Abstract
Protein kinase CK2 (CK2) influences one-fifth of the cellular phosphoproteome. It regulates almost all cellular pathways and is thus a critical switch between biological processes within a cell. Inhibition of CK2 reverses oncogene addiction of tumor and alters tumor microenvironment. The success of this strategy and its clinical translation opens new opportunities. Targeting CK2 in musculoskeletal disorders is promising. Clinical manifestations of these disorders include dysfunctional inflammation, dysregulated cell differentiation, and senescence. Processes regulated by CK2 include all of these. Its emerging role in senescence also indicates its function’s centrality in cellular metabolism. This review summarizes considerations for targeting CK2 in musculoskeletal disorders. We have discussed the implications of CK2-regulated processes in musculoskeletal disorders.
1. Introduction
Musculoskeletal disorders affect skeletal muscle, bone, cartilage, and connective tissue. Disorders such as osteoporosis (OP), osteoarthritis (OA), and rheumatoid arthritis (RA) can cause chronic pain, hamper mobility, and reduce quality of life. They are also among the costliest co-morbidities [1]. Traditionally, musculoskeletal disorders were identified as structural dysfunctions of respective physiologies. However, recently, metabolic dysregulation has come up as a significant risk factor in these diseases. Metabolic dysregulation is caused by the defective regulation of biochemical pathways required for homeostasis [2,3]. Emerging studies are investigating the underlying mechanism by which metabolic disorders increase the severity of musculoskeletal disorders. Rheumatoid arthritis (RA) is an autoimmune disorder affecting joints. Researchers are exploring the link between autoimmunity and altered metabolism [4,5]. Likewise, osteoarthritis (OA) is characterized by the progressive degeneration of joints. The pathogenesis of OA is affected by metabolic disorders [6,7]. Small molecular metabolites are assessed for OA-associated biomarker identification [8]. Altered lipid metabolism and immune response during obesity increase the severity of OA symptoms [9]. Osteoporosis (OP) is a disorder associated with low bone mineral density. Several studies investigate the complex relationship between bone homeostasis and metabolic disorders, especially in the case of OP [10,11,12,13,14,15]. Patients diagnosed with OP are at a higher risk of fractures. An impaired immune system affects the natural process of fracture healing [16]. Defects in fracture healing in obese and insulin-resistant individuals are attributed to the dysregulated innate immunity during the healing-associated inflammatory response [17].
Protein kinase CK2 (formerly called “Casein Kinase 2”) is a ubiquitous serine/threonine kinase. The kinase has no extracellular activator [18]. It is not regulated hierarchically by other kinases of vertical signaling cascades. Thus, the regulatory role of CK2 is not limited to the activation of molecular signaling from the plasma membrane to the nucleus. Instead, it crosswise associates and integrates multiple pathways [19,20]. Hence, it is a ‘Lateral player’ in many biochemical pathways [21,22]. Expression and activity of CK2 are dysregulated in musculoskeletal disorders [23]. Deviation in metabolic processes affects tissue homeostasis and response to endocrine regulation [24]. The role of the inflammatory system and senescence in these diseases is also linked with metabolic dysregulation [25,26]. Moreover, the role of CK2 in modulating immune response, adipogenesis, and mitochondrial function has long been studied [27,28,29]. The kinase can identify and phosphorylate a vast range of substrates, estimated to be more than 400 today [30,31]. It is predicted to account for almost 20% of the phosphoproteome of cells [32]. Since CK2 is a lateral player, the effects of its dysregulation are manifold.
CK2 is ubiquitously present and expressed in almost all types of cells. The mechanism of pathway regulation by CK2 is different from that of other kinases. The following factors summarize the complexity of CK2’s function. First, while most kinases are activated in response to particular stimuli, CK2 is constitutively active [33] and requires no specific stimulus [18,34,35]. Second, the localization and form of CK2 change based on cell type and metabolic status [36]. CK2 has two catalytic subunits, CK2α and CK2α’, and a regulatory CK2β subunit. In vivo, it exists as a heterotetramer [37]. The tetramer form of CK2 is also termed CK2 holoenzyme. Holoenzyme comprises catalytic subunits α and α’ and two regulatory β subunits. The kinetics of holoenzyme formation and cellular distribution of these subunits change under different stimuli [38]. Third, the β subunit is not strictly a regulatory subunit. It is required to recognize substrates and structural stability of catalytic subunits. However, few substrates are phosphorylated without the β subunit, which makes its role elusive. Fourth, the catalytic subunits CK2α and CK2α’ are almost the same in structure, but their involvement in catalytic activity is different [39]. One is preferred over the other in some processes. Finally, the expression of subunits is regulated by various growth factors, and a single subunit’s expression level affects the expression and activity of other subunits.
The only clinically approved small molecular inhibitor of CK2, Silmitasertib (CX-4945), is a first-in-class small molecule of its kind [40]. It is an ATP-competitive inhibitor with the highest level of selectivity for CK2. Its use in cancer therapies is under investigation. However, like most currently available CK2 inhibitors, it inhibits CK2 catalytic activity completely. Controlling the inhibition of CK2 activity during specific cellular processes while sparing other activity is not possible. Furthermore, complete inhibition of the catalytic activity of CK2 causes cytotoxicity. This is due to the cells’ dependence on the basal phosphorylation level of CK2 substrates for survival [35]. Cytotoxicity is not always desirable, especially in treating musculoskeletal disorders. The technology of modulating the phosphorylation of CK2 substrates taking part in disease pathogenesis rather than inhibiting overall kinase activity is emerging. Focusing on specific CK2–substrate interaction will help bring a pathology-tailored approach for musculoskeletal disorders [41].
One such peptide inhibitor of CK2 named CIGB300 inhibits phosphorylation of specific CK2 substrates. This synthetic peptide binds to the phosphoacceptor domain of CK2 [42]. It was first discovered as an effective peptide in treating cervical cancer. It is also effective in treating Large Cell Lung Carcinoma (LCLC), Non-Small Cell Lung Cancer (NSCLC), advanced cervical cancer, and acute myeloid leukemia [43,44,45,46]. Due to its substrate-specific inhibition, it is highly effective and has mild side effects. This drug is under investigation in Phase 2 clinical trials, and studies about its effectiveness will soon be available [29]. The substrate-specific rather than global inhibitory effect of CIGB300 is thus emerging in cancer treatment.
Disease-modifying drugs help reverse the symptoms of disorders; they do so because they are designed to target molecular interactions critical for pathogenesis [47]. This approach re-directs metabolic pathways towards homeostasis. Few disease-modifying drugs, like Sprifermin (recombinant human FGF18, rhFGF18) for OA and methotrexate for RA, are available to treat musculoskeletal diseases [48,49]. Interventions that can restore dysregulated metabolism in these diseases are necessary [47]. CK2 is a lateral player, restoring its activity will effectively re-direct the metabolism to a healthy state. However, Targeting CK2 is challenging due to the complexity of its function [21,22,50]. Efforts to reveal the complexities of CK2 function in musculoskeletal disorders will help design disease-modifying therapeutics.
For the development of new drugs, the success rate of candidate molecules to move from bench to bedside is only 10%, despite the robustness of preclinical studies and perfect administration of clinical trials. Moreover, an excellent pre-clinical performer may be withdrawn due to the long processing time between drug discovery and commercial production. Substantial costs are involved with this process, and more importantly, the high failure rate delays the advances in better treatment for affected individuals. Lack of biomarker identification, off-target activity, and excess toxicity are a few of the challenges identified that can be addressed while designing new candidates. This process can utilize advanced knowledge of the molecular interactions between the drug and target [51]. In the case of CK2, many small molecular inhibitors have been discovered, most of which are investigated for the treatment of cancers. Yet, very few are approved for clinical use, partly due to their high cytotoxicity. Re-purposing the knowledge about these CK2 inhibitory molecules for treating musculoskeletal disorders is possible. Understanding the critical interactions of CK2 with its substrates in musculoskeletal diseases becomes essential.
In this review, first, we discuss the implications of CK2 in musculoskeletal disorders. Next, we discuss mechanisms of target identification by CK2 and the distinct functions of its subunits. Then, we describe currently available inhibitors of CK2 and their mechanism of action. Finally, we discuss a novel disease-modifying approach for targeting CK2–substrate interactions. This approach achieves CK2 activity modulation using biomimetic peptides targeting specific interactions of CK2 within the signal transduction cascade.
2. Implication of CK2 in Musculoskeletal Disorders
2.1. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is an autoimmune disorder affecting the joints. The exact mechanisms through which RA develops have yet to be fully understood [52,53]. One leading hypothesis is that the interactions between epigenetic modifications and environmental influences facilitate self-antigens production and induce autoimmunity. Self-antigens are then modified by citrullination [5]. As the body cannot recognize citrullinated antigens, the modified self-antigens are then recognized by antigen-presenting cells, which transport them to the lymph nodes. In the lymph nodes, helper T cells and B cells are activated through co-stimulation. The B cells differentiate into plasma cells that produce antibodies against the self-antigens [54]. In this manner, the body produces antibodies against self-antigens and induces tissue inflammation [55]. Synovial hyperplasia and similar disorders may also release cytokines contributing to joint inflammation and autoimmunity. Specifically, the synovial fluid and joint capsule become inflamed during RA, contributing to bone and cartilage erosion [56]. During RA, the balance between the Type I T Helper (Th1) and Type 2 T Helper (Th2) cells is disrupted in favor of the Th1 cells, causing inflammation. Type 17 T Helper (Th17) cells induce the proliferation of fibroblast-like synoviocytes (FLS) [57]. FLSs contribute to the progression of RA. FLSs are found in the synovial intimal lining of healthy tissue; they secrete products that contribute to the synovial fluid and articular cartilage. In RA, they secrete cytokines that promote inflammation and proteases that degrade cartilage [58].
Protein kinase CK2 plays a role in RA through CK2α’s control of the Interleukin-12 (IL-12) receptor, which regulates the IL-STAT4 signaling pathway through which Th1 cell differentiation is promoted. The IL-STAT4 pathway is regulated primarily by the expression of IL-12R in CD4+ T cells, though it is also regulated, to a lesser extent, by interferon-α (IFN-α) and interleukin 23 (IL-23). The phosphorylation of the STAT4 pathway induces downstream signaling that ultimately facilitates Th1 cell differentiation [59]. CK2α, CK2α’, and CK2β promote Th17 cell differentiation. CK2α also regulates Th1 cell differentiation and suppresses the activity of Th2 cells [59,60].
2.2. Osteoarthritis
Osteoarthritis (OA) is a degenerative disease of the articular cartilage, which is increasingly common with advanced age, especially in women. OA is associated with loss in the number and activity of chondrocytes through increased apoptosis. Reduced activity of chondrocytes reduces the repair of the collagen matrix and diminishes the structural integrity of the cartilage. Oxidative stress is another hallmark of OA. Metabolic disorders affect the severity of OA. Increased serum glucose levels due to type 2 diabetes and metabolic syndrome increase advanced glycation products (AGESs) levels. The AGES are associated with the collagen matrix of cartilage and cause structural damage. They also interact with receptors of AGES called RAGE and cause oxidative stress in chondrocytes [61]. Excess reactive oxygen species (ROS) production leads to oxidative stress response, increasing chondrocyte senescence [62].
CK2 is an inhibitor of apoptosis, and its expression is reduced in chondrocytes of patients diagnosed with OA. Inhibition of CK2, sensitized chondrocytes to Tumor Necrosis Factor-alpha (TNFα)-mediated cell death [63]. Parathyroid hormone-related protein (PTHrP) is a parathyroid hormone homolog. It protects chondrocytes from undergoing excessive apoptosis [64]. PTHrP exerts its protective activity against mitochondria-dependent apoptotic pathway via upregulation of CK2 activity [65,66]. PTHrP is localized to nuclei due to its mid-region bipartite nuclear targeting sequence (NTS) [67]. In one study, treatment with synthetic peptide PTHrP-NTS (the NTS region of PTHrP) protected HEK293 cells from TNFα-activated apoptosis. Exogenous NTS increased nuclear accumulation of CK2. Nuclear retention and activity of CK2 were enhanced. Similar results were obtained in cells treated with intact PTHrP [68]. CK2 is also involved in the oxidative stress response. CK2 signaling is essential for activating transcription factor NF-E2-related factor 2 (Nrf2). Nrf 2 signaling is crucial for redox balancing within cells [69]. It reduces cellular ROS levels and suppresses nuclear factor-κB (NF-κB) localization in the nucleus. Nrf2 upregulates expression of Heme oxygenase-1 (HO-1). Enzyme HO-1 is vital in heme degradation and countering oxidative stress response [70]. This enzyme becomes activated in chondrocytes in response to peroxynitrite-induced oxidative stress [71]. In another study, inhibition of the catalytic activity of CK2 with 4,5,6,7-terabromo-2-azabenzimidazole (TBB) and 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB) induced senescence and apoptosis in chondrocytes. Overexpression of HO-1 reduced the TBB-induced senescence. Chondrocytes overexpressing HO-1 had reduced sensitivity towards TBB-induced senescence. The knockdown of CK2 reduced type II collagen and increased β catenin expression [72]. The small heat shock protein αB-crystallin is a mediator for the expression of chondrogenic Bone Morphogenetic Protein-2 and collagen type II. Its expression is downregulated in OA [73]. Chondrocytes treated with CK2 inhibitors and αB-crystallin siRNA were sensitized to apoptosis. However, αB-crystallin had a protective effect. Its expression was modulated, and cellular localization was changed after CK2 inhibition [74]. The role of αB-crystallin in aiding CK2 to prevent chondrocyte apoptosis should be explored further. Thus, evidence suggests that CK2 protects chondrocytes from oxidative stress and apoptosis.
2.3. Bone Fracture
Healthy bone is excellent at self-healing in the event of injury. The process occurs roughly through the following phases. The first is the inflammatory phase and hematoma formation. Physical rupture of the bone causes infiltration of immunoregulatory cells, such as Macrophages, at the site. These cells secrete inflammatory cytokines IL-1, IL-6, and TNFα during the early phases of healing [75]. These factors recruit mesenchymal stem cells (MSC) to the fracture site. MSCs undergo chondrogenic and osteogenic differentiation through secreted growth factors. Next, angiogenesis and soft callus formation begin, at which point the central region of the fracture undergoes endochondral ossification during this phase [76]. The MSCs undergo differentiation into osteo-chondroprogenitor cells [77]. Thus, formed chondrocytes form the cartilaginous matrix. Angiogenesis and calcification of the cartilaginous matrix by hypertrophic chondrocytes marks the start of the next phase of hard bone formation. In this phase, the cartilage is replaced by bone via hard calcification. Here, the activity of osteoblasts increases. Finally, in the last phase, bone remodeling occurs. Here, the coordinated function between bone matrix-secreting osteoblasts and bone-resorbing osteoclasts takes place [64]. Bone remodeling is essential in maintaining bone shape. During aging, macrophages, chondrocytes, and osteocytes secrete senescence-associated factors [78]. Their secretion prolongs the inflammatory phase and hampers the bone healing process [79,80].
The bone morphogenic protein (BMP) pathway is essential for fracture healing [81,82]. BMP ligands are critical growth factors for bone, cartilage, joint development, and homeostasis. They drive MSC differentiation. BMP-2 stimulation of senescent bone marrow mesenchymal stem cells (BMSCs) activated adipogenic and cell death pathways like NF-kB or p38-mitogen activated protein kinase (MAPK). In non-senescent bone marrow mesenchymal stem cells (BMSCs), BMP-2 activated bone-forming pathways such as SMAD, BMP, and TGFβ [83]. CK2 negatively regulates BMP-2-activated signaling [84].
Casein kinase 2 interacting protein-1 (CKIP-1) is a negative regulator of BMP signaling [85]. CKIP-1 interacts with the CK2α subunit and regulates signaling between CK2α and effector molecules of BMP signaling [86,87]. CKIP-1 is known to inhibit BMP signaling by promoting Smad1 ubiquitination. The suppression of BMP signaling leads to suppressed osteogenic differentiation [23]. The CKIP-1 knockout mice had abnormally high levels of bone mass. The effects of CKIP-1 on bone growth and repair were age-dependent, with CKIP-1 having a more substantial impact on the bone mass of 18-month-old mice and comparatively little effect on 2-month-old mice. CKIP-1 is a negative regulator of osteogenic differentiation, and age-related inflammation causes an upregulation of CKIP-1, although the exact mechanisms of this upregulation remain unclear [88,89]. Inhibition of interaction between CK2α and CKIP-1 is a possible therapeutic strategy for bone fractures.
2.4. Osteoporosis
Osteoporosis (OP) is a degenerative disease that, as of 2023, affects more than 200 million people. The incidence rate of OP is 1.7% of men and 26% of women over age 50 worldwide [65]. OP is associated with a loss of bone mass and mineral density. This weakens the bones and predisposes them to fractures. It is a musculoskeletal disorder that is very closely related to metabolic dysregulation. OP is characterized by a disruption in the balance between osteoblasts and osteoclasts, cells that generate bone and cells that degrade bone, respectively. One common type of OP is glucocorticoid-induced osteoporosis (GIO). GIO is diagnosed in patients undergoing treatment with glucocorticoids for inflammatory or auto-immune diseases. Glucocorticoids inhibit osteoblast function and decrease the vascularity of the bone [66]. In one study, primary cells from human patients and mice with GIO had elevated levels of CKIP-1 and reduced levels of Smad1/5 compared to controls. CKIP-1 is a non-enzymatic protein that regulates the CK2α subunit of protein kinase CK2. It can be utilized in the localization of protein kinase CK2 in cell [67]. In vitro, elevated expression and activity of CKIP-1 in osteoblasts inhibited Smad-dependent BMP signaling [23]. CKIP-1 regulates BMP signaling and can recruit CK2α to the plasma membrane. Hence, its overexpression can affect the activation of BMP signaling in osteoblasts and osteoclasts. CKIP-1’s suppression of BMP signaling inhibits and affects the differentiation of MSC into osteoblasts. These alterations contribute to the reduction in bone mineralization observed in GIO patients. Suppressing the interaction between CKIP-1 and interaction with CK2 could be a novel therapeutic strategy for treating GIO. Upregulation of BMP signaling can increase bone formation and repair [16]. Blocking of the interaction between CK2 and CKIP-1 may induce BMP signaling [68]. A challenge in creating osteoporosis therapeutics is maintaining the optimal balance between osteoblasts and osteoclasts, cells that generate and degrade bone, respectively. Ideally, a therapeutic for OP would increase osteoblast activity while decreasing osteoclast activity, inducing bone formation and repair.
3. Inhibition of CK2
3.1. Molecular Function of CK2
3.1.1. Structure of CK2 and Substrate Recognition
CK2 was discovered as a mitochondrial kinase in 1954; since then, its function has been studied in various biological processes [90]. Recent research has identified its new substrates and allowed us to expand our knowledge about its regulation of biological processes.
The catalytic subunits of CK2 are acidophilic. Its substrates often have acidic amino acids near the phosphorylation site. The consensus sequence on the phosphorylation target ([pS/pT]-(P)-x-[E/D] or [pS/pT]-(P)-x-pS is identified as the minimum identification sequence required for target identification [30,91]. However, due to the short length of this sequence and flexibility for amino acids in the second and third positions, the frequency of occurrence of this sequence across all proteins is high. Using the consensus prediction software Prosite (http://prosite.expasy.org, accessed on 29 January 2024), the total number of sequences containing CK2 phosphorylation motifs comes out as 8640. However, the predicted number of targets of CK2 was much higher than the actual number of targets [92]. Also, CK2 phosphorylates a few substrates that do not have the consensus sequence. Thus, target validation is essential in determining the bona-fide substrates of CK2. It also helps target interactions where alternate kinases cannot nullify the drug’s effect. Approaches like phospho-proteomics have helped determine the number of targets this kinase affects.
Chonjowski et al. developed a method for validating genuine targets of CK2, specifically by taking advantage of the co-substrate specificity of CK2. The co-substrate specificity of CK2 means that the kinase can use either adenosine triphosphate (ATP) or guanosine triphosphate (GTP) to phosphorylate its substrates. Briefly, an analog of GTP–guanosine 5′-[γ-thio]triphosphate (GTPγS) is incorporated into the cell suspension. Other endogenous kinases do not use this substrate. Thus, substrates phosphorylated with CK2 are identified by the presence of ‘PγS’ using Mass Spectrometry. Furthermore, validation of CK2 targets with co-immunoprecipitation is possible with substrate-specific antibodies and pull-down assays. This method allows the use of versatile biological samples and helps capture phosphorylation reactions while the cells are still alive [31,93].
Inhibition of CK2 kinase activity by ATP competitive inhibitors does not alter the phosphorylation of all its targets. Changes in phosphorylation levels of CK2 targets by two CK2 inhibitors, Silmitasertib and GO289, were different. Both inhibitors have very low off-target activity and have an almost overlapping set of affected CK2 phosphorylation targets. However, the extent of reduction in phosphorylation of its targets is very different between these two. Both inhibitors bind the same site on CK2α and cause very different effects. This difference could be attributed to the ability of CK2 to identify its targets under various conditions [18,94].
To identify substrates exclusively phosphorylated by CK2, inhibitor-resistant mutants of CK2 were created. The kinase is known to phosphorylate only 20% of the substrates containing the minimal recognition motif. With the mutants and subsequent use of inhibitors, the expanse of substrates was estimated in C2C12 cell lines. Studies using lysates do not consider the temporal and spatial factors for CK2–substrate interactions. Instead, these studies used the stable isotope labeling using amino acids in cell culture (SILAC) approach. This allowed the study of interaction when the cells performed their normal function. The disparity was observed within popular CK2 inhibitors, with only a proportion of inhibitors showing dose-dependent inhibition of CK2 activity [95].
3.1.2. Function of Subunits
CK2 has catalytic subunits, CK2α and CK2α’, as well as regulatory CK2β subunits. The enzyme functions as a holoenzyme, and each subunit can also work in isolation [37]. Individual subunits of CK2 seem to play diverse roles exclusive to the holoenzyme form. There are many instances where individual subunits of CK2 perform distinct functions [96]. Inhibition of a particular subunit or its depletion in cellular compartments affects the expression and activity of other subunits. Additionally, by performing subunit knockout, the extent of the contribution of individual subunits was determined. This approach is helpful in the creation of subunit-specific drugs. In most cases, the abrogation of catalytic subunits impacts the expression and function of other subunits. These are explained using the following examples.
A coordinated regulation mechanism between the three holoenzyme subunits was observed during skeletal muscle differentiation. The mechanism involved regulating early myogenic regulator genes such as myoblast determination protein 1 (MyoD) and myogenin, followed by the expression of muscle-specific genes such as caveolin-3, troponin, and myomixer. The holoenzyme’s individual subunits and catalytic activity also coordinated plasma membrane fusion and the shutting of embedded proteins. In individual knockouts of subunits in C2C12 cells with CRISPR/Cas9, the deletion of one subunit affected the expression of other subunits, demonstrating their interdependence. Deletion of the CK2α subunit downregulated CK2β expression, whereas deletion of the CK2β subunit upregulated CK2α expression. However, deletion of CK2α’ did not alter the levels of the other two units. In knockouts of CK2α and CK2β, MyoD expression was reduced and abrogated, respectively, whereas the deletion of CK2α’ did not have an effect. The reduction in MyoD expression in CK2α deletion was due to the downregulation of CK2β in these mutants. In CK2β null mutants, the expression of this early myogenic regulatory factor (MRF) was not activated until the re-induction of the subunit in CK2β knockdown C2C12 cells already expressing low levels of MyoD. With increasing expression of CK2β in these cells, the level of MyoD increased, which was also reflected as an increase in its mRNA at the transcription level. This indicates that the CK2β with other epigenetic modifiers activate the expression of MyoD. The level of MyoD expression was not affected by varying the catalytic activity of CK2 by pharmacological inhibition or changes in the expression of CK2α and CK2α’ subunits. During the later stages of myogenic differentiation, the levels of MyoD reduce, whereas the expression of muscle-specific genes increases in WT C2C12 cells. Here, pharmacological inhibition of kinase activity in CK2α’ knockout mutants reduced the expression of these markers. This evidence indicates the role of the other catalytic subunit CK2α in the expression of muscle-specific markers in later stages. The role of CK2α’ during myotube formation was indispensable. With the deletion of this subunit, there was a reduction in the number and size of myotubes compared to WT. Exogenous CK2α’ is sufficient to rescue reduced fusogenic activity of muscle fibers because of CK2 deletion [78].
One of the two catalytic subunits, either CK2α or CK2α’, is more critical in some biological processes. This is the case during skeletal muscle development and homeostasis. Here, the activity of the CK2α subunit is crucial [97]. Skeletal muscle specific CK2α knockout mice were produced by breeding floxed mice with human skeletal actin Cre reporter mice. The grip strength of the CK2α knockout mice was decreased compared to the controls, and the difference was more significant in older mice. CK2β expression levels were significantly reduced in the CK2α knockout mice, but expression of CK2α’decreased slightly. Phospho-Akt1 serine 129 and 473 proteins were upregulated in the knockout mice. There was no difference in gastrocnemius and soleus muscles between the CK2α knockout mice and the controls with Hematoxylin and Eosin staining. The muscle sections from the knockout mice had higher levels of centralized nuclei than the controls, indicating their higher propensity for muscular regeneration. The soleus fibers of the knockout mice had lower levels of slow type I muscle fibers but increased levels of fast type II fibers compared to the control. This evidence suggests that CK2α is essential in skeletal muscle regeneration. CK2α knockout mice had elevated expression of several mitophagy markers, including sequestosome-1(SQSTM1), optineurin (OPTN), and microtubule associated protein 1 light chain 3 beta (MAP1LC3B-II). The upregulation of these markers suggests that autophagic processes were increased in the absence of CK2 catalytic activity. These mice also had decreased levels of mitochondrial genomic DNA. Knockout of CK2α also impaired neuromuscular function. The expression of various proteins essential to skeletal muscle function was aberrant [98]. Thus, deleting one catalytic subunit alone was sufficient to hamper the process.
Knockout of CK2 catalytic and regulatory subunits in myogenic C2C12 cells had very different effects. With the knockout of CK2α and CK2α’ subunits, there was a substantial reduction in phosphorylation of predicted CK2 targets (18 out of 24). Expression of the CK2β subunit was reduced with this deletion. One possible explanation could be an increased rate of degradation of CK2β. Phosphorylation was upregulated in some of the non-CK2 phosphorylation targets, which could be explained by changes in the expression and activity of other kinases in the absence of CK2 catalytic subunits. In the knockout of CK2β subunits, there was a proportion of reduction in phosphorylation that was slightly lower than that of the catalytic units (9 out of 15). Here, the catalytic subunit expression was increased compared to the wild type. For some of the CK2 substrates, such as Cdc37 (pS13), the knockout of CK2 did not affect phosphorylation. However, for the substrate, Akt S129, the phosphorylation was drastically reduced to the extent it was almost absent with the knockout of either the catalytic or regulatory subunit. These differential effects on substrates shed light on the function of CK2 subunits as compensatory to each other in some cases or the strict requirements of holoenzymes in others. Changes in the phosphorylation status of predicted CK2 targets and non-targets based on sequence prediction indicated that the consensus sequence alone is not sufficient or essential for target identification and phosphorylation to occur [31].
3.2. Small Molecular Inhibitors of CK2 and Their Mechanism of Action
A critical application of small molecule inhibitors of CK2 is within the treatment of cancer [99,100]. For example, osteosarcoma, a bone malignancy that originates in the osteoblasts, is one such cancer in which these inhibitors are an effective treatment. In osteosarcoma, CK2 is overexpressed. In cloned human osteosarcoma 143B, cell line CK2α and CK2β are upregulated compared to healthy osteoblasts and MSCs. The osteosarcoma cells treated with the CK2 inhibitor Silmitasertib suppressed growth and proliferation and increased apoptosis. Interestingly, the proliferation and differentiation of MSCs were unaffected. The siRNA-induced knockdown of the CK2α and CK2β subunits in vitro suppressed the growth and proliferation of osteosarcoma cells. The 143B osteosarcoma cells were xenografted into mice and treated with Silmitasertib. Similar to in vitro findings, Silmitasertib inhibited the proliferation of the tumor cells and, therefore, constrained the growth of the tumor. These findings support the idea that Silmitasertib is a potential therapeutic for osteosarcoma. It inhibits CK2, which induces the apoptosis of sarcoma cells while having little to no effect on MSCs [74]. The implication of CK2 inhibition in osteosarcoma is explained in Figure 1.
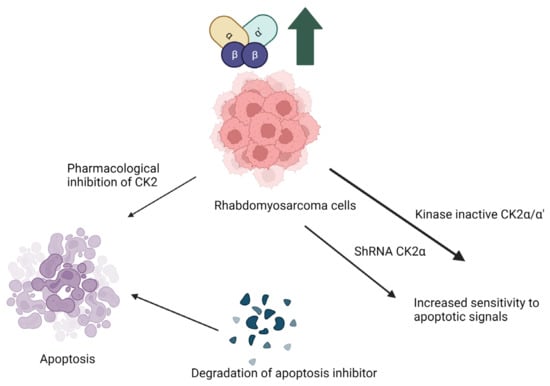
Figure 1.
Implication of CK2 in rhabdomyosarcoma and effect of inhibition of CK2 kinase activity. Pharmacological inhibition of CK2 makes the cells more sensitive to apoptotosis due to degradation of apoptosis inhibitor proteins. Abrogation of CK2α by generation of kinase inactive mutants or knockdown with ShRNA make the cells more sensitive apoptotic signals. (Image is created with https://www.biorender.com/ accessed on 9 January 2024).
Rhabdomyosarcoma, a cancer initiated in the skeletal muscle. Treatment of rhabdomyosarcoma is another example of the therapeutic implications of CK2 inhibitors. CK2 is upregulated in rhabdomyosarcoma cells. Current research on the pathophysiology of rhabdomyosarcoma uses cloned human tumor cell lines JR1 and Rh30. When these cell lines were treated with the CK2 inhibitor 5,6-dichloro benzimidazole (DRB), the proliferation of pro-apoptotic signals commenced, such as cytochrome c and Smad/DIABLO. This caused widespread cell death. JR1 and Rh30 cells transfected with either short hairpin RNA targeted to CK2α or kinase inactive CK2α/CK2α’, which led to heightened sensitivity to pro-apoptotic signals due to the suppressed CK2 activity. Inhibitors of CK2 have the potential as therapeutics to prevent the growth and metastasis of rhabdomyosarcoma cells [73]. Here, inhibition of kinase activity and knockdown of the α/α’ subunit increased apoptosis.
Additionally, research supports the utility of CK2 inhibition in treating rheumatoid arthritis (RA). In one study, human primary CD4+T lymphocytes isolated from patients diagnosed with RA were treated with the CK2 inhibitor Silmitasertib. Cellular responses in Th1 and Th17 cells were suppressed compared to controls. However, the Th2 cellular activity was enhanced. Th1 cells secrete proinflammatory factors, such as TNF-α and IL-2, while Th2 cells secrete anti-inflammatory factors, such as IL-3, IL-4, and IL-5 [101]. In the same study, for the collagen-induced RA mouse model, the effects of CK2 inhibition were similar [102]. Mice were treated with Silmitasertib via ingestion. The Th1 and Th17 cell responses were inhibited, and their RA symptoms were reduced [57,102].
Another important cellular component in inflammatory pathologies is the primary fibroblast-like synoviocyte (FLS). These are abundant in synovial tissue and function to regulate synovial fluid as well as the extracellular matrix [57]. In one study, primary FLSs from patients diagnosed with RA showed higher levels of CK2 expression and activity than primary FLSs from patients diagnosed with OA, the control. Stimulation of the RA primary FLSs with Silmitasertib suppressed the cell cycle by inhibiting CK2, a key component in regulating the cell cycle. Furthermore, the drug’s inhibition of CK2 decreased migration and proliferation of RA-FLSs, ultimately attenuating symptoms of RA, such as inflammation and joint pain [57]. Figure 2 summarizes in vitro and in vivo findings from these two studies.
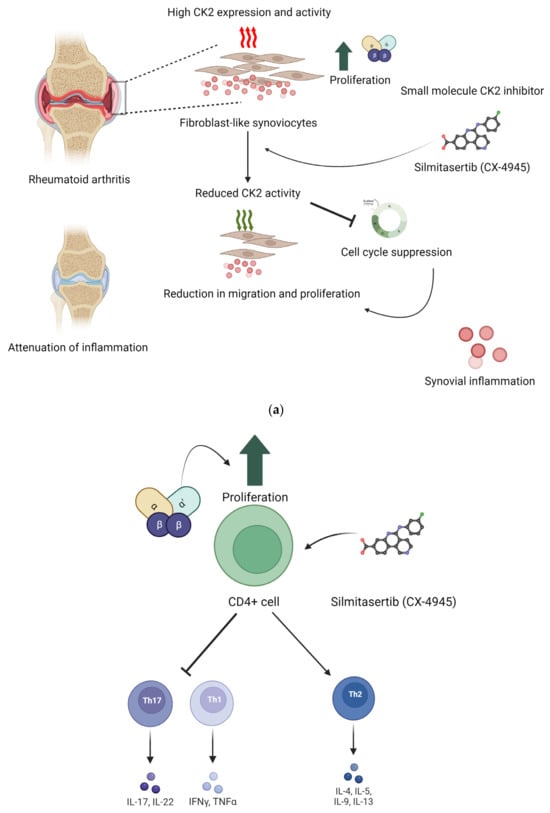
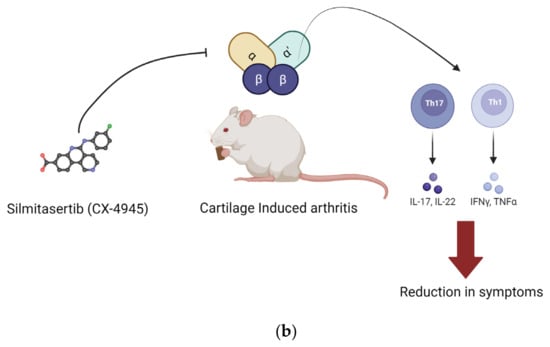
Figure 2.
Implication of CK2 in RA and effect of inhibition of CK2 kinase activity. In RA, the expression of CK2 is upregulated. This upregulation increases proliferation and survival of CD4+ cells. Silmitasertib is a small molecule inhibitor of CK2 kinase activity. (a) Inhibition of CK2 in fibroblast-like synoviocytes reduced their proliferation and hence reduced the synovial inflammation. (b) In RA, CD4+ cells treated with CK2 inhibitors had a reduction in activation of Th1 and Th17 T cell response, and it activated Th2 type cell response. Inhibition of CK2 in CD4+ cells reduced their proliferation, and hence, Th1 and Th17 T cell response was activated. These effects were seen in in vivo conditions as well. (Image created with https://www.biorender.com/ accessed on 9 January 2024).
The two most promising CK2 inhibitors include small molecule Silmitasertib and peptide inhibitor CIBG-300 [29]. Both are under clinical investigation for their use in cancer treatment. The ATP-competitive CK2-specific small molecule inhibitor exploits the differences in this kinase’s ATP-binding domain with others. The ATP-binding domain in the CK2α subunit is small compared to other kinases in this family.
Silmitasertib is highly specific for CK2 amongst small molecule inhibitors of CK2, but the drug is not without adverse effects [98]. The inhibitor caused abnormal alternative splicing in CK2 independent manner [103], the CK2α transcript was affected as well [97]. In Cholangiocarcinoma, the drug induced methuosis and caused cell death in a CK2-independent manner. In pancreatic ductal adenocarcinoma (PDAC), the responsiveness to Silmitasertib was tested in different cell lines. Expression of CK2 varied in the cell lines tested, although it was elevated in all of them compared to non-neoplastic cells. Elevated expression of CK2 in tumor cells’ responsiveness to Silmitasertib was not correlated to the extent of CK2 upregulation within the cell lines tested [104]. Additionally, inhibition of total CK2 activity within the cell was found to cause uncontrollable events in non-tumor cells [104,105,106].
Silmitasertib also has off target effects. Silmitasertib inhibited dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) and glycogen synthase kinase-3β (GSK3β) kinases [107]. Off-target effects are also seen in the case of inhibition of Cdc2-like kinases (Clks). Furthermore, the efficacy of Silmitasertib in cancer is also lower than that of several other inhibitors of CK2, which can be ascribed to its moderate selectivity [99]. The off-target activity of Silmitasertib is reported in the literature [86]. Because Silmitasertib has many off-target effects, a more specific molecule based on the drug’s structure is now under development [107].
The utility of CK2 inhibition by Silmitasertib for treating a non-neoplastic disease-osteoporosis was tested. Here, the effect of sub-cytotoxic concentrations of Silmitasertib was tested on osteoblast and osteoclast differentiation. It inhibited differentiation of bone-marrow-derived macrophages (BMM) into osteoclasts through RANKL activation. Inhibition of CK2 reduced phosphorylation of Akt, which caused downregulation in the expression of the transcription factor Nuclear Factor of Activated T Cells 1 (NFATc1), an effector of the Akt signaling cascade. Inhibition of CK2 also reduced RANKL-induced formation of tartrate-resistant acid phosphatase (TRAP), positive multinucleated cell formation, and TRAP activity, which is essential for osteoclast formation. On the other hand, BMP-2-mediated differentiation of myoblastic C2C12 cells into osteoblasts was enhanced with CK2 inhibition. Alkaline Phosphatase (ALP) activity and nuclear smad1/5/8 accumulation increased with increasing Silmitasertib concentration in differentiating cells. There was also an increase in Extracellular receptor Kinase (ERK) 1/2 phosphorylation. Thus, inhibition of CK2 accelerated osteoblast differentiation via the ERK1/2–Smad signaling axis [108].
Allosteric inhibitors bind at a site other than the enzyme’s active site. They cause a change in the active conformation of the enzyme, which hampers substrate binding. Thus, they are not competitive for the substrate of the enzyme [109]. Allosteric inhibitors of CK2 are non-ATP competitive [110]. Allosteric inhibitors of CK2 can bind to three different sites [111]. Most effective inhibitors bind at the α and β unit interface [112]. This approach retains the activity of the catalytic subunits and hence only affects the phosphorylation of CK2β-dependent substrates [29]. The regulatory β subunits act as docking sites for a few CK2 substrates and protect the catalytic subunits from degradation. The other two sites where allosteric inhibitors can bind are the αD pocket, the linker region in the ATP binding site [113], and the αC helix region, which is rich in glycine. Detailed descriptions of various CK2 inhibitors and their strategies can be found in the current literature [29]. The structure-activity relation of CK2 inhibitors is reviewed in the literature [29,114,115,116]. Many of these are either not highly specific or have low cell permeability. Some compounds known to inhibit CK2 do not show cytotoxicity in cancer cell lines. However, their utility in treating musculoskeletal disorders is worth investigating. In these diseases, re-directing pathways involving deregulated CK2 is more critical than causing general cytotoxicity.
Further studies are needed for clinical translation of these inhibitors. Examples of small molecular CK2 inhibitors are included in Table 1. Based on kinase panel assay, their selectivity in inhibiting CK2 is mentioned as CK2 selectivity. The pre-clinical efficacy is described for each one of these.

Table 1.
Small molecular inhibitors of CK2.
It is an effective strategy to target CK2 with other kinases that are either phosphorylated by it or act on the same substrate. Here, Bromodomain-Containing Protein 4 (BRD4) is a kinase that, along with CK2, is highly expressed in several different types of cancer. BRD4 is a transcriptional and epigenetic regulatory factor that belongs to the bromodomain and extraterminal (BET) family of chromatin adapter proteins. Its localization and stability are regulated by its phosphorylation by CK2. The use of inhibitors of BRD4 in cancer treatment is limited due to drug resistance [140]. In lung adenocarcinoma, the mechanism of drug resistance against BRD4 inhibitors was CK2-dependent [141]. A dual inhibitor for CK2 and BRD4 was more effective than targeting CK2 alone. Similarly, proviral integration of Moloney virus-1 (PIM) kinases and CK2 interacts with Myc [142,143]. These two kinases also have functional and structural similarities. In diseases with high expression of PIM and CK2, it was more effective to target both kinases. Both kinases are upregulated in hypoxic conditions in tumors [144]. SR-protein-specific kinase 1 (SRPK1) is involved in angiogenesis and overexpressed in several cancers. SRPIN803 inhibited both SRPK1 and CK2. Its ophthalmic application was also studied. TNIK and DYRK1 help maintain the phosphorylation of key regulators in cancer stem cells. Compound 108600 inhibited TNIK, DYRK1, and CK2. However, there was also an allosteric change in the CK2 α subunit that inhibited holoenzyme formation [109]. Table 2 summarizes the clinical efficacy of dual inhibitors of CK2.
Table 2.
Multitarget inhibitors of CK2.
The synthetic peptide CIBG-300 is effective in the treatment of cervical cancer. It is currently in phase 1 and 2 clinical trials [150]. It was also found to be anti-angiogenic [151]. The drug is derived from a cyclic nine-mer peptide. This peptide inhibited phosphorylation of human papillomavirus type-16 E7 (HPV-16 E7) oncoprotein phosphorylation in a phage display assay. Upon conjugating with cell-penetrating peptide Tat (amino acids 48–68) to increase the cell permeability of the peptide, it inhibited phosphorylation of the HPV-16 E7 oncoprotein by CK2 and induced apoptosis in cell cultures [42]. This peptide was then studied in different types of cancer [152]. CIGB-300 interacted more with catalytic subunits and interacted less with the regulatory subunit. Also, it responded with the CK2α’ more than the CK2α subunit [43]. The target of this peptide was B23/nucleophosmin, a nucleolar protein that selectively modulates specific genes. It affected protein synthesis, energy metabolism, and biogenesis of ribosomes [140]. These findings have led to a challenge in the defining mechanism of action of CIBG-300. Previously termed as a CK2 inhibitor, its specificity in targeting selected phosphosites rather than generalized inhibition has gained attention.
Inhibitors with a higher specificity can target specific subunits or forms of the CK2 holoenzyme. Some of them, like CIBG-300, are capable of inhibiting selective phosphosites. They are tested for their use in cancer treatment as well as treatment of other diseases. These are listed in Table 3.
Table 3.
CK2 inhibitors with pathway or subunit-specific mechanism of action.
4. Targeting CK2 Interactions with Its Substrates–Novel Approach
Targeting CK2–substrates interaction critical for pathogenesis for musculoskeletal disorders can be carried out to engineer metabolic pathways to a healthy state. Interfering peptides inspired by the structure of the CK2 substrate can be used for this purpose.
Such biomimetic peptides are designed based on protein-protein interactions (PPIs). PPIs are essential to cellular functioning, and any dysfunction could represent an opportunity for specific targeting [156]. The nature of these interactions is being established using cutting-edge computational methods paired with existing knowledge. These studies aid in the design of drugs targeting very specific PPIs implicated in disease pathology [157]. Peptides and monoclonal antibodies have been widely used to produce new therapeutics to target extracellular PPIs. Still, the development of peptides targeting intracellular PPIs has been far more limited due to the challenges of maintaining the peptide’s stability, administration, and bioavailability. Interfering peptides can interfere with PPIs and have been the focus of increasing attention by researchers. The large contact surface associated with PPIs has previously been an obstacle in the development of therapeutics, but interfering peptides have been found to interact more efficiently with large protein surfaces [158]. Peptides offer several advantages when used as therapeutics compared to small molecular drugs. First, they can cover more surface area in the PPI they are inhibiting. Then, their physical properties are easily modifiable based on their application. Moreover, they can be designed to be highly selective and can cover more surface area as inhibitors. Also, they have fewer toxicological effects [159]. Importantly, peptides can be developed to penetrate specific plasma membranes, which aids in targeting them to specific intracellular compartments, a long-standing challenge in therapeutic development [160,161,162,163]. Several studies prove that interfering peptides alter cellular processes [155,164,165,166]. Peptides can be produced in several ways, the simplest being procuring naturally occurring peptides [167]. Another method is to develop large numbers of peptides synthetically and then screen them to evaluate their therapeutic potential. A third method is to use peptides corresponding to fragments at the ends of the PPIs [168].
Disease-Modifying Peptide Drugs-CK2.1 and CK2.3
One of the most integral ways in which CK2 regulates the function of the musculoskeletal system is through the Bone Morphogenetic Protein (BMP) signaling pathway. The BMP signaling pathway regulates chondrogenesis and osteogenesis and maintains connective tissue homeostasis. The BMP-2 ligand interacts with BMPRIa and BMPRII receptors to activate the BMP pathway. CK2 interacts with the BMPRIa receptor in the absence of the BMP-2 ligand. Upon binding the BMP-2 ligand, the BMPRIa receptor undergoes heterodimerization with the BMPRII receptor. Formation of this complex releases of CK2. The release of CK2 initiates the subsequent phosphorylation of downstream signaling, which can cause the differentiation of MSCs into adipocytes, osteoblasts, and chondrocytes, depending on the pathway initiated. However, controlling the cell’s fate via the signaling cascade activated by BMP2 is impossible. This led to the design of interfering peptides activating specific branches of the BMP pathway [169]. Three BMPRIa mimetic peptides were synthesized to initiate the BMP signaling pathway without BMP2. These peptides contained one of three CK2 phosphorylation sites on BMPRIa (SLKD, SYED, or SLYD). At the N-terminus, there is the Antennapedia Homeodomain sequence for cellular uptake. Flanking the CK2 phosphorylation motif, there is a BMPRIa homologous sequence. These peptides are named CK2.1, CK2.2, and CK2.3 based on the phosphorylation site they contain [170]. CK2.1 induced chondrogenesis in vitro in mouse MSC line C3H10T1/2 and in vivo in the Destabilized Medial Meniscus (DMM) mouse model. CK2.1 increased proteoglycan synthesis and elevated levels of collagen type II without causing chondrocyte hypertrophy, a frequent occurrence with BMP2-induced signaling [171,172]. Rabbit MSCs were treated with a bilayer peptide-loaded scaffold consisting of CK2.1-coated β-glycerophosphate/chitosan, which resulted in chondrogenesis and osteoblastogenesis. This technique could be beneficial for treating articular osteochondral defects, common in OA patients, involving damage to the cartilage and underlying bone [23]. CK2.2 induced both adipogenesis and osteogenesis, while CK2.3 induced osteogenesis. CK2.3 would be preferred in treating OP and other disorders that reduce bone mass and mineral density because it does not simultaneously induce adipogenesis, as does CK2.2 [171]. CK2.3 activates ERK phosphorylation by preventing CK2 binding to the BMRPIa–BMPRII receptor complex. CK2.3 increases bone area, bone mass, and mineral density [173]. In C2C12 cloned mouse myoblast cells, CK2.3 decreased osteoclastogenesis, increased osteoblastogenesis, and increased mineralization compared to controls. CK2.3 has been tested in an aging mouse model. In vivo, injections of CK2.3 via calvarial injection in mice increased bone area, density, and growth [170]. In 6-to-9-day-old C57BL/6J mice injected with CK2.3 via the calvaria and 8-week-old mice injected via the tail vein, mice showed increased bone formation and increased bone mineral density. At the same time, osteoclast activity appeared to be suppressed, indicating CK2.3 has the potential to alleviate the symptoms of OP by simultaneously enhancing osteoblast activity and inhibiting osteoclast activity, inducing bone growth and repair [174,175,176]. The principle of design of BMPRIa biomimetic peptides CK2.1 and CK2.3 and their effects are summarized in Figure 3.
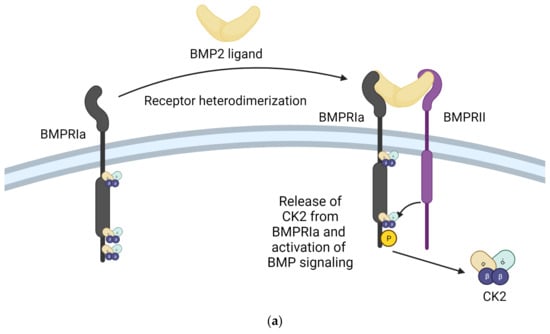
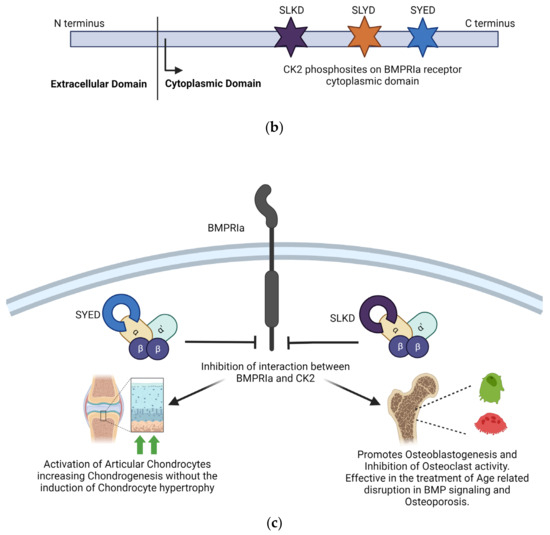
Figure 3.
Inhibition of the interaction between CK2 and BMPRIa is an effective strategy for treating Osteoarthritis and Osteoporosis. (a) Stimulation with BMP2 ligand causes heterodimerization of BMPRIa receptor with BMPRII. Release of CK2 from BMPRIa and its phosphorylation by BMPRII activates the BMP signaling pathway. (b) BMPRIa receptor has three phosphosites on the cytoplasmic/intracellular domain. These phosphosites were validated using CK2 mutants. (c) BMPRIa mimetic peptides containing phosphosite sequences corresponding to ‘SYED’ activate articular chondrocytes to repair osteoarthritic lesions in knee articular cartilage. The BMPRIa mimetic peptide containing phosphosite sequence corresponding to ‘SLKD’ corrects the BMP pathway in senile mice. It helps regain balance between Osteoblastogenesis and Osteoclastic activity, which is affected in Osteoporosis. (Image created with https://www.biorender.com/ accessed on 9 January 2024).
5. Discussion
Inhibition of CK2 is proving to be highly successful in treating cancer. In musculoskeletal disorders, the influence of metabolic disorders is a research focus. The kinase CK2 is highly involved in the regulation of cellular metabolism. In rheumatoid arthritis, elevated CK2 activity promotes the inflammatory pathways. In osteoarthritis, a reduction in the activity of CK2 reduces the viability of chondrocytes. In osteoporosis, the imbalance in BMP signaling is due to the dysregulated function of CK2. During fracture healing, aging and senescence cause defective bone formation. CK2 is essential to cell survival and helps the cell counter senescence. Studying the role of CK2 in these diseases will demystify several mechanisms involved.
The pathogenesis of most musculoskeletal disorders has a component of dysregulated cell differentiation, senescence, and inflammation. Causing cytotoxicity is not always the best course of action for these disorders. Instead, correction of underlying biochemical imbalances is an effective treatment. The role of CK2 as the lateral player makes it an ideal protein target for these disorders.
The evolution of small molecular inhibitors of CK2 is mainly focused on their use as cancer therapeutics. Improved specificity and cell permeability are the two main achievements. These inhibitors are commonly used in combination with targeted therapy. However, their use in treating non-neoplastic disorders like musculoskeletal disorders is not common. Also, targeting the kinase activity is only sometimes the most effective method for this group of disorders. The entire phosphoproteome of CK2 is still being experimentally validated. Targeting of redundant interactions and false positive substrates thus stands as a possibility. The peptide drugs target the identified interaction between CK2 and BMPRIa. These are also seen to be highly cell-permeable. Further investigation into their tissue-specific drug delivery and toxicity needs to be undertaken. Evidence of their ability to drive the differentiation of MSCs into different lineages opens many possibilities. Drugs highly specific to kinase substrate interaction can be a molecular engineering tool. It can direct the molecular pathways towards the desired lineage. Targeting the pleiotropic kinase CK2 in this manner is an emerging prospect for designing a new generation of drugs. Figure 4 illustrates the current advances in CK2 inhibitors and the aspects to be addressed by upcoming drugs targeting CK2–substrate interactions.
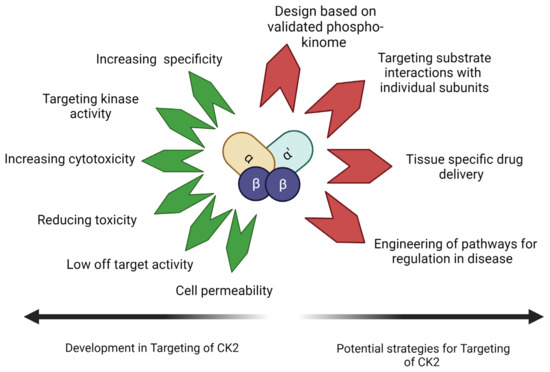
Figure 4.
CK2 inhibitors have been designed with increased specificity for CK2, reducing off-target effects and increasing cell permeability. CK2 inhibitors have been found to increase cytotoxicity in anti-cancer drugs. With knowledge of the phosphosites of CK2 and the role of CK2 subunits and molecular forms, future CK2 inhibitors can be designed to target pathology-specific substrate interactions (Image Created with https://www.biorender.com/ accessed on 9 January 2024).
Author Contributions
Conceptualization, V.P. and A.N.; methodology, V.P.; software, V.P.; validation, A.N., V.P. and K.D.; formal analysis, V.P.; investigation, V.P.; resources, A.N.; data curation, V.P. and K.D.; writing—original draft preparation, V.P.; writing—review and editing, A.N., V.P. and K.D.; visualization, V.P. and K.D.; supervision, A.N.; project administration, A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors would like to acknowledge all members of the Nohe Lab, the University of Delaware, and the Delaware Biotechnology Institute for their continued support during this project. The authors thank Daniel Halloran and Kelechi Chukwuocha for their immense help and valuable suggestions in writing this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- El Batawi, M.A. Work-related diseases. A new program of the World Health Organization. Scand. J. Work. Environ. Health 1984, 10, 341–346. [Google Scholar] [CrossRef]
- Khan, M.A.; Zubair, H.; Anand, S.; Srivastava, S.K.; Singh, S.; Singh, A.P. Dysregulation of metabolic enzymes in tumor and stromal cells: Role in oncogenesis and therapeutic opportunities. Cancer Lett. 2020, 473, 176–185. [Google Scholar] [CrossRef]
- Egami, R.; Kokaji, T.; Hatano, A.; Yugi, K.; Eto, M.; Morita, K.; Ohno, S.; Fujii, M.; Hironaka, K.-I.; Uematsu, S.; et al. Trans-omic analysis reveals obesity-associated dysregulation of inter-organ metabolic cycles between the liver and skeletal muscle. iScience 2021, 24, 102217. [Google Scholar] [CrossRef]
- Comertpay, B.; Gov, E. Immune cell-specific and common molecular signatures in rheumatoid arthritis through molecular network approaches. Biosystems 2023, 234, 105063. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, S.; Cao, N.; Wang, Q.; Liu, Y.; Xu, Q.; Zhang, L.; Sun, C.; Xiao, X.; Yao, J. Intestinal flora, intestinal metabolism, and intestinal immunity changes in complete Freud’s adjuvant-rheumatoid arthritis C57BL/6 mice. Int. Immunopharmacol. 2023, 125 Pt A, 111090. [Google Scholar] [CrossRef]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Cho, Y.; Jeong, S.; Kim, H.; Kang, D.; Lee, J.; Kang, S.-B.; Kim, J.-H. Disease-modifying therapeutic strategies in osteoarthritis: Current status and future directions. Exp. Mol. Med. 2021, 53, 1689–1696. [Google Scholar] [CrossRef]
- Liao, Z.; Han, X.; Wang, Y.; Shi, J.; Zhang, Y.; Zhao, H.; Zhang, L.; Jiang, M.; Liu, M. Differential Metabolites in Osteoarthritis: A Systematic Review and Meta-Analysis. Nutrients 2023, 15, 4191. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Zong, Z.; Deng, J.; Huang, J.; Liu, G.; Wei, B.; Cui, L.; Li, G.; Zhong, H.; Lin, S. Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration. Nutrients 2022, 14, 3984. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shen, J.; Li, X.; Bao, Y.; Zhao, T.; Li, B.; Qin, D. Regulatory Effects of Quercetin on Bone Homeostasis: Research Updates and Future Perspectives. Am. J. Chin. Med. 2023, 51, 2077–2094. [Google Scholar] [CrossRef]
- Robin, F.; Chappard, D.; Leroyer, P.; Latour, C.; Mabilleau, G.; Monbet, V.; Cavey, T.; Horeau, M.; Derbré, F.; Roth, M.; et al. Differences in bone microarchitecture between genetic and secondary iron-overload mouse models suggest a role for hepcidin deficiency in iron-related osteoporosis. FASEB J. 2023, 37, e23245. [Google Scholar] [CrossRef] [PubMed]
- Alghadir, A.H.; Gabr, S.A.; Iqbal, A. Hand grip strength, vitamin D status, and diets as predictors of bone health in 6–12 years old school children. BMC Musculoskelet. Disord. 2023, 24, 830. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.S.; Venturini, L.G.; Speck-Hernandez, C.A.; Alabarse, P.V.; Xavier, T.; Taira, T.M.; Duffles, L.F.; Cunha, F.Q.; Fukada, S.Y. AMPKα1 negatively regulates osteoclastogenesis and mitigates pathological bone loss. J. Biol. Chem. 2023, 299, 105379. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, M.; Zhang, Y.; Wu, J.; Gao, M.; Lei, T.; Huang, F.; Chen, H.; Wu, M. Risk factors for the comorbidity of osteoporosis/osteopenia and kidney stones: A cross-sectional study. Arch. Osteoporos. 2023, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Sung, E.; Kang, J.-H.; Kim, C.-H.; Shin, H.; Yoo, E.; Kim, M.; Lee, M.Y.; Shin, S. Association between body fat and bone mineral density in Korean adults: A cohort study. Sci. Rep. 2023, 13, 17462. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, D.K.; Reider, I.; Kamal, F.; Norbury, C.C.; Elbarbary, R.A. Distinct defects in early innate and late adaptive immune responses typify impaired fracture healing in diet-induced obesity. Front. Immunol. 2023, 14, 1250309. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’amore, C.; Cesaro, L.; Sarno, S.; Pinna, L.A.; Ruzzene, M.; Salvi, M. How can a traffic light properly work if it is always green? The paradox of CK2 signaling. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 321–359. [Google Scholar] [CrossRef]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- St-Denis, N.A.; Litchfield, D.W. Protein kinase CK2 in health and disease: From birth to death: The role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef]
- Halloran, D.; Pandit, V.; Nohe, A. The Role of Protein Kinase CK2 in Development and Disease Progression: A Critical Review. J. Dev. Biol. 2022, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, C.; Wu, X.; Zhang, Z.; Li, J.; Guo, B.; Li, D.; Liang, C.; Dang, L.; Pan, X.; et al. Targeting osteoblastic casein kinase-2 interacting protein-1 to enhance Smad-dependent BMP signaling and reverse bone formation reduction in glucocorticoid-induced osteoporosis. Sci. Rep. 2017, 7, 41295. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Song, J.; Bae, Y.S. CK2 Down-Regulation Increases the Expression of Senescence-Associated Secretory Phenotype Factors through NF-κB Activation. Int. J. Mol. Sci. 2021, 22, 406. [Google Scholar] [CrossRef]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of Protein Kinase CK2 Inhibitors Derived from 4,5,6,7-Tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef]
- Sarno, S.; Pinna, L.A. Protein kinase CK2 as a druggable target. Mol. Biosyst. 2008, 4, 889–894. [Google Scholar] [CrossRef]
- Borgo, C.; Ruzzene, M. Protein kinase CK2 inhibition as a pharmacological strategy. Adv. Protein Chem. Struct. Biol. 2021, 124, 23–46. [Google Scholar] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Borgo, C.; Cesaro, L.; Zaramella, S.; Vilardell, J.; Salvi, M.; Arrigoni, G.; Pinna, L.A. Re-evaluation of protein kinase CK2 pleiotropy: New insights provided by a phosphoproteomics analysis of CK2 knockout cells. Cell. Mol. Life Sci. 2018, 75, 2011–2026. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Cesaro, L.; Pinna, L.A. Variable contribution of protein kinases to the generation of the human phosphoproteome: A global weblogo analysis. Biomol. Concepts 2010, 1, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Guerra, B.; Niefind, K.; Issinger, O.G. Structural basis of the constitutive activity of protein kinase CK2. Methods Enzymol. 2010, 484, 515–529. [Google Scholar] [PubMed]
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115 Pt 20, 3873–3878. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A. Protein kinase CK2. Int. J. Biochem. Cell Biol. 1997, 29, 551–554. [Google Scholar] [CrossRef]
- Filhol, O.; Cochet, C. Protein kinase CK2 in health and disease: Cellular functions of protein kinase CK2: A dynamic affair. Cell Mol. Life Sci. 2009, 66, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef]
- Olsten, M.E.; Litchfield, D.W. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem. Cell Biol. 2004, 82, 681–693. [Google Scholar] [CrossRef]
- Montenarh, M.; Götz, C. Protein Kinase CK2α’, More than a Backup of CK2α. Cells 2023, 12, 2834. [Google Scholar] [CrossRef]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-Chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic Acid (CX-4945), the First Clinical Stage Inhibitor of Protein Kinase CK2 for the Treatment of Cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Zanin, S.; Sandre, M.; Cozza, G.; Ottaviani, D.; Marin, O.; Pinna, L.A.; Ruzzene, M. Chimeric peptides as modulators of CK2-dependent signaling: Mechanism of action and off-target effects. Biochim. Biophys. Acta 2015, 1854 Pt B, 1694–1707. [Google Scholar] [CrossRef]
- Perea, S.E.; Baladrón, I.; Valenzuela, C.; Perera, Y. CIGB-300: A peptide-based drug that impairs the Protein Kinase CK2-mediated phosphorylation. Semin. Oncol. 2018, 45, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Pérez, G.V.; Rosales, M.; Ramón, A.C.; Rodríguez-Ulloa, A.; Besada, V.; González, L.J.; Aguilar, D.; Vázquez-Blomquist, D.; Falcón, V.; Caballero, E.; et al. CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model. Biomedicines 2022, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Cirigliano, S.M.; Bessone, M.I.D.; Berardi, D.E.; Flumian, C.; Bal de Kier Joffé, E.D.; Perea, S.E.; Farina, H.G.; Todaro, L.B.; Urtreger, A.J. The synthetic peptide CIGB-300 modulates CK2-dependent signaling pathways affecting the survival and chemoresistance of non-small cell lung cancer cell lines. Cancer Cell Int. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Sarduy, M.R.; García, I.; Coca, M.A.; Perera, A.; Torres, L.A.; Valenzuela, C.M.; Baladrón, I.; Solares, M.; Reyes, V.; Hernández, I.; et al. Optimizing CIGB-300 intralesional delivery in locally advanced cervical cancer. Br. J. Cancer 2015, 112, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Blomquist, D.; Ramón, A.C.; Rosales, M.; Pérez, G.V.; Rosales, A.; Palenzuela, D.; Perera, Y.; Perea, S.E. Gene expression profiling unveils the temporal dynamics of CIGB-300-regulated transcriptome in AML cell lines. BMC Genom. 2023, 24, 373. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Merchan, E.C. The Current Role of Disease-modifying Osteoarthritis Drugs. Arch. Bone Jt. Surg. 2023, 11, 11–22. [Google Scholar]
- Song, Z.; Li, Y.; Shang, C.; Shang, G.; Kou, H.; Li, J.; Chen, S.; Liu, H. Sprifermin: Effects on Cartilage Homeostasis and Therapeutic Prospects in Cartilage-Related Diseases. Front. Cell Dev. Biol. 2021, 9, 786546. [Google Scholar] [CrossRef]
- Maksimovic, V.; Pavlovic-Popovic, Z.; Vukmirovic, S.; Cvejic, J.; Mooranian, A.; Al-Salami, H.; Mikov, M.; Golocorbin-Kon, S. Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol. Biol. Rep. 2020, 47, 4699–4708. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef]
- Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A Synopsis. Am. J. Manag. Care 2014, 20, S128–S135. [Google Scholar]
- Huang, J.; Fu, X.; Chen, X.; Li, Z.; Huang, Y.; Liang, C. Promising Therapeutic Targets for Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 686155. [Google Scholar] [CrossRef]
- Mcinnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.; Bungao, S.G. Management of Rheumatoid Arthritis: An Overview. Cells 2021, 10, 2857. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lei, Y.; Guo, X.; Zhu, D.; Zhang, H.; Guo, Z.; Xu, Z.; Zhao, H.; Xi, Y.; Peng, X.; et al. CX-4945 inhibits fibroblast-like synoviocytes functions through the CK2-p53 axis to reduce rheumatoid arthritis disease severity. Int. Immunopharmacol. 2023, 119, 110163. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2009, 1, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gibson, S.A.; Yan, Z. Protein kinase 2 (CK2) controls CD4(+) T cell effector function in the pathogenesis of colitis. Mucosal Immunol. 2020, 13, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Klein, M.; Reuters, S. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat. Immunol. 2015, 16, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Kurz, B.; Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: Friends or foes? Osteoarthr. Cartil. 2005, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Song, Y.S.; Lee, S.Y.; Yoon, Y.G.; Lee, S.H.; Park, B.S.; Yun, I.; Choi, H.; Kim, K.; Chung, W.T.; et al. Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-α-mediated chondrocyte death through apoptosis and autophagy. PLoS ONE 2011, 6, e19163. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, S.; Lai, F.; Shiraishi, M.; Kobayashi, T.; Kozhemyakina, E.; Yao, T.-P.; Lassar, A.B.; Kronenberg, H.M. PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. J. Clin. Investig. 2019, 4, e97903. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; A Young, D.; Rowan, A.D.; Xu, X.; E Cawston, T.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, M.; Naselli, F.; Abruscato, G.; Luparello, C.; Caradonna, F. Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases. Biology 2023, 12, 950. [Google Scholar] [CrossRef]
- Aarts, M.M.; Levy, D.; He, B.; Stregger, S.; Chen, T.; Richard, S.; Henderson, J.E. Parathyroid Hormone-related Protein Interacts with RNA. J. Biol. Chem. 1999, 274, 4832–4838. [Google Scholar] [CrossRef]
- Okoumassoun, L.E.; Russo, C.; Denizeau, F.; Averill-Bates, D.; Henderson, J.E. Parathyroid hormone-related protein (PTHrP) inhibits mitochondrial-dependent apoptosis through CK2. J. Cell. Physiol. 2007, 212, 591–599. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.-Y.; Bach, F.H. Heme Oxygenase-1 Modulates the Expression of Adhesion Molecules Associated with Endothelial Cell Activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef]
- Park, Y.C.; Kim, K.M.; Song, J.D.; Chung, H.T. Protein kinase CK2 mediates peroxynitrite-induced heme oxygenase-1 expression in articular chondrocytes. Int. J. Mol. Med. 2012, 29, 1039–1044. [Google Scholar] [CrossRef]
- Kim, K.M.; Sohn, D.H.; Kim, K.; Park, Y.C. Inhibition of protein kinase CK2 facilitates cellular senescence by inhibiting the expression of HO-1 in articular chondrocytes. Int. J. Mol. Med. 2019, 43, 1033–1040. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Applegate, V.; Nickelsen, A.; Klußmann, M.; Neundorf, I.; Götz, C.; Jose, J.; Niefind, K. Molecular Plasticity of Crystalline CK2α′ Leads to KN2, a Bivalent Inhibitor of Protein Kinase CK2 with Extraordinary Selectivity. J. Med. Chem. 2022, 65, 1302–1312. [Google Scholar] [CrossRef]
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Yoo, S.H.; Kim, H.Y.; Chung, W.T.; Yoo, Y.H. Alpha B-Crystallin Protects Rat Articular Chondrocytes against Casein Kinase II Inhibition-Induced Apoptosis. PLoS ONE 2016, 11, e0166450. [Google Scholar] [CrossRef]
- Muire, P.J.; Mangum, L.H.; Wenke, J.C. Time Course of Immune Response and Immunomodulation During Normal and Delayed Healing of Musculoskeletal Wounds. Front. Immunol. 2020, 11, 1056. [Google Scholar] [CrossRef]
- Prein, C.; Beier, F. ECM signaling in cartilage development and endochondral ossification. Curr. Top. Dev. Biol. 2019, 133, 25–47. [Google Scholar] [PubMed]
- PPajarinen, J.; Lin, T.; Gibon, E.; Kohno, Y.; Maruyama, M.; Nathan, K.; Lu, L.; Yao, Z.; Goodman, S.B. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials 2019, 196, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Recknagel, S.D.; Bindl, R.; Brochhausen, C.; Göckelmann, M.D.; Wehner, T.; Schoengraf, P.; Huber-Lang, M.; Claes, L.; Ignatius, A.D. Systemic inflammation induced by a thoracic trauma alters the cellular composition of the early fracture callus. J. Trauma Inj. Infect. Crit. Care 2013, 74, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Khosla, S. Fracture Healing in the Setting of Endocrine Diseases, Aging, and Cellular Senescence. Endocr. Rev. 2022, 43, 984–1002. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; La Rochelle, W.J.; Anver, M.; E Bockman, D.; Merlino, G. Scatter factor/hepatocyte growth factor as a regulator of skeletal muscle and neural crest development. Proc. Natl. Acad. Sci. USA 1996, 93, 5866–5871. [Google Scholar] [CrossRef]
- Majidinia, M.; Sadeghpour, A.; Yousefi, B. The roles of signaling pathways in bone repair and regeneration. J. Cell Physiol. 2018, 233, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Dumic-Cule, I.; Peric, M.; Kucko, L.; Grgurevic, L.; Pecina, M.; Vukicevic, S. Bone morphogenetic proteins in fracture repair. Int. Orthop. 2018, 42, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lee, J.H.; Lee, K.M.; Lee, C.-K.; Shin, D.-M. BMP-2 Induced Signaling Pathways and Phenotypes: Comparisons Between Senescent and Non-senescent Bone Marrow Mesenchymal Stem Cells. Calcif. Tissue Int. 2021, 110, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; King, D.; Young, K.; Litchfield, D.W.; Petersen, N.O.; Nohe, A. Casein kinase 2 beta-subunit is a regulator of bone morphogenetic protein 2 signaling. Biophys. J. 2010, 99, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Huo, L.; Liu, Y.; Deng, P.; Szymanski, J.; Li, J.; Luo, X.; Hong, C.; Lin, J.; Wang, C.Y. PGC-1α Controls Skeletal Stem Cell Fate and Bone-Fat Balance in Osteoporosis and Skeletal Aging by Inducing TAZ. Cell Stem Cell 2018, 23, 193–209.e5. [Google Scholar] [CrossRef]
- Nie, J.; Liu, L.; He, F.; Fu, X.; Han, W.; Zhang, L. CKIP-1: A scaffold protein and potential therapeutic target integrating multiple signaling pathways and physiological functions. Ageing Res. Rev. 2013, 12, 276–281. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, L. Physiological functions of CKIP-1: From molecular mechanisms to therapy implications. Ageing Res. Rev. 2019, 53, 100908. [Google Scholar] [CrossRef]
- Yuan, Y.; Sun, J.; Zhou, H.; Wang, S.; He, C.; Chen, T.; Fang, M.; Li, S.; Kang, S.; Huang, X.; et al. The effect of QiangGuYin on osteoporosis through the AKT/mTOR/autophagy signaling pathway mediated by CKIP-1. Aging 2022, 14, 892–906. [Google Scholar] [CrossRef]
- Peng, X.; Wu, X.; Zhang, J.; Zhang, G.; Li, G.; Pan, X. The role of CKIP-1 in osteoporosis development and treatment. Bone Jt. Res. 2018, 7, 173–178. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369 Pt 1, 1–15. [Google Scholar] [CrossRef]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 847–859. [Google Scholar] [CrossRef]
- Franchin, C.; Borgo, C.; Zaramella, S.; Cesaro, L.; Arrigoni, G.; Salvi, M.; Pinna, L.A. Exploring the CK2 Paradox: Restless, Dangerous, Dispensable. Pharmaceuticals 2017, 10, 11. [Google Scholar] [CrossRef]
- Chojnowski, J.E.; Mcmillan, E.A.; Strochlic, T.I. Identification of Novel CK2 Kinase Substrates Using a Versatile Biochemical Approach. J. Vis. Exp. 2019, 21, e59037. [Google Scholar]
- Borgo, C.; Cesaro, L.; Hirota, T.; Kuwata, K.; D’amore, C.; Ruppert, T.; Blatnik, R.; Salvi, M.; Pinna, L.A. Comparing the efficacy and selectivity of Ck2 inhibitors a phosphoproteomics approach. Eur. J. Med. Chem. 2021, 214, 113217. [Google Scholar] [CrossRef] [PubMed]
- Gyenis, L.; Menyhart, D.; Cruise, E.S.; Jurcic, K.; Roffey, S.E.; Chai, D.B.; Trifoi, F.; Fess, S.R.; Desormeaux, P.J.; Díaz, T.N.d.V.; et al. Chemical Genetic Validation of CSNK2 Substrates Using an Inhibitor-Resistant Mutant in Combination with Triple SILAC Quantitative Phosphoproteomics. Front. Mol. Biosci. 2022, 9, 909711. [Google Scholar] [CrossRef]
- Borgo, C.; Cesaro, L.; Hirota, T.; Kuwata, K.; D’Amore, C.; Ruppert, T.; Blatnik, R.; Salvi, M.; Pinna, L.A. Analysis of the phosphoproteome of CK2α((-/-))/Δα’ C2C12 myoblasts compared to the wild-type cells. Open Biol. 2023, 13, 220220. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.-Y.; Chi, S.-W.; Lee, M.-S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a Novel Function of CX-4945 as a Splicing Regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef] [PubMed]
- Merholz, M.; Jian, Y.; Wimberg, J.; Gessler, L.; Hashemolhosseini, S. In Skeletal Muscle Fibers, Protein Kinase Subunit CSNK2A1/CK2α Is Required for Proper Muscle Homeostasis and Structure and Function of Neuromuscular Junctions. Cells 2022, 11, 3962. [Google Scholar] [CrossRef]
- Salvi, M.; Borgo, C.; Pinna, L.A.; Ruzzene, M. Targeting CK2 in cancer: A valuable strategy or a waste of time? Cell Death Discov. 2021, 7, 325. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Kren, B.T.; Afzal, M.; Scaria, G.A.; Klein, M.A.; Ahmed, K. Protein kinase CK2—Diverse roles in cancer cell biology and therapeutic promise. Mol. Cell. Biochem. 2022, 478, 899–926. [Google Scholar] [CrossRef]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [Google Scholar] [CrossRef]
- Ye, H.; Fu, D.; Fang, X.; Xie, Y.; Zheng, X.; Fan, W.; Hu, F.; Li, Z. Casein Kinase II exacerbates rheumatoid arthritis via promoting Th1 and Th17 cell inflammatory responses. Expert. Opin. Ther. Targets 2021, 25, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yun, J.-S.; Kim, W.-K.; Chun, H.-S.; Jin, H.; Cho, S.; Chang, J.H. Structural Basis for the Selective Inhibition of Cdc2-Like Kinases by CX-4945. BioMed Res. Int. 2019, 2019, 6125068. [Google Scholar] [CrossRef]
- Ma, Y.; Sender, S.; Sekora, A.; Kong, W.; Bauer, P.; Ameziane, N.; Krake, S.; Radefeldt, M.; Al-Ali, R.; Weiss, F.U.; et al. Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 4409. [Google Scholar] [CrossRef]
- Lertsuwan, J.; Lertsuwan, K.; Sawasdichai, A.; Tasnawijitwong, N.; Lee, K.Y.; Kitchen, P.; Afford, S.; Gaston, K.; Jayaraman, P.S.; Satayavivad, J. CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers 2018, 10, 283. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Villar, P.; Trigo, C.; Caamaño, E.; Niechi, I.; Pérez, P.; Muñoz, J.P.; Aguayo, F.; Burzio, V.A.; Varas-Godoy, M.; et al. CK2 inhibition with silmitasertib promotes methuosis-like cell death associated to catastrophic massive vacuolization of colorectal cancer cells. Cell Death Dis. 2019, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Xu, G.; Gou, S. Novel CK2-Specific Pt(II) Compound Reverses Cisplatin-Induced Resistance by Inhibiting Cancer Cell Stemness and Suppressing DNA Damage Repair in Non-small Cell Lung Cancer Treatments. J. Med. Chem. 2021, 64, 4163–4178. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.H.; Moon, S.H.; Kim, J. The Protein Kinase 2 Inhibitor CX-4945 Regulates Osteoclast and Osteoblast Differentiation In Vitro. Mol. Cells 2013, 36, 417–423. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Wang, D.; Chen, Y.; Zhang, N. Recent Advances in the Discovery of CK2 Allosteric Inhibitors: From Traditional Screening to Structure-Based Design. Molecules 2020, 25, 870. [Google Scholar] [CrossRef]
- Moucadel, V.; Prudent, R.; Sautel, C.F.; Teillet, F.; Barette, C.; Lafanechere, L.; Receveur-Brechot, V.; Cochet, C. Antitumoral activity of allosteric inhibitors of protein kinase CK2. Oncotarget 2011, 2, 997–1010. [Google Scholar] [CrossRef]
- Li, C.; Zhang, X.; Zhang, N.; Zhou, Y.; Sun, G.; Zhao, L.; Zhong, R. Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules 2020, 25, 237. [Google Scholar] [CrossRef]
- Brear, P.; North, A.; Iegre, J.; Georgiou, K.H.; Lubin, A.; Carro, L.; Green, W.; Sore, H.F.; Hyvönen, M.; Spring, D.R. Novel non-ATP competitive small molecules targeting the CK2 α/β interface. Bioorg Med. Chem. 2018, 26, 3016–3020. [Google Scholar] [CrossRef] [PubMed]
- Iegre, J.; Brear, P.; De Fusco, C.; Yoshida, M.; Mitchell, S.L.; Rossmann, M.; Carro, L.; Sore, H.F.; Hyvönen, M.; Spring, D.R. Second-generation CK2α inhibitors targeting the αD pocket. Chem. Sci. 2018, 9, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Zhang, X.; Attarian, D.; Kraus, V.B. Synergistic roles of CBX4 chromo and SIM domains in regulating senescence of primary human osteoarthritic chondrocytes. Arthritis Res. Ther. 2023, 25, 197. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Niwa, Y.; Kuwata, K.; Srivastava, A.; Hyoda, T.; Tsuchiya, Y.; Kumagai, M.; Tsuyuguchi, M.; Tamaru, T.; Sugiyama, A.; et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci. Adv. 2019, 5, eaau9060. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Sheth, P.R.; Basso, A.D.; Paliwal, S.; Gray, K.; Fischmann, T.O.; Le, H.V. Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett. 2011, 585, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, E.L.; Iegre, J.; Brear, P.; A Zhabina, E.; Hyvönen, M.; Spring, D. Downfalls of Chemical Probes Acting at the Kinase ATP-Site: CK2 as a Case Study. Molecules 2021, 26, 1977. [Google Scholar] [CrossRef]
- Schneider, C.C.; Hessenauer, A.; Götz, C.; Montenarh, M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol. Rep. 2009, 21, 1593–1597. [Google Scholar]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: A novel powerful and selective inhibitor of protein kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an Anthraquinone Derivative Isolated from the Rhizomes of Rheum palmatum, Selectively Inhibits the Activity of Casein Kinase II as a Competitive Inhibitor. Planta Medica 1999, 65, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, S.L.; Dou, W.; Zhang, S.; Chen, J.H.; Shen, Y.; Shen, J.H.; Leng, Y. Emodin, a natural product, selectively inhibits 11beta-hydroxysteroid dehydrogenase type 1 and ameliorates metabolic disorder in diet-induced obese mice. Br. J. Pharmacol. 2010, 161, 113–126. [Google Scholar] [CrossRef]
- Sarno, S.; Moro, S.; Meggio, F.; Zagotto, G.; Ben, D.D.; Ghisellini, P.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol. Ther. 2002, 93, 159–168. [Google Scholar] [CrossRef]
- Cozza, G.; Mazzorana, M.; Papinutto, E.; Bain, J.; Elliott, M.; di Maira, G.; Gianoncelli, A.; Pagano, M.A.; Sarno, S.; Ruzzene, M.; et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem. J. 2009, 421, 387–395. [Google Scholar] [CrossRef]
- Franchin, C.; Cesaro, L.; Salvi, M.; Millioni, R.; Iori, E.; Cifani, P.; James, P.; Arrigoni, G.; Pinna, L. Quantitative analysis of a phosphoproteome readily altered by the protein kinase CK2 inhibitor quinalizarin in HEK-293T cells. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2014, 1854, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Debreczeni, J.; Fedorov, O.Y.; Nelson, A.; Marsden, B.D.; Knapp, S. Structural Basis of Inhibitor Specificity of the Human Protooncogene Proviral Insertion Site in Moloney Murine Leukemia Virus (PIM-1) Kinase. J. Med. Chem. 2005, 48, 7604–7614. [Google Scholar] [CrossRef]
- Mathison, C.J.N.; Chianelli, D.; Rucker, P.V.; Nelson, J.; Roland, J.; Huang, Z.; Yang, Y.; Jiang, J.; Xie, Y.F.; Epple, R.; et al. Efficacy and Tolerability of Pyrazolo [1,5-a]pyrimidine RET Kinase Inhibitors for the Treatment of Lung Adenocarcinoma. ACS Med. Chem. Lett. 2020, 11, 558–565. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Krämer, A.; Kurz, C.G.; Berger, B.-T.; Celik, I.E.; Tjaden, A.; Greco, F.A.; Knapp, S.; Hanke, T. Optimization of pyrazolo[1,5-a]pyrimidines lead to the identification of a highly selective casein kinase 2 inhibitor. Eur. J. Med. Chem. 2020, 208, 112770. [Google Scholar] [CrossRef]
- Licciardello, M.P.; Workman, P. A New Chemical Probe Challenges the Broad Cancer Essentiality of CK2. Trends Pharmacol. Sci. 2021, 42, 313–315. [Google Scholar] [CrossRef]
- Wells, C.I.; Drewry, D.H.; Pickett, J.E.; Tjaden, A.; Krämer, A.; Müller, S.; Gyenis, L.; Menyhart, D.; Litchfield, D.W.; Knapp, S.; et al. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem. Biol. 2021, 28, 546–558.e10. [Google Scholar] [CrossRef]
- Yao, K.; Youn, H.; Gao, X.; Huang, B.; Zhou, F.; Li, B.; Han, H. Casein kinase 2 inhibition attenuates androgen receptor function and cell proliferation in prostate cancer cells. Prostate 2012, 72, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Poletto, G.; Di Maira, G.; Cozza, G.; Ruzzene, M.; Sarno, S.; Bain, J.; Elliott, M.; Moro, S.; Zagotto, G.; et al. Tetrabromocinnamic Acid (TBCA) and Related Compounds Represent a New Class of Specific Protein Kinase CK2 Inhibitors. ChemBioChem 2007, 8, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Golub, A.G.; Bdzhola, V.G.; Kyshenia, Y.V.; Sapelkin, V.M.; Prykhod’ko, A.O.; Kukharenko, O.P.; Ostrynska, O.V.; Yarmoluk, S.M. Structure-based discovery of novel flavonol inhibitors of human protein kinase CK2. Mol. Cell. Biochem. 2011, 356, 107–115. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.-G. Protein Kinase CK2 in Human Diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pagano, M.A.; Moro, S.; Zagotto, G.; Ruzzene, M.; Sarno, S.; Cozza, G.; Bain, J.; Elliott, M.; Deana, A.D.; et al. Inhibition of Protein Kinase CK2 by Condensed Polyphenolic Derivatives. An in Vitro and in Vivo Study. Biochemistry 2004, 43, 12931–12936. [Google Scholar] [CrossRef] [PubMed]
- Brear, P.; De Fusco, C.; Georgiou, K.H.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvönen, M. Specific inhibition of CK2α from an anchor outside the active site. Chem. Sci. 2016, 7, 6839–6845. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Gou, S. A platinum(II) complex HY1-Pt overcomes cisplatin-induced resistance and attenuates metastasis of epithelial ovarian cancer by cancer cell stemness inhibition. Int. J. Biochem. Cell Biol. 2023, 157, 106395. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, P.; Zou, L.; Zhang, J.; Chen, J.; Yang, C.; He, G.; Liu, B.; Liu, J.; Chiang, C.M.; et al. Discovery of Novel Dual-Target Inhibitor of Bromodomain-Containing Protein 4/Casein Kinase 2 Inducing Apoptosis and Autophagy-Associated Cell Death for Triple-Negative Breast Cancer Therapy. J. Med. Chem. 2021, 64, 18025–18053. [Google Scholar] [CrossRef]
- Calder, J.; Nagelberg, A.; Luu, J.; Lu, D.; Lockwood, W.W. Resistance to BET inhibitors in lung adenocarcinoma is mediated by casein kinase phosphorylation of BRD4. Oncogenesis 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Channavajhala, P.; Seldin, D.C. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 2002, 21, 5280–5288. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, J.; Roh, M.; O E Franco, O.; Hayward, S.W.; Wills, M.L.; A Abdulkadir, S. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef]
- Wińska, P.; Wielechowska, M.; Koronkiewicz, M.; Borowiecki, P. Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1. Pharmaceutics 2023, 15, 1991. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Sautel, C.F.; Barette, C.; Lafanechère, L.; Mouawad, L.; Grierson, D.; Schmidt, F.; Filippakopoulos, P.; Bullock, A.N.; et al. New potent dual inhibitors of CK2 and Pim kinases: Discovery and structural insights. FASEB J. 2010, 24, 3171–3185. [Google Scholar]
- Sarno, S.; Mazzorana, M.; Traynor, R.; Ruzzene, M.; Cozza, G.; Pagano, M.A.; Meggio, F.; Zagotto, G.; Battistutta, R.; Pinna, L.A. Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: Role of a nitro group that discriminates between CK2 and DYRK1A. Cell. Mol. Life Sci. 2011, 69, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Łukowska-Chojnacka, E.; Wińska, P.; Wielechowska, M.; Poprzeczko, M.; Bretner, M. Synthesis of novel polybrominated benzimidazole derivatives-potential CK2 inhibitors with anticancer and proapoptotic activity. Bioorg Med. Chem. 2016, 24, 735–741. [Google Scholar] [CrossRef]
- Morooka, S.; Hoshina, M.; Kii, I.; Okabe, T.; Kojima, H.; Inoue, N.; Okuno, Y.; Denawa, M.; Yoshida, S.; Fukuhara, J.; et al. Identification of a Dual Inhibitor of SRPK1 and CK2 That Attenuates Pathological Angiogenesis of Macular Degeneration in Mice. Mol. Pharmacol. 2015, 88, 316–325. [Google Scholar] [CrossRef]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.R.C.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 inhibition by 108600 suppresses triple negative breast cancer stem cells and chemotherapy-resistant disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef]
- Perera, Y.; Ramos, Y.; Padrón, G.; Caballero, E.; Guirola, O.; Caligiuri, L.G.; Lorenzo, N.; Gottardo, F.; Farina, H.G.; Filhol, O.; et al. CIGB-300 anticancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 2020, 470, 63–75. [Google Scholar] [CrossRef]
- Farina, H.G.; Acero, F.B.; Perera, Y.; Rodríguez, A.; Perea, S.E.; Castro, B.A.; Gomez, R.; Alonso, D.F.; Gomez, D.E. CIGB-300, a proapoptotic peptide, inhibits angiogenesis in vitro and in vivo. Exp. Cell Res. 2011, 317, 1677–1688. [Google Scholar] [CrossRef]
- Gottardo, M.F.; Capobianco, C.S.; Sidabra, J.E.; Garona, J.; Perera, Y.; Perea, S.E.; Alonso, D.F.; Farina, H.G. Preclinical efficacy of CIGB-300, an anti-CK2 peptide, on breast cancer metastasic colonization. Sci. Rep. 2020, 10, 14689. [Google Scholar] [CrossRef]
- Zien, P.; Duncan, J.S.; Skierski, J.; Bretner, M.; Litchfield, D.W.; Shugar, D. Tetrabromobenzotriazole (TBBt) and tetrabromobenzimidazole (TBBz) as selective inhibitors of protein kinase CK2: Evaluation of their effects on cells and different molecular forms of human CK2. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2005, 1754, 271–280. [Google Scholar] [CrossRef]
- Koo, J.-H.; Yu, H.C.; Nam, S.; Kim, D.-C.; Lee, J.H. Casein Kinase 2 Alpha Inhibition Protects against Sepsis-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2023, 24, 9783. [Google Scholar] [CrossRef]
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2014, 34, 3688–3699. [Google Scholar] [CrossRef]
- Wang, T.; Yang, N.; Liang, C.; Xu, H.; An, Y.; Xiao, S.; Zheng, M.; Liu, L.; Wang, G.; Nie, L. Detecting Protein-Protein Interaction Based on Protein Fragment Complementation Assay. Curr. Protein Pept. Sci. 2020, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Bailey-Kellogg, C. Protein interaction interface region prediction by geometric deep learning. Bioinformatics 2021, 37, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Berillo, D.; Yeskendir, A.; Zharkinbekov, Z.; Raziyeva, K.; Saparov, A. Peptide-Based Drug Delivery Systems. Medicina 2021, 57, 1209. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Zhang, L.; Chen, P. Membrane Internalization Mechanisms and Design Strategies of Arginine-Rich Cell-Penetrating Peptides. Int. J. Mol. Sci. 2022, 23, 9038. [Google Scholar] [CrossRef]
- De Oliveira, E.C.L.; da Costa, K.S.; Taube, P.S.; Lima, A.H.; Junior, C.D.S.D.S. Biological Membrane-Penetrating Peptides: Computational Prediction and Applications. Front. Cell Infect. Microbiol. 2022, 12, 838259. [Google Scholar] [CrossRef]
- Lai, Y.; Fois, G.; Flores, J.R.; Tuvim, M.J.; Zhou, Q.; Yang, K.; Leitz, J.; Peters, J.; Zhang, Y.; Pfuetzner, R.A.; et al. Inhibition of calcium-triggered secretion by hydrocarbon-stapled peptides. Nature 2022, 603, 949–956. [Google Scholar] [CrossRef]
- Dong, C.Z.; Bruzzoni-Giovanelli, H.; Yu, Y.; Dorgham, K.; Parizot, C.; Zini, J.M.; Brossas, J.Y.; Tuffery, P.; Rebollo, A. Identification of peptides interfering with the LRRK2/PP1 interaction. PLoS ONE 2020, 15, e0237110. [Google Scholar] [CrossRef]
- Dergunova, L.V.; Filippenkov, I.B.; Limborska, S.A.; Myasoedov, N.F. Neuroprotective Peptides and New Strategies for Ischemic Stroke Drug Discoveries. Genes 2023, 14, 953. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Loisel, S.; Sidorenko, V.; Scodeller, P.; Parizot, C.; Savier, E.; Haute, T.; Teesalu, T.; Rebollo, A. Preclinical Validation of Tumor-Penetrating and Interfering Peptides against Chronic Lymphocytic Leukemia. Mol. Pharm. 2022, 19, 895–903. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; Chen, Z.; Chen, Y.; Lin, Q.; Liang, Y. Exogenous Bioactive Peptides Have a Potential Therapeutic Role in Delaying Aging in Rodent Models. Int. J. Mol. Sci. 2022, 23, 1421. [Google Scholar] [CrossRef]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein-protein interactions: The next generation of drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Lisberg, A.; Ellis, R.; Nicholson, K.; Moku, P.; Swarup, A.; Dhurjati, P.; Nohe, A. Mathematical modeling of the effects of CK2.3 on mineralization in osteoporotic bone. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; Gurski, L.; Bonor, J.; Price, C.; Wang, L.; Beamer, W.G.; Nohe, A. Casein kinase 2 regulates in vivo bone formation through its interaction with bone morphogenetic protein receptor type Ia. Bone 2011, 49, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Akkiraju, H.; Bonor, J.; Nohe, A. CK2.1, a novel peptide, induces articular cartilage formation in vivo. J. Orthop. Res. 2016, 35, 876–885. [Google Scholar] [CrossRef]
- Akkiraju, H.; Srinivasan, P.P.; Xu, X.; Jia, X.; Safran, C.B.K.; Nohe, A. CK2.1, a bone morphogenetic protein receptor type Ia mimetic peptide, repairs cartilage in mice with destabilized medial meniscus. Stem Cell Res. Ther. 2017, 8, 82. [Google Scholar] [CrossRef]
- Takahashi, K.; Setoguchi, T.; Tsuru, A.; Saitoh, Y.; Nagano, S.; Ishidou, Y.; Maeda, S.; Furukawa, T.; Komiya, S. Inhibition of casein kinase 2 prevents growth of human osteosarcoma. Oncol. Rep. 2016, 37, 1141–1147. [Google Scholar] [CrossRef]
- Halloran, D.; Pandit, V.; MacMurray, C.; Stone, V.; DeGeorge, K.; Eskander, M.; Root, D.; McTague, S.; Pelkey, H.; Nohe, A. Age-Related Low Bone Mineral Density in C57BL/6 Mice Is Reflective of Aberrant Bone Morphogenetic Protein-2 Signaling Observed in Human Patients Diagnosed with Osteoporosis. Int. J. Mol. Sci. 2022, 23, 11205. [Google Scholar] [CrossRef] [PubMed]
- Vrathasha, V.; Weidner, H.; Nohe, A. Mechanism of CK2.3, a Novel Mimetic Peptide of Bone Morphogenetic Protein Receptor Type IA, Mediated Osteogenesis. Int. J. Mol. Sci. 2019, 10, 2500. [Google Scholar] [CrossRef] [PubMed]
- Weidner, H.; Gao, V.Y.; Dibert, D.; McTague, S.; Eskander, M.; Duncan, R.; Wang, L.; Nohe, A. CK2.3, a Mimetic Peptide of the BMP Type I Receptor, Increases Activity in Osteoblasts over BMP2. Int. J. Mol. Sci. 2019, 20, 5877. [Google Scholar] [CrossRef] [PubMed]
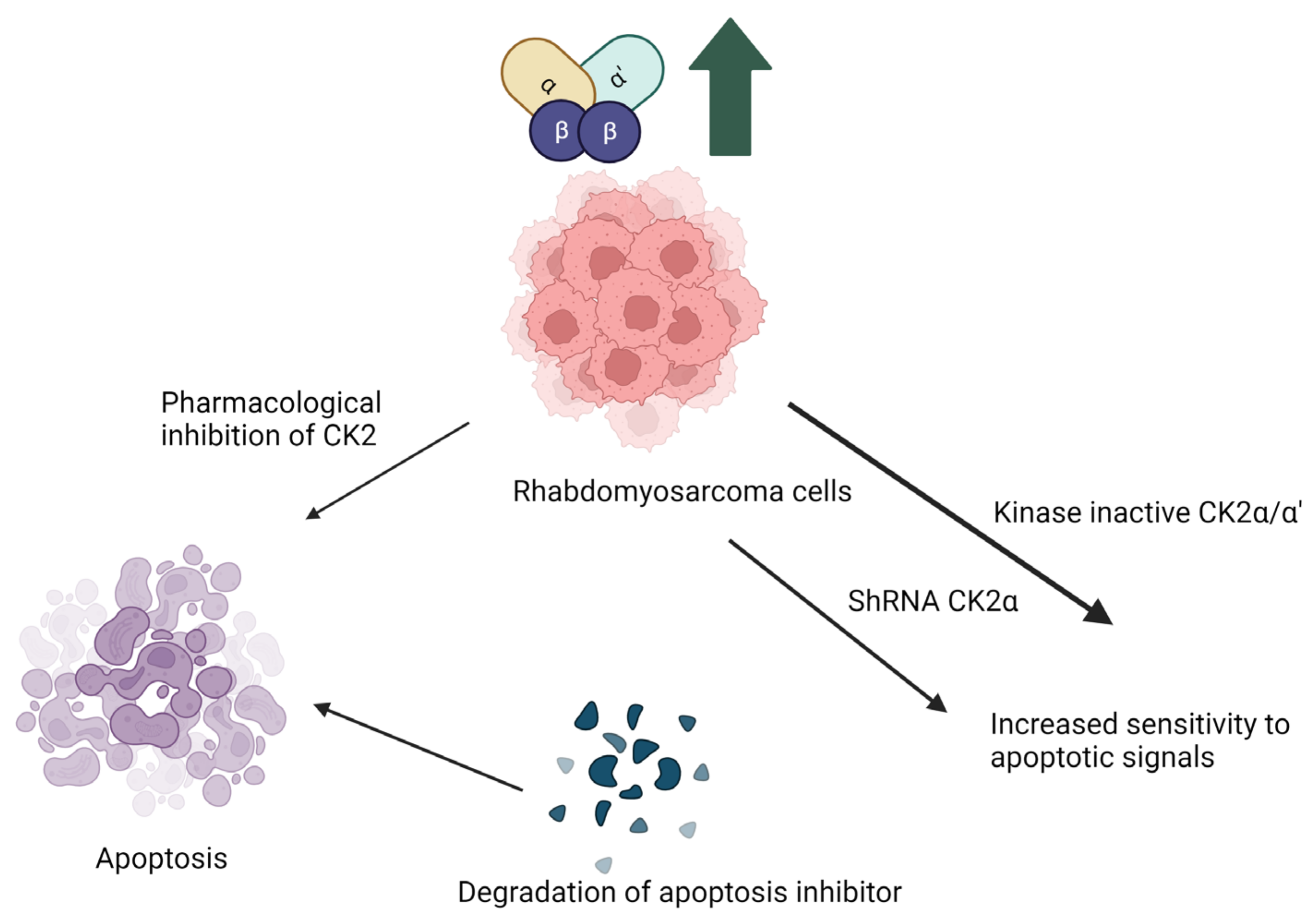
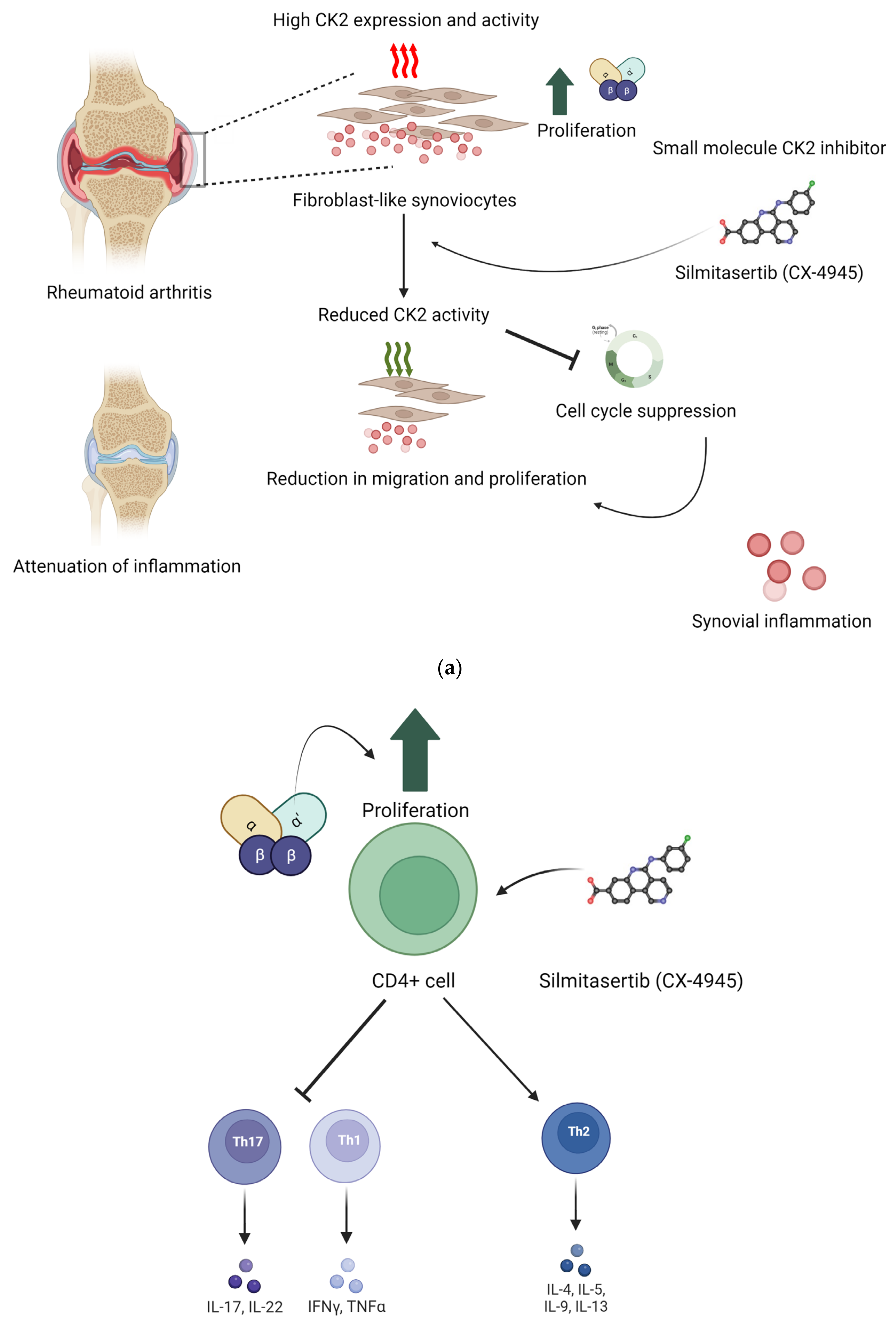
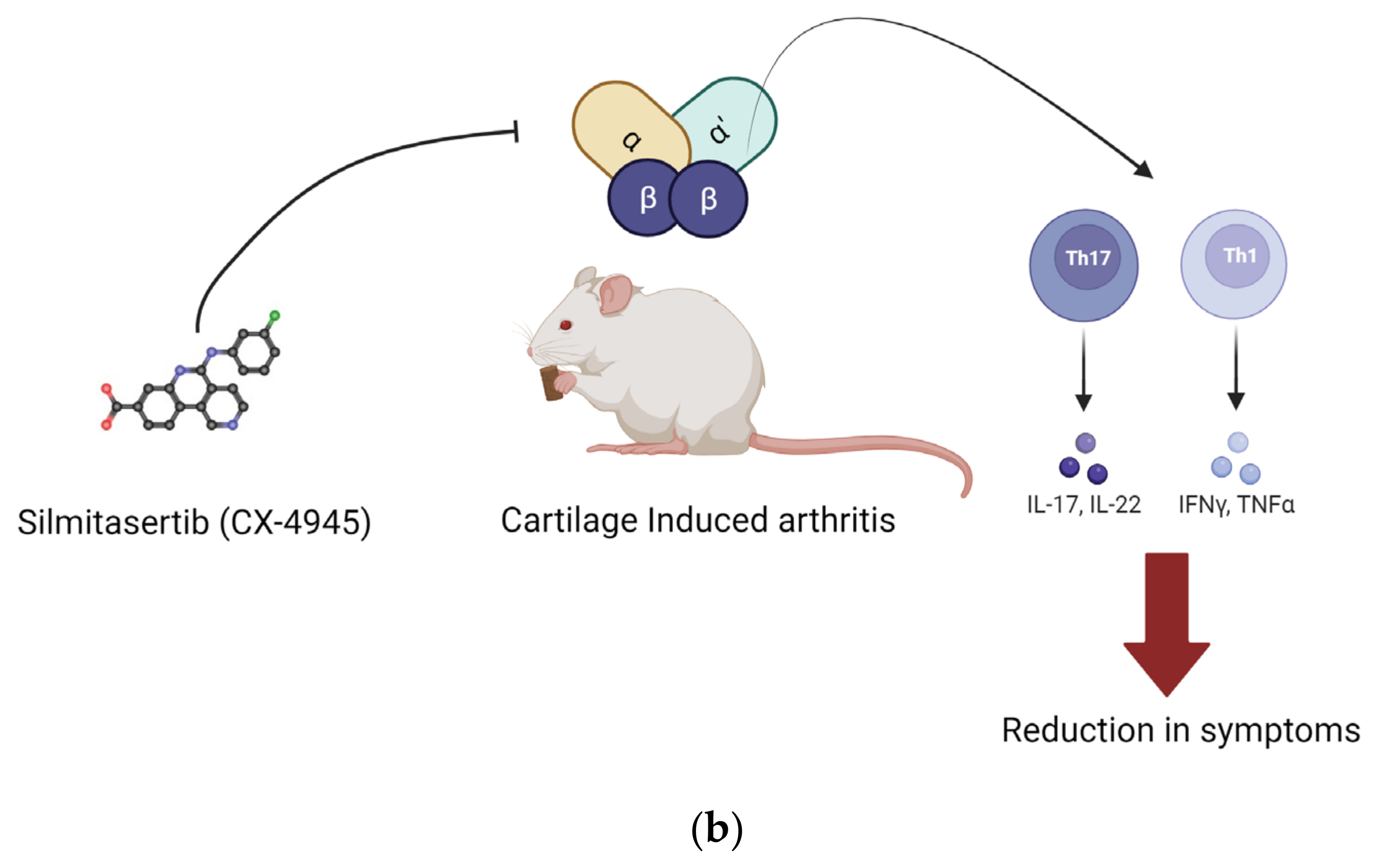
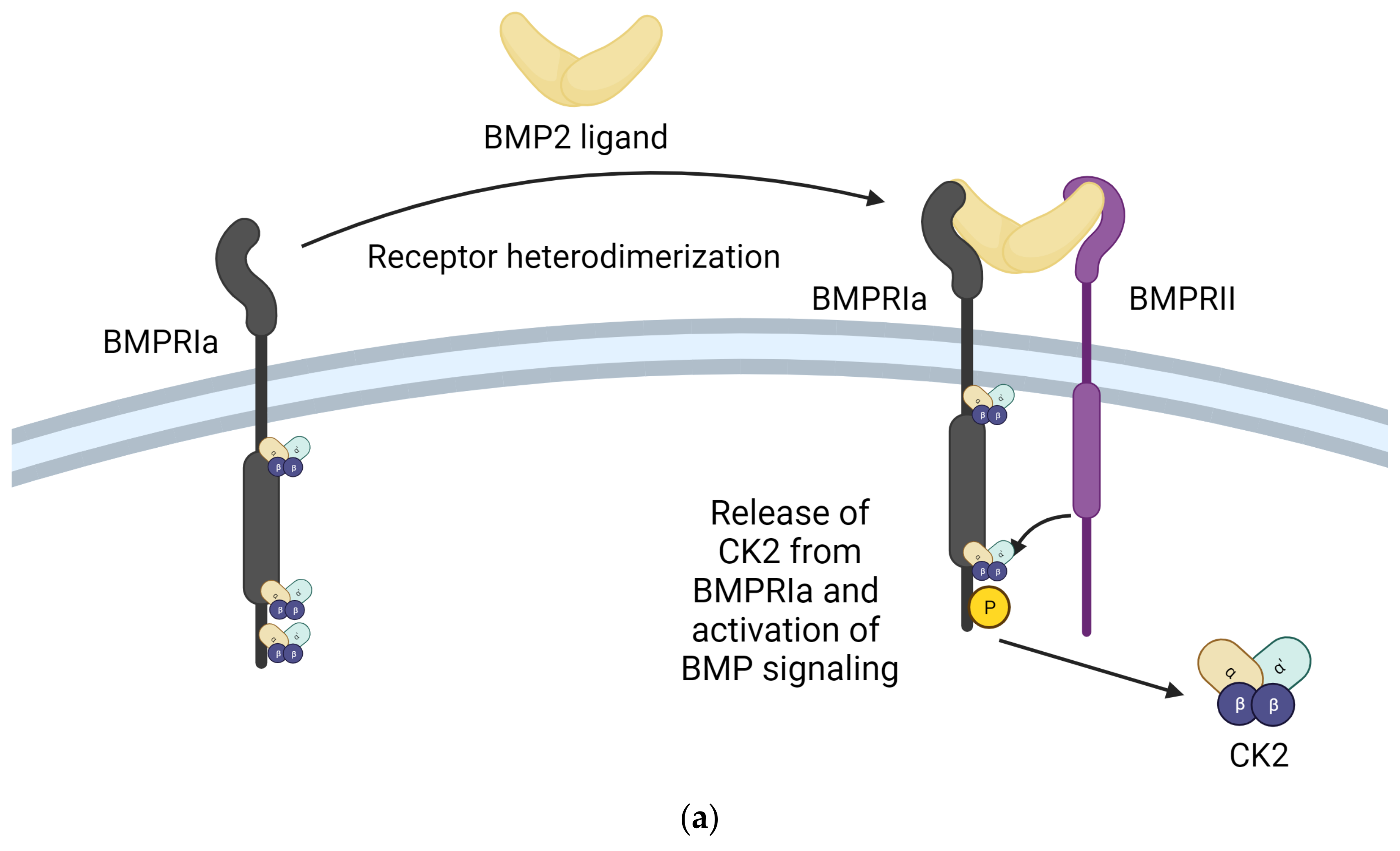
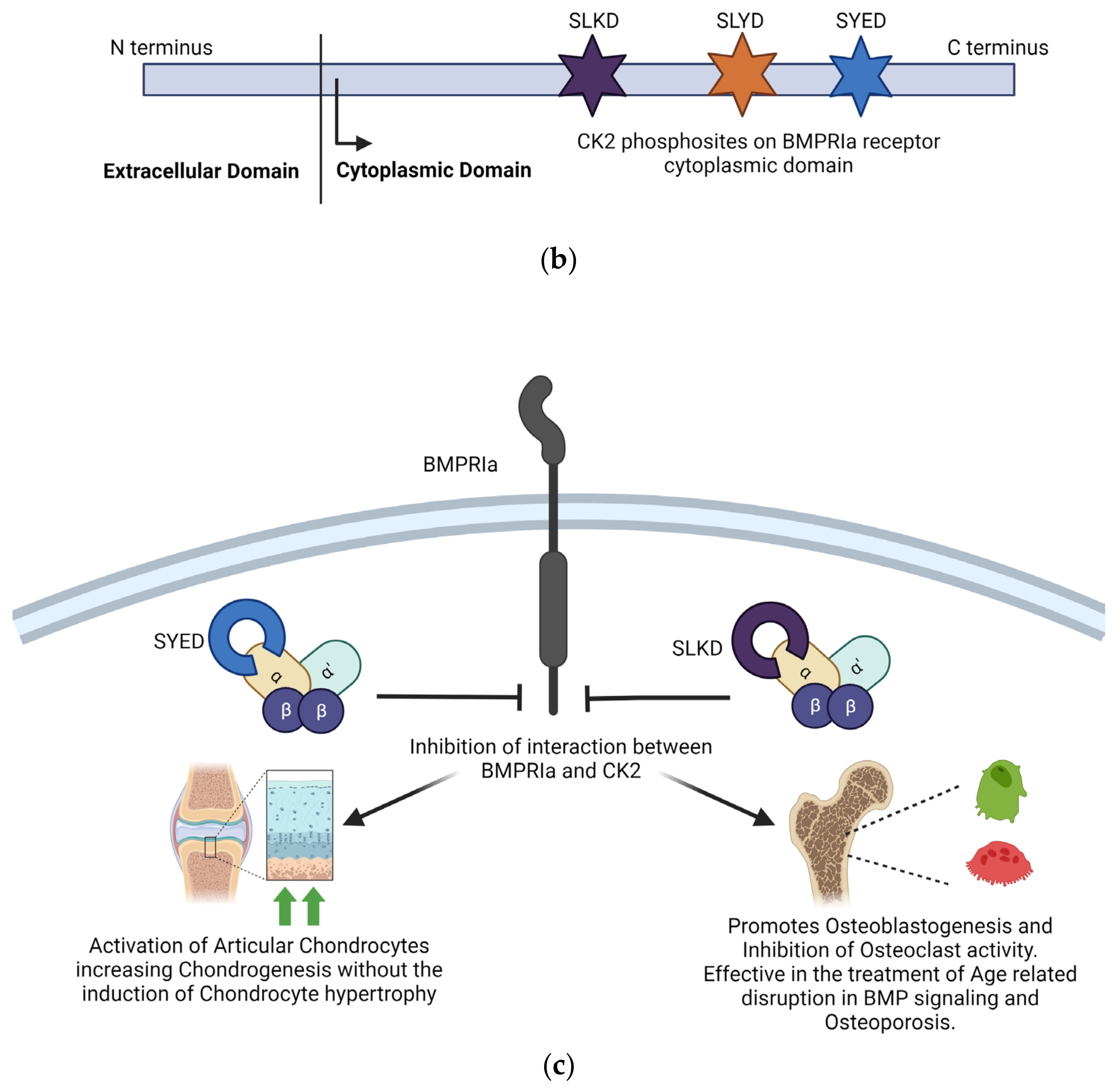
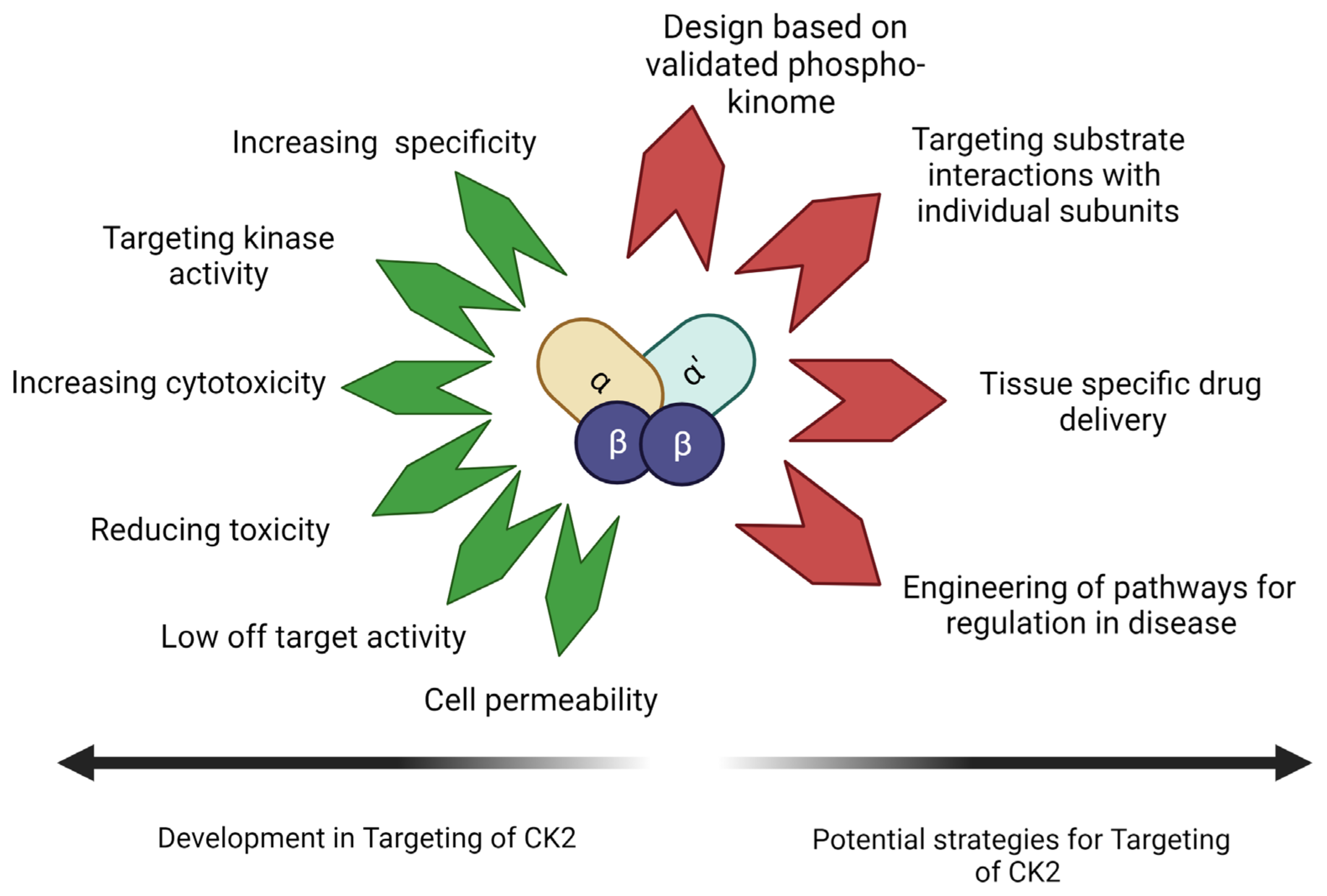
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).