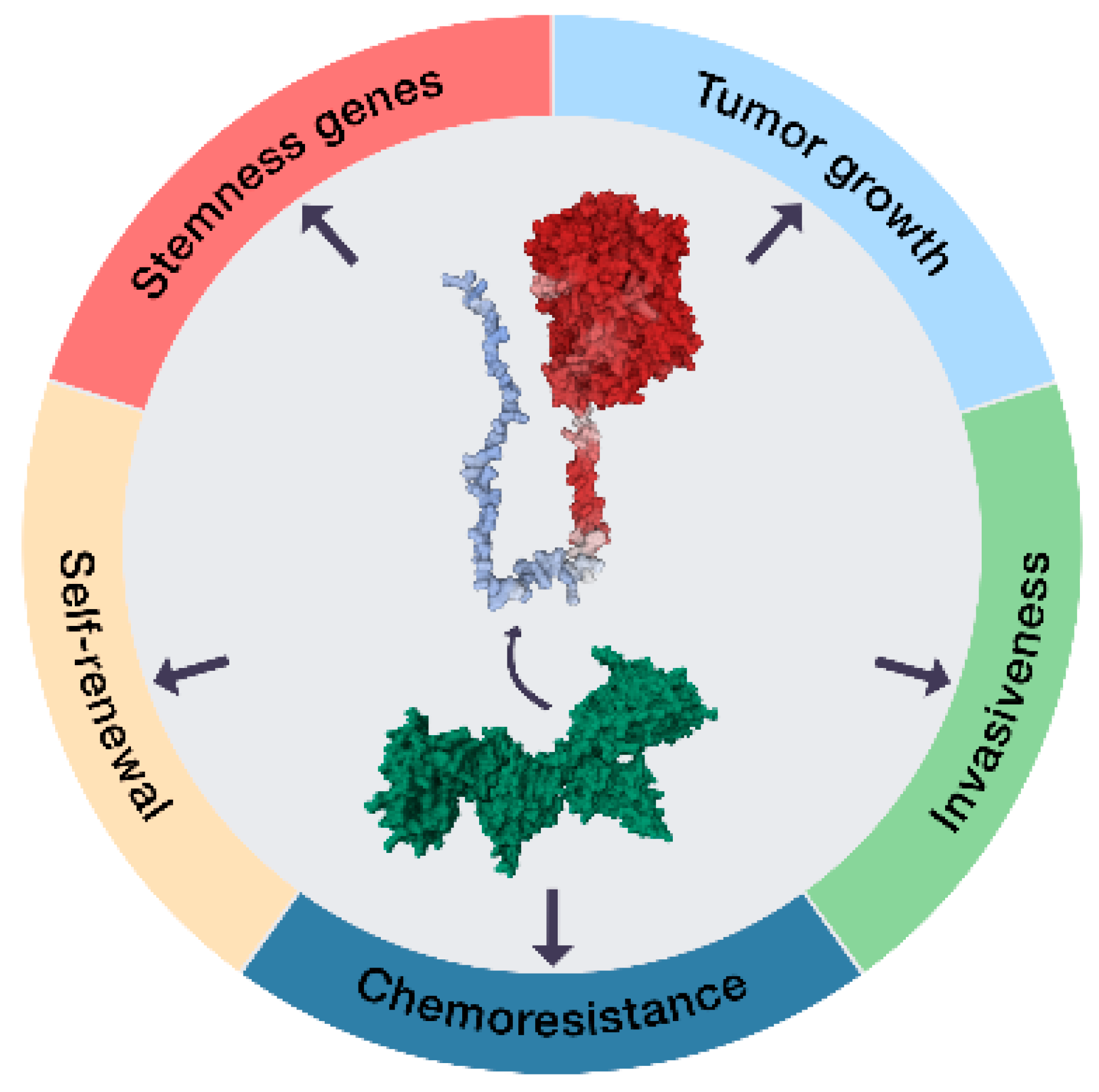
2. Results and Discussion
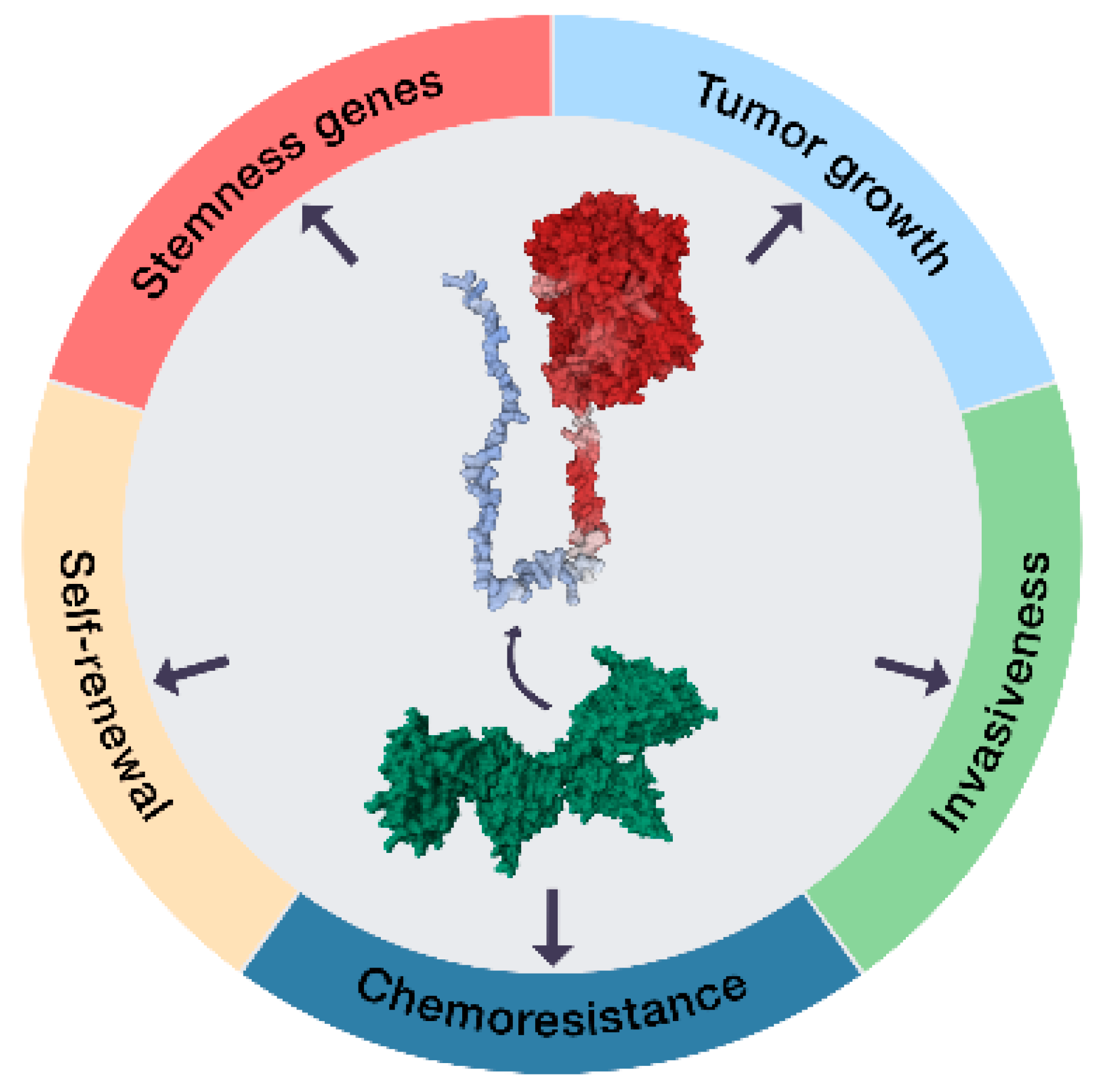
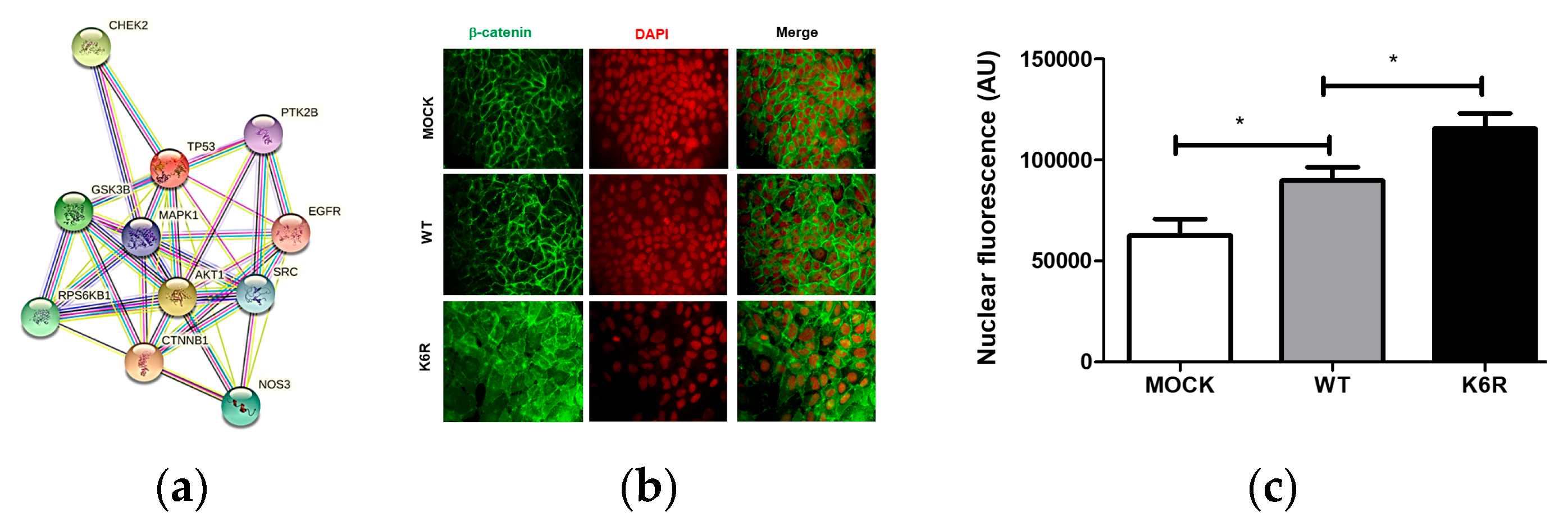
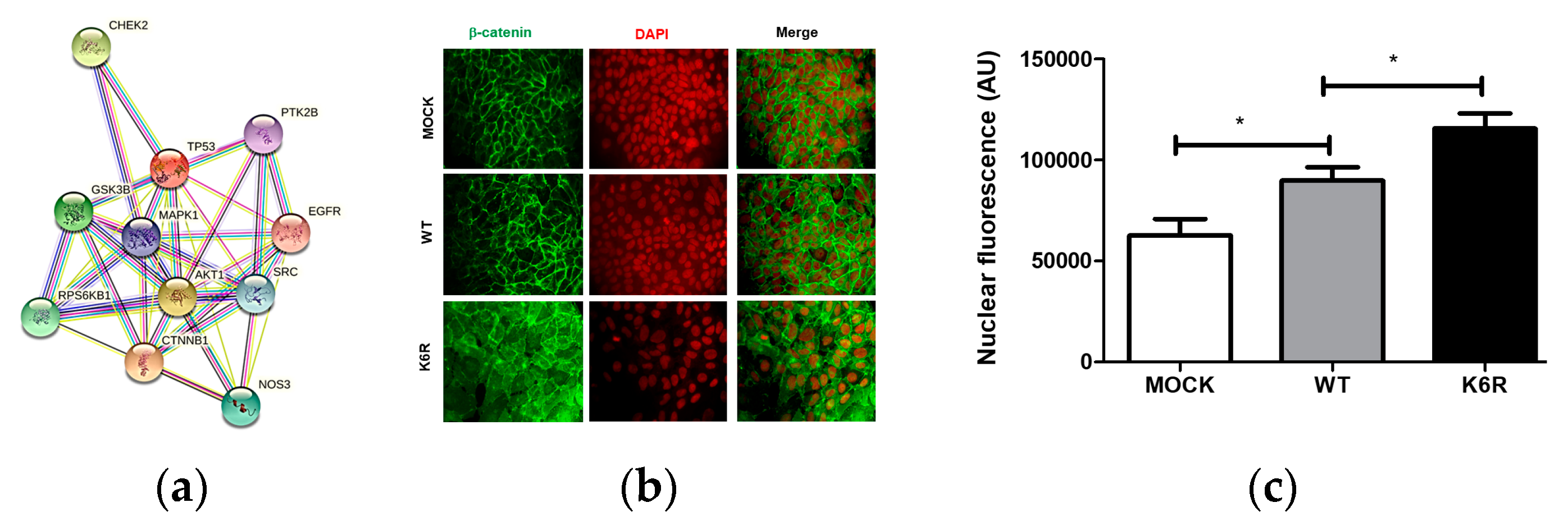
2.1. The ECE1c-Dependent Acquisition of a CSC Phenotype
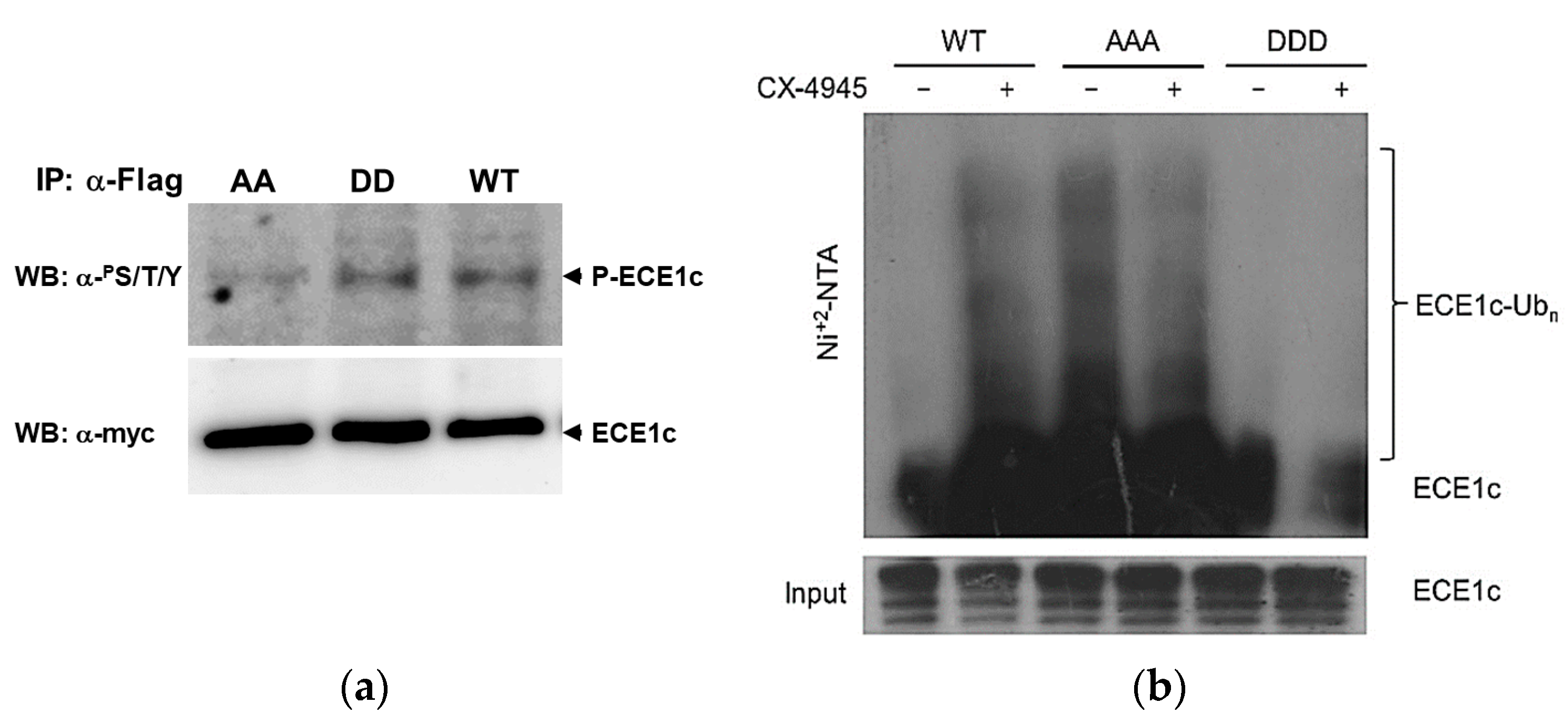
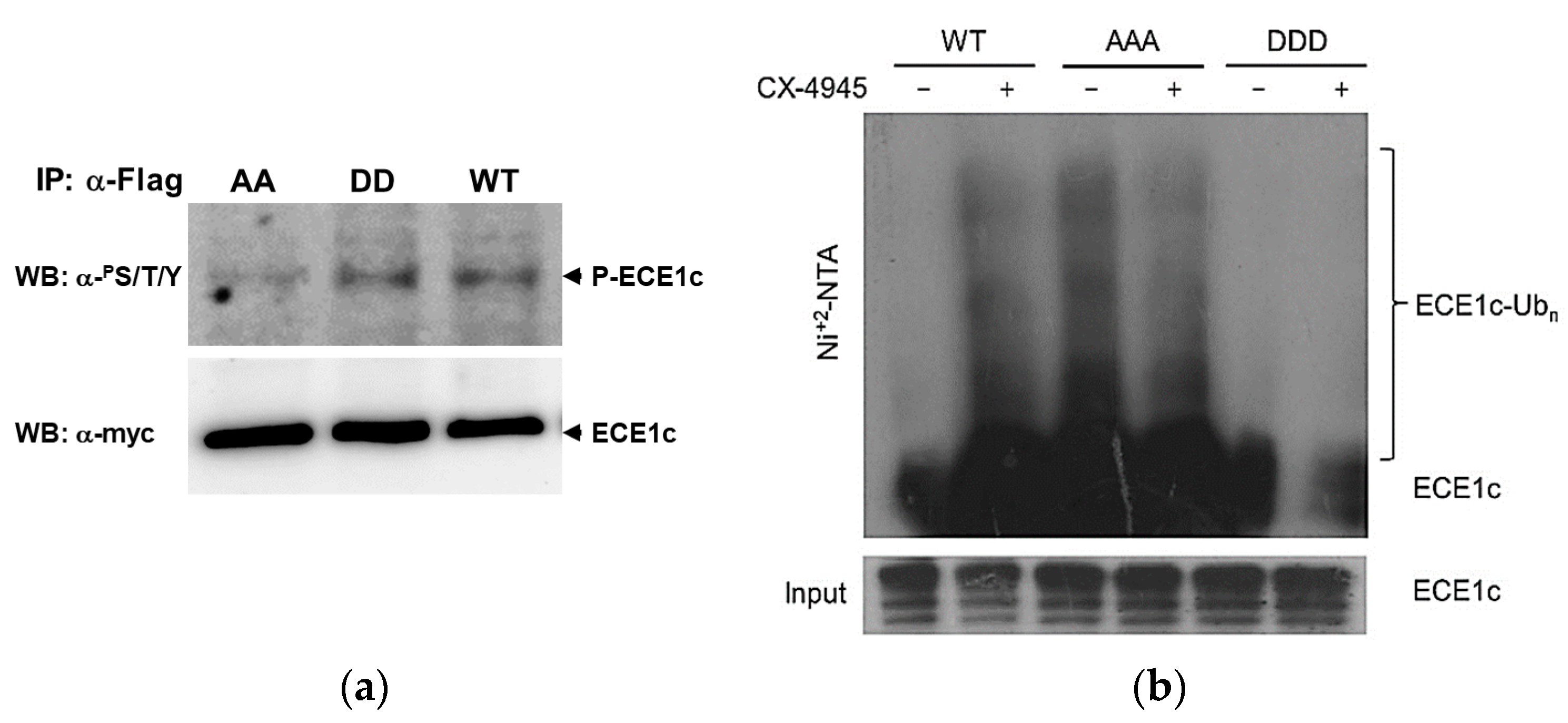
2.2. CK2’s Phosphorylation of ECE1c and Aggressiveness
2.3. The Non-Canonical ET1 Regulation of Signaling Proteins
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; Van Es, J.H.; Van De Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol. 2014, 20, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Yanagisawa, M. Endothelin: 20 Years from Discovery to Therapy. Can J. Physiol. Pharmacol. 2008, 86, 485–498. [Google Scholar] [CrossRef]
- Kawanabe, Y.; Nauli, S.M. Endothelin. Cell. Mol. Life Sci. 2011, 68, 195–203. [Google Scholar] [CrossRef]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L. The endothelin axis in cancer. Int. J. Biochem. Cell Biol. 2008, 40, 1443–1451. [Google Scholar] [CrossRef]
- Khimji, A.-K.; Rockey, D.C. Endothelin—Biology and disease. Cell. Signal. 2010, 22, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Jafri, F.; Ergul, A. Nuclear Localization of Endothelin-Converting Enzyme-1: Subisoform Specificity. Arter. Thromb. Vasc. Biol. 2003, 23, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Funke-Kaiser, H.; Thomas, A.; Bremer, J.; Kovacevic, S.D.; Scheuch, K.; Bolbrinker, J.; Theis, S.; Lemmer, J.; Zimmermann, A.; Zollmann, F.S.; et al. Regulation of the major isoform of human endothelin-converting enzyme-1 by a strong housekeeping promoter modulated by polymorphic microsatellites. J. Hypertens. 2003, 21, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Lindenau, S.; von Langsdorff, C.; Saxena, A.; Paul, M.; Orzechowski, H.-D. Genomic organisation of the mouse gene encoding endothelin-converting enzyme-1 (ECE-1) and mRNA expression of ECE-1 isoforms in murine tissues. Gene 2006, 373, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Niechi, I. Endothelin-converting enzyme-1 in cancer aggressiveness. Cancer Lett. 2019, 452, 152–157. [Google Scholar] [CrossRef]
- Arun, C.; London, N.; Hemingway, D. Prognostic Significance of Elevated Endothelin-1 Levels in Patients with Colorectal Cancer. Int. J. Biol. Markers 2004, 19, 32–37. [Google Scholar] [CrossRef]
- Simpson, R.A.; Dickinson, T.; Porter, K.E.; London, N.J.M.; Hemingway, D.M. Raised levels of plasma big endothelin 1 in patients with colorectal cancer. Br. J. Surg. 2000, 87, 1409–1413. [Google Scholar] [CrossRef]
- Arun, C.; Swift, B.; Porter, K.; West, K.; London, N.; Hemingway, D. The Role of Big Endothelin-1 in Colorectal Cancer. Int. J. Biol. Markers 2002, 17, 268–274. [Google Scholar] [CrossRef]
- Lambert, L.A.; Whyteside, A.R.; Turner, A.J.; Usmani, B.A. Isoforms of endothelin-converting enzyme-1 (ECE-1) have opposing effects on prostate cancer cell invasion. Br. J. Cancer 2008, 99, 1114–1120. [Google Scholar] [CrossRef]
- Niechi, I.; Silva, E.; Cabello, P.; Huerta, H.; Carrasco, V.; Villar, P.; Cataldo, L.R.; Marcelain, K.; Armisen, R.; Varas-Godoy, M.; et al. Colon cancer cell invasion is promoted by protein kinase CK2 through increase of endothelin-converting enzyme-1c protein stability. Oncotarget 2015, 6, 42749–42760. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta BBA Proteins Proteom. 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Perez-Oliva, A.B.; Cozza, G.; Gourlay, R.; Weidlich, S.; Campbell, D.G.; Pinna, L.A.; Sapkota, G.P. Casein kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser 16 to trigger its nuclear localization. Sci. Signal. 2015, 8, ra35. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Litchfield, D.W. From Birth to Death: The Role of Protein Kinase CK2 in the Regulation of Cell Proliferation and Survival. Cell. Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta BBA—Proteins Proteom. 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Qu, L.; Ding, Y. Protein and mRNA Characterization in Human Colorectal Carcinoma Cell Lines with Different Metastatic Potentials. Cancer Investig. 2007, 25, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-Y.; Tai, C.; Hsu, J.-C.; Li, C.-F.; Fang, C.-L.; Lai, H.-C.; Hseu, Y.-C.; Lin, Y.-F.; Uen, Y.-H. Overexpression of Nuclear Protein Kinase CK2 α Catalytic Subunit (CK2α) as a Poor Prognosticator in Human Colorectal Cancer. PLoS ONE 2011, 6, e17193. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Ruzzene, M. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 287. [Google Scholar] [CrossRef]
- Borgo, C.; D’amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Pérez-Moreno, P.; Indo, S.; Niechi, I.; Huerta, H.; Cabello, P.; Jara, L.; Aguayo, F.; Varas-Godoy, M.; Burzio, V.A.; Tapia, J.C. Endothelin-converting enzyme-1c promotes stem cell traits and aggressiveness in colorectal cancer cells. Mol. Oncol. 2020, 14, 347–362. [Google Scholar] [CrossRef]
- Niechi, I.; Erices, J.I.; Carrillo-Beltrán, D.; Uribe-Ojeda, A.; Torres, Á.; Rocha, J.D.; Uribe, D.; Toro, M.A.; Villalobos-Nova, K.; Gaete-Ramírez, B.; et al. Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells. Cells 2023, 12, 506. [Google Scholar] [CrossRef] [PubMed]
- Almarza, C.; González, M.; Toro, M.; Villalobos-Nova, K.; Niechi, I.; Silva-Pavez, E.; López-Muñoz, R.A.; Varas-Godoy, M.; Burzio, V.A.; Jara, L.; et al. Cisplatin-Resistance and Aggressiveness Are Triggered upon Expression of a Super-Stable Endothelin-Converting Enzyme-1c in Lung Cancer Cells. Unpublished manuscript.
- Pérez-Moreno, P.; Quezada-Meza, C.; Chavez-Almarza, C.; Niechi, I.; Silva-Pavez, E.; Trigo-Hidalgo, C.; Aguayo, F.; Jara, L.; Cáceres-Verschae, A.; Varas-Godoy, M.; et al. Phosphorylation of Endothelin-Converting Enzyme-1c at Serines 18 and 20 by CK2 Promotes Aggressiveness Traits in Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Marin, O.; Sarno, S.; Boschetti, M.; Pagano, M.A.; Meggio, F.; Ciminale, V.; D’Agostino, D.M.; Pinna, L.A. Unique features of HIV-1 Rev protein phosphorylation by protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 2000, 481, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Bolanos-Garcia, V.M.; Sayed, M.; Allende, C.C.; Allende, J.E. Cell cycle regulatory protein p27KIP1 is a substrate and interacts with the protein kinase CK2. J. Cell. Biochem. 2004, 91, 865–879. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Smerdon, S.J.; Yaffe, M.B. Recognition of Phospho-Serine/Threonine Phosphorylated Proteins by Phos-pho-Serine/Threonine-Binding Domains. In Handbook of Cell Signaling, 2nd ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 2, pp. 539–550. [Google Scholar] [CrossRef]
- Yaffe, M.B.; Smerdon, S.J. PhosphoSerine/Threonine Binding Domains: You Can’t pSERious? Structure 2001, 9, R33–R38. [Google Scholar] [CrossRef]
- Almawi, A.W.; Matthews, L.A.; Guarné, A. FHA domains: Phosphopeptide binding and beyond. Prog. Biophys. Mol. Biol. 2017, 127, 105–110. [Google Scholar] [CrossRef]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 Proteins: Structure, Function, and Regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef]
- Wang, B.; Li, X.; Liu, L.; Wang, M. β-Catenin: Oncogenic role and therapeutic target in cervical cancer. Biol. Res. 2020, 53, 33. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, J.; Fukunaga, Y.; Abelardo, E.; Nagafuchi, A. Defining the function of β-catenin tyrosine phosphorylation in cadherin-mediated cell–cell adhesion. Genes Cells 2008, 13, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Su, M.; Zhu, Y.; Zhou, Y.; Soomro, S.H.; Fu, H. Protein Kinase CK2, a Potential Therapeutic Target in Carcinoma Management. Asian Pac. J. Cancer Prev. 2019, 20, 23–32. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalobos-Nova, K.; Toro, M.d.l.Á.; Pérez-Moreno, P.; Niechi, I.; Tapia, J.C. The CK2/ECE1c Partnership: An Unveiled Pathway to Aggressiveness in Cancer. Kinases Phosphatases 2024, 2, 1-8. https://doi.org/10.3390/kinasesphosphatases2010001
Villalobos-Nova K, Toro MdlÁ, Pérez-Moreno P, Niechi I, Tapia JC. The CK2/ECE1c Partnership: An Unveiled Pathway to Aggressiveness in Cancer. Kinases and Phosphatases. 2024; 2(1):1-8. https://doi.org/10.3390/kinasesphosphatases2010001
Chicago/Turabian StyleVillalobos-Nova, Karla, María de los Ángeles Toro, Pablo Pérez-Moreno, Ignacio Niechi, and Julio C. Tapia. 2024. "The CK2/ECE1c Partnership: An Unveiled Pathway to Aggressiveness in Cancer" Kinases and Phosphatases 2, no. 1: 1-8. https://doi.org/10.3390/kinasesphosphatases2010001
APA StyleVillalobos-Nova, K., Toro, M. d. l. Á., Pérez-Moreno, P., Niechi, I., & Tapia, J. C. (2024). The CK2/ECE1c Partnership: An Unveiled Pathway to Aggressiveness in Cancer. Kinases and Phosphatases, 2(1), 1-8. https://doi.org/10.3390/kinasesphosphatases2010001