


In Silico Therapeutic Study: The Next Frontier in the Fight against SARS-CoV-2 and Its Variants



Abstract
1. Introduction
2. Modeling for CADD with Small Molecules
2.1. Computational Peptide Inhibition Studies
2.2. Quantitative Structure–Activity Relationships (QSAR) Mapping
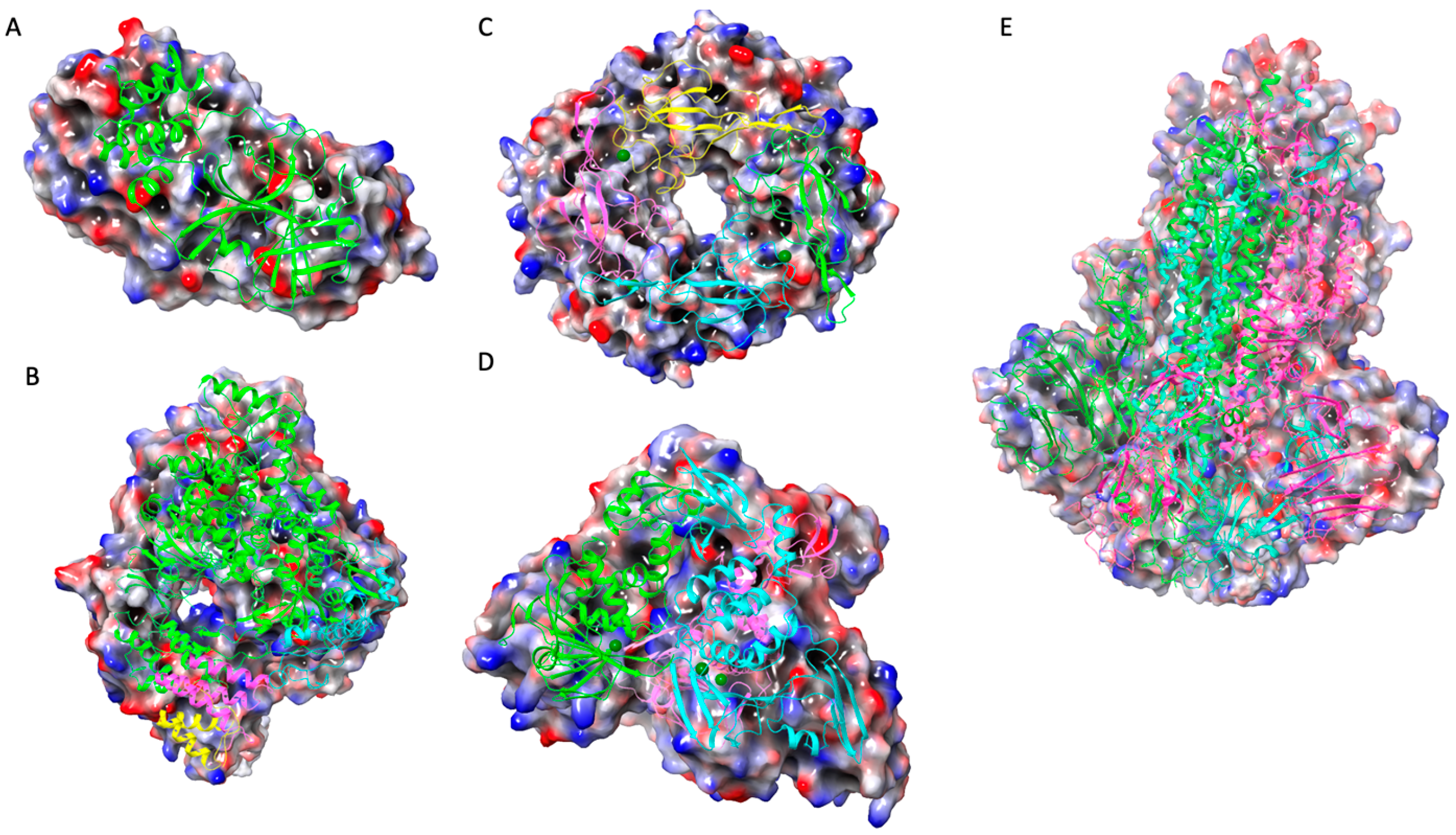
2.3. Antibody Docking
2.4. ML for Inhibitor Screening
2.5. ML for Antibody Screening
2.6. Nanoinformatics for SARS-CoV-2 Inhibition
3. Future Prospects of Computational Therapeutic Study for SARS-CoV-2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment out Coronavirus (COVID-19); StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Niknam, Z.; Jafari, A.; Golchin, A.; Danesh Pouya, F.; Nemati, M.; Rezaei-Tavirani, M.; Rasmi, Y.J. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 2022, 27, 6. [Google Scholar] [CrossRef] [PubMed]
- Chera, A.; Tanca, A. Remdesivir: The first FDA-approved anti-COVID-19 Treatment for Young Children. Discoveries 2022, 10, e151. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S. Remdesivir for the treatment of COVID-19. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Rezagholizadeh, A.; Khiali, S.; Sarbakhsh, P.; Entezari-Maleki, T. Remdesivir for treatment of COVID-19; an updated systematic review and meta-analysis. Eur. J. Pharmacol. 2021, 897, 173926. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsalam, S.; Salama, M.; Soliman, S.; Naguib, A.M.; Ibrahim, I.S.; Torky, M.; Abd El Ghafar, M.S.; Abdul-Baki, E.A.-R.M.; Elhendawy, M. Remdesivir efficacy in COVID-19 treatment: A randomized controlled trial. Am. J. Trop. Med. Hyg. 2022, 106, 886. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, A.M.; Eckert, I.; Tramujas, L.; Butler-Laporte, G.; McDonald, E.G.; Brophy, J.M.; Lee, T.C. Effect of tocilizumab, sarilumab, and baricitinib on mortality among patients hospitalized for COVID-19 treated with corticosteroids: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2023, 29, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Baghi, H.B.; Nahand, J.S.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to Covid-19 therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Toussi, S.S.; Hammond, J.L.; Gerstenberger, B.S.; Anderson, A.S. Therapeutics for COVID-19. Nat. Microbiol. 2023, 8, 771–786. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An updated review of computer-aided drug design and its application to COVID-19. BioMed. Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef]
- Keshavarzi Arshadi, A.; Webb, J.; Salem, M.; Cruz, E.; Calad-Thomson, S.; Ghadirian, N.; Collins, J.; Diez-Cecilia, E.; Kelly, B.; Goodarzi, H. Artificial intelligence for COVID-19 drug discovery and vaccine development. Front. Artif. Intell. 2020, 65, 1–13. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, D.; Zhang, X.; Shi, Y.; Han, J.; Zhou, L.; Wu, L.; Ma, M.; Li, J.; Peng, S. D3AI-CoV: A deep learning platform for predicting drug targets and for virtual screening against COVID-19. Brief. Bioinform. 2022, 23, bbac147. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiao, Y.; Shi, C.; Zhang, Y.J. Deep learning-based molecular dynamics simulation for structure-based drug design against SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 5014–5027. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.-T.; Pandey, M.; Smith, J.R.; Ban, F.; Fernandez, M.; Cherkasov, A. Targeting SARS-CoV-2 papain-like protease in the post-vaccine era. Trends Pharmacol. Sci. 2022, 43, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Réau, M.; Langenfeld, F.; Zagury, J.-F.; Lagarde, N.; Montes, M. Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef]
- Jain, H.; Meshram, D.; Desai, S. Introduction and Application of Quantitative Structure Activity Relationship: A Review. Syst. Rev. Pharm. 2023, 14, 465. [Google Scholar]
- Nogueira, M.S.; Koch, O. The development of target-specific machine learning models as scoring functions for docking-based target prediction. J. Chem. Inf. Model. 2019, 59, 1238–1252. [Google Scholar] [CrossRef]
- Da Silva Rocha, S.F.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M. Virtual screening techniques in drug discovery: Review and recent applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Braga, R.C.; Alves, V.M.; Silva, A.C.; Nascimento, M.N.; Silva, F.C.; Liao, L.M.; Andrade, C.J.H. Virtual screening strategies in medicinal chemistry: The state of the art and current challenges. Curr. Top. Med. Chem. 2014, 14, 1899–1912. [Google Scholar] [CrossRef]
- Ghosh, D.; Ghosh Dastidar, D.; Roy, K.; Ghosh, A.; Mukhopadhyay, D.; Sikdar, N.; Biswas, N.K.; Chakrabarti, G.; Das, A. Computational prediction of the molecular mechanism of statin group of drugs against SARS-CoV-2 pathogenesis. Sci. Rep. 2022, 12, 6241. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Pathania, S.; Rawal, R. Exploring RdRp–remdesivir interactions to screen RdRp inhibitors for the management of novel coronavirus 2019-nCoV. SAR QSAR Environ. Res. 2020, 31, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Saloni; Kumari, D.; Ranjan, P.; Chakraborty, T. A computational study of potential therapeutics for COVID-19 invoking conceptual density functional theory. Struct. Chem. 2022, 33, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, F.; Xu, X.; Gao, X. Impact of computational approaches in the fight against COVID-19: An AI guided review of 17,000 studies. Brief. Bioinform. 2022, 23, bbab456. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Fuochi, V.; Furneri, P.M.; Rescifina, A.J. Natural Substances in the Fight of SARS-CoV-2: A Critical Evaluation Resulting from the Cross-Fertilization of Molecular Modeling Data with the Pharmacological Aspects. Curr. Med. Chem. 2021, 28, 8333–8383. [Google Scholar] [CrossRef]
- Bhavaniramya, S.; Ramar, V.; Vishnupriya, S.; Palaniappan, R.; Sibiya, A.; Vaseeharan, B. Comprehensive analysis of SARS-COV-2 drug targets and pharmacological aspects in treating the COVID-19. Curr. Mol. Pharmacol. 2022, 15, 393–417. [Google Scholar] [CrossRef]
- Yang, H.; Bartlam, M.; Rao, Z.J. Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr. Pharm. Des. 2006, 12, 4573–4590. [Google Scholar] [CrossRef]
- Kar, S.; Leszczynski, J.J. Drug databases for development of therapeutics against coronaviruses. In In Silico Modeling of Drugs Against Coronaviruses: Computational Tools; Roy, K., Ed.; Humana: New York, NY, USA, 2021; pp. 761–780. [Google Scholar]
- Saikia, S.; Bordoloi, M. Molecular docking: Challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Keith, J.A.; Vassilev-Galindo, V.; Cheng, B.; Chmiela, S.; Gastegger, M.; Müller, K.-R.; Tkatchenko, A. Combining machine learning and computational chemistry for predictive insights into chemical systems. Chem. Rev. 2021, 121, 9816–9872. [Google Scholar] [CrossRef]
- Serafim, M.S.; Gertrudes, J.C.; Costa, D.M.; Oliveira, P.R.; Maltarollo, V.G.; Honorio, K.M.J. Knowing and combating the enemy: A brief review on SARS-CoV-2 and computational approaches applied to the discovery of drug candidates. Biosci. Rep. 2021, 41, BSR20202616. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kovalenko, S.; Bhardwaj, S.; Sethi, A.; Gorobets, N.Y.; Desenko, S.M.; Rathi, B. Drug repurposing against SARS-CoV-2 using computational approaches. Drug Discov. Today 2022, 27, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, K.; Yazdanpanah, N.; Saghazadeh, A.; Rezaei, N. Computational drug discovery and repurposing for the treatment of COVID-19: A systematic review. Bioorg. Chem. 2021, 106, 104490. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Villoutreix, B.O. Resources and computational strategies to advance small molecule SARS-CoV-2 discovery: Lessons from the pandemic and preparing for future health crises. Comput. Struct. Biotechnol. J. 2021, 19, 2537–2548. [Google Scholar] [CrossRef]
- Prasad, K.; Kumar, V. Artificial intelligence-driven drug repurposing and structural biology for SARS-CoV-2. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100042. [Google Scholar] [CrossRef]
- Arora, G.; Joshi, J.; Mandal, R.S.; Shrivastava, N.; Virmani, R.; Sethi, T. Artificial intelligence in surveillance, diagnosis, drug discovery and vaccine development against COVID-19. Pathogens 2021, 10, 1048. [Google Scholar] [CrossRef]
- Lv, H.; Shi, L.; Berkenpas, J.W.; Dao, F.-Y.; Zulfiqar, H.; Ding, H.; Zhang, Y.; Yang, L.; Cao, R. Application of artificial intelligence and machine learning for COVID-19 drug discovery and vaccine design. Brief. Bioinform. 2021, 22, bbab320. [Google Scholar] [CrossRef]
- Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.; Idroes, R.; Tallei, T.; Emran, T. Interactions of the receptor binding domain of SARS-CoV-2 variants with hACE2: Insights from molecular docking analysis and molecular dynamic simulation. Biology 2021, 10, 880. [Google Scholar] [CrossRef]
- Arun, K.; Sharanya, C.; Abhithaj, J.; Francis, D.; Sadasivan, C.J. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 2021, 39, 4647–4658. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Rensi, S.; Keys, A.; Lo, Y.-C.; Derry, A.; McInnes, G.; Liu, T.; Altman, R. Homology modeling of TMPRSS2 yields candidate drugs that may inhibit entry of SARS-CoV-2 into human cells. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef] [PubMed]
- Chikhale, R.V.; Gupta, V.K.; Eldesoky, G.E.; Wabaidur, S.M.; Patil, S.A.; Islam, M.A. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2021, 39, 6660–6675. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.M.; Ismail, M.I.; Bauer, M.R.; Bekhit, A.A.; Boeckler, F.M. Supporting SARS-CoV-2 papain-like protease drug discovery: In silico methods and benchmarking. Front. Chem. 2020, 8, 592289. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Abubaker, J.; Al-Mulla, F. Structural modelling of SARS-CoV-2 alpha variant (B.1.1.7) suggests enhanced furin binding and infectivity. Virus Res. 2021, 303, 198522. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Javali, P.S.; Pandianb, C.J.; Ali, M.A.; Srivastava, V.; Jeyaraman, J. Computational investigation of the increased virulence and pathogenesis of SARS-CoV-2 lineage B. 1.1. 7. Phys. Chem. Chem. Phys. 2022, 24, 20371–20380. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Lee, S.-S.; Seo, E.-M.; Chakraborty, C.B. 1.1. 7 (Alpha) variant is the most antigenic compared to Wuhan strain, B. 1.351, B. 1.1. 28/triple mutant and B. 1.429 variants. Front. Microbiol. 2022, 13, 895695. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA. 1) and sub-variants (BA. 1.1, BA. 2, and BA. 3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Ovchynnykova, O.; Kapusta, K.; Sizochenko, N.; Sukhyy, K.M.; Kolodziejczyk, W.; Hill, G.A.; Saloni, J. Homology modeling and molecular dynamics-driven search for natural inhibitors that universally target receptor-binding domain of spike glycoprotein in SARS-CoV-2 variants. Molecules 2022, 27, 7336. [Google Scholar] [CrossRef]
- Eweas, A.F.; Osman, H.-E.H.; Naguib, I.A.; Abourehab, M.A.; Abdel-Moneim, A.S. Virtual screening of repurposed drugs as potential spike protein inhibitors of different SARS-CoV-2 variants: Molecular Docking Study. Curr. Issues Mol. Biol. 2022, 44, 3018–3029. [Google Scholar] [CrossRef]
- Yang, Q.; Jian, X.; Syed, A.A.S.; Fahira, A.; Zheng, C.; Zhu, Z.; Wang, K.; Zhang, J.; Wen, Y.; Li, Z.; et al. Structural comparison and drug screening of spike proteins of ten SARS-CoV-2 variants. Research 2022, 2022, 9781758. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Zhao, X.; Luo, S.; Zhang, J.Z.; Duan, L. Molecular mechanism of the non-covalent orally targeted SARS-CoV-2 Mpro inhibitor S-217622 and computational assessment of its effectiveness against mainstream variants. J. Phys. Chem. Lett. 2022, 13, 8893–8901. [Google Scholar] [CrossRef] [PubMed]
- Quan, B.-X.; Shuai, H.; Xia, A.-J.; Hou, Y.; Zeng, R.; Liu, X.-L.; Lin, G.-F.; Qiao, J.-X.; Li, W.-P.; Wang, F.-L. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 2022, 7, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Costa, L.S.; Dos Santos, K.B.; Karl, A.L.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2021, 11, 5543. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Olson, K.M.; Wotring, J.W.; Sexton, J.Z.; Carlson, H.A.; Traynor, J.R. In silico analysis of SARS-CoV-2 proteins as targets for clinically available drugs. Sci. Rep. 2022, 12, 5320. [Google Scholar] [CrossRef]
- Liscano, Y.; Oñate-Garzón, J.; Ocampo-Ibáñez, I.D. In silico discovery of antimicrobial peptides as an alternative to control SARS-CoV-2. Molecules 2020, 25, 5535. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Jamal, Q.M.S.; Rehman, S.; Almatroudi, A.; Alzohairy, M.A.; Alomary, M.N.; Tripathi, T.; Alharbi, A.H.; Adil, S.F.; Khan, M.J.; et al. TAT-peptide conjugated repurposing drug against SARS-CoV-2 main protease (3CLpro): Potential therapeutic intervention to combat COVID-19. Arab. J. Chem. 2020, 13, 8069–8079. [Google Scholar] [CrossRef]
- Balmeh, N.; Mahmoudi, S.; Fard, N.A. Manipulated bio antimicrobial peptides from probiotic bacteria as proposed drugs for COVID-19 disease. Inform. Med. Unlocked 2021, 23, 100515. [Google Scholar] [CrossRef]
- Mohammadi, M.; Rajabi, S.; Piroozmand, A.; Mirhosseini, S.A. In silico study of pacific oyster antiviral polypeptides as potential inhibitory compounds for SARS-CoV-2 main protease. Jentashapir J. Cell. Mol. Biol. 2020, 11, e108932. [Google Scholar] [CrossRef]
- Stoddard, S.V.; Wallace, F.E.; Stoddard, S.D.; Cheng, Q.; Acosta, D.; Barzani, S.; Bobay, M.; Briant, J.; Cisneros, C.; Feinstein, S. In silico design of peptide-based SARS-CoV-2 fusion inhibitors that target wt and mutant versions of SARS-CoV-2 HR1 Domains. Biophysica 2021, 1, 311–327. [Google Scholar] [CrossRef]
- Tallei, T.E.; Fatimawali; Adam, A.A.; Elseehy, M.M.; El-Shehawi, A.M.; Mahmoud, E.A.; Tania, A.D.; Niode, N.J.; Kusumawaty, D.; Rahimah, S. Fruit bromelain-derived peptide potentially restrains the attachment of SARS-CoV-2 variants to hACE2: A pharmacoinformatics approach. Molecules 2022, 27, 260. [Google Scholar] [CrossRef] [PubMed]
- Rajpoot, S.; Solanki, K.; Kumar, A.; Zhang, K.Y.; Pullamsetti, S.S.; Savai, R.; Faisal, S.M.; Pan, Q.; Baig, M.S. In-silico design of a novel tridecapeptide targeting spike protein of SARS-CoV-2 variants of concern. Int. J. Pept. Res. Ther. 2022, 28, 28. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Banavath, H.N.; Godara, P.; Naik, B.; Srivastava, V.; Prusty, D. Identification of antiviral peptide inhibitors for receptor binding domain of SARS-CoV-2 omicron and its sub-variants: An in-silico approach. 3 Biotech 2022, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Ničkčović, V.P.; Nikolić, G.R.; Nedeljković, B.M.; Mitić, N.; Danić, S.F.; Mitić, J.; Marčetić, Z.; Sokolović, D.; Veselinović, A.M. In silico approach for the development of novel antiviral compounds based on SARS-CoV-2 protease inhibition. Chem. Pap. 2022, 76, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Oubahmane, M.; Hdoufane, I.; Delaite, C.; Sayede, A.; Cherqaoui, D.; El Allali, A. Design of Potent Inhibitors Targeting the Main Protease of SARS-CoV-2 Using QSAR Modeling, Molecular Docking, and Molecular Dynamics Simulations. Pharmaceuticals 2023, 16, 608. [Google Scholar] [CrossRef] [PubMed]
- Tamilarasi, W.; Balamurugan, B.J. ADMET and quantitative structure property relationship analysis of anti-Covid drugs against omicron variant with some degree-based topological indices. Int. J. Quantum Chem. 2022, 122, e26967. [Google Scholar] [CrossRef]
- Costa, A.S.; Martins, J.P.A.; de Melo, E.B. SMILES-based 2D-QSAR and similarity search for identification of potential new scaffolds for development of SARS-CoV-2 MPRO inhibitors. Struct. Chem. 2022, 33, 1691–1706. [Google Scholar] [CrossRef]
- Zaki, M.E.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Bajaj, S.O.; El-Sayed, N.N.; Ghosh, A.; Lewaa, I. Identification of anti-SARS-CoV-2 compounds from food using QSAR-based virtual screening, molecular docking, and molecular dynamics simulation analysis. Pharmaceuticals 2021, 14, 357. [Google Scholar] [CrossRef]
- Masand, V.H.; Akasapu, S.; Gandhi, A.; Rastija, V.; Patil, M.K. Structure features of peptide-type SARS-CoV main protease inhibitors: Quantitative structure activity relationship study. Chemom. Intell. Lab. Syst. 2020, 206, 104172. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Z.; Vijver, M.G.; Peijnenburg, W.J. Probing nano-QSAR to assess the interactions between carbon nanoparticles and a SARS-CoV-2 RNA fragment. Ecotoxicol. Environ. Saf. 2021, 219, 112357. [Google Scholar] [CrossRef]
- Park, T.; Lee, S.-Y.; Kim, S.; Kim, M.J.; Kim, H.G.; Jun, S.; Kim, S.I.; Kim, B.T.; Park, E.C.; Park, D. Spike protein binding prediction with neutralizing antibodies of SARS-CoV-2. BioRxiv 2020, 2020-02. [Google Scholar] [CrossRef]
- Cheng, M.H.; Krieger, J.M.; Banerjee, A.; Xiang, Y.; Kaynak, B.; Shi, Y.; Arditi, M.; Bahar, I. Impact of new variants on SARS-CoV-2 infectivity and neutralization: A molecular assessment of the alterations in the spike-host protein interactions. Iscience 2022, 25, 103939. [Google Scholar] [CrossRef] [PubMed]
- Das, N.C.; Chakraborty, P.; Bayry, J.; Mukherjee, S. Comparative binding ability of human monoclonal antibodies against Omicron variants of SARS-CoV-2: An in silico investigation. Antibodies 2023, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Elend, L.; Jacobsen, L.; Cofala, T.; Prellberg, J.; Teusch, T.; Kramer, O.; Solov’Yov, I.A. Design of SARS-CoV-2 main protease inhibitors using artificial intelligence and molecular dynamic simulations. Molecules 2022, 27, 4020. [Google Scholar] [CrossRef] [PubMed]
- Gawriljuk, V.O.; Zin, P.P.K.; Puhl, A.C.; Zorn, K.M.; Foil, D.H.; Lane, T.R.; Hurst, B.; Tavella, T.A.; Costa, F.T.M.; Lakshmanane, P. Machine learning models identify inhibitors of SARS-CoV-2. J. Chem. Inf. Model. 2021, 61, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Gao, K.; Chen, J.; Wang, R.; Wei, G.-W. Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning. Chem. Sci. 2020, 11, 12036–12046. [Google Scholar] [CrossRef]
- Haneczok, J.; Delijewski, M. Machine learning enabled identification of potential SARS-CoV-2 3CLpro inhibitors based on fixed molecular fingerprints and Graph-CNN neural representations. J. Biomed. Inform. 2021, 119, 103821. [Google Scholar] [CrossRef]
- Qu, H.; Wang, S.; He, M.; Wu, Y.; Yan, F.; Liu, T.; Zhang, M. A Novel de novo Design Study of Potent SARS-CoV-2 Main Protease Inhibitors Based on Reinforcement Learning and Molecular Docking. ResearchSquare 2023, 1–22. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, Y.; Li, Z.; Chen, Z.; Wang, X.; Li, Z.; Yu, L.; Cheng, X.; Li, W.; Jiang, W.-J. A deep learning-based drug repurposing screening and validation for anti-SARS-CoV-2 compounds by targeting the cell entry mechanism. bioRxiv 2023, 675, 113–121. [Google Scholar] [CrossRef]
- Kumar, A.; Loharch, S.; Kumar, S.; Ringe, R.P.; Parkesh, R.J. Exploiting cheminformatic and machine learning to navigate the available chemical space of potential small molecule inhibitors of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 424–438. [Google Scholar] [CrossRef]
- Jukič, M.; Škrlj, B.; Tomšič, G.; Pleško, S.; Podlipnik, Č.; Bren, U. Prioritisation of compounds for 3CLpro inhibitor development on SARS-CoV-2 variants. Molecules 2021, 26, 3003. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Saha, I.; Gambin, A. Interactome-Based Machine Learning Predicts Potential Therapeutics for COVID-19. ACS Omega 2023, 8, 13840–13854. [Google Scholar] [CrossRef] [PubMed]
- Magar, R.; Yadav, P.; Barati Farimani, A. Potential neutralizing antibodies discovered for novel corona virus using machine learning. Sci. Rep. 2021, 11, 5261. [Google Scholar] [CrossRef] [PubMed]
- Desautels, T.; Zemla, A.; Lau, E.; Franco, M.; Faissol, D. Rapid in silico design of antibodies targeting SARS-CoV-2 using machine learning and supercomputing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Frei, L.; Gao, B.; Han, J.; Taft, J.M.; Irvine, E.B.; Weber, C.R.; Kumar, R.; Eisinger, B.; Reddy, S.T. Deep learning-guided selection of antibody therapies with enhanced resistance to current and prospective SARS-CoV-2 Omicron variants. bioRxiv 2023, 1–38. [Google Scholar] [CrossRef]
- Abo-Zeid, Y.; Ismail, N.S.; McLean, G.R.; Hamdy, N.M. A molecular docking study repurposes FDA approved iron oxide nanoparticles to treat and control COVID-19 infection. Eur. J. Pharm. Sci. 2020, 153, 105465. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, M.; Abdel-Bar, H.M.; Elmowafy, E.; El-Khouly, A.; Mansour, M.; Awad, G.A. Investigating the internalization and COVID-19 antiviral computational analysis of optimized nanoscale zinc oxide. ACS Omega 2021, 6, 6848–6860. [Google Scholar] [CrossRef]
- Skariyachan, S.; Gopal, D.; Deshpande, D.; Joshi, A.; Uttarkar, A.; Niranjan, V. Carbon fullerene and nanotube are probable binders to multiple targets of SARS-CoV-2: Insights from computational modeling and molecular dynamic simulation studies. Infect. Genet. Evol. 2021, 96, 105155. [Google Scholar] [CrossRef]
- Aallaei, M.; Molaakbari, E.; Mostafavi, P.; Salarizadeh, N.; Maleksah, R.E.; Afzali, D. Investigation of Cu metal nanoparticles with different morphologies to inhibit SARS-CoV-2 main protease and spike glycoprotein using Molecular Docking and Dynamics Simulation. J. Mol. Struct. 2022, 1253, 132301. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Abelyan, N.; Abdelgawad, M.A.; Musa, A.; Ghoneim, M.M.; Al-Warhi, T.; Aljaeed, N.; Alotaibi, O.J.; Alnusaire, T.S.; Abdelwahab, S.F. Strawberry and ginger silver nanoparticles as potential inhibitors for SARS-CoV-2 assisted by in silico modeling and metabolic profiling. Antibiotics 2021, 10, 824. [Google Scholar] [CrossRef]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering anti-cancer drugs via computational methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, R.; Dangi, M.; Khichi, A.; Chhillar, A.K. Relevance of molecular docking studies in drug designing. Curr. Bioinform. 2020, 15, 270–278. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Aguilar-Toala, J.E.; Liceaga, A.M. Integration of molecular docking analysis and molecular dynamics simulations for studying food proteins and bioactive peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudi, S.; Taghavi Shahraki, B.; Rameh, F.; Nazarabi, M.; Fatahi, Y.; Akhavan, O.; Rabiee, M.; Mostafavi, E.; Lima, E.C.; Saeb, M.R. A review on computer-aided chemogenomics and drug repositioning for rational COVID-19 drug discovery. Chem. Biol. Drug Des. 2022, 100, 699–721. [Google Scholar] [CrossRef]
- Rudrapal, M.; Chetia, D. Virtual screening, molecular docking and QSAR studies in drug discovery and development programme. J. Drug Deliv. Ther. 2020, 10, 225–233. [Google Scholar] [CrossRef]
- Guarra, F.; Colombo, G. Computational Methods in Immunology and Vaccinology: Design and Development of Antibodies and Immunogens. J. Chem. Theory Comput. 2023, 19, 5315–5333. [Google Scholar] [CrossRef]
- Jain, K.K. Personalized therapy of Infectious Diseases. In Textbook of Personalized Medicine; Springer: Cham, Switzerland, 2021; pp. 325–341. [Google Scholar]
- Patil, A.; Singh, N.; Bange, K. Antiviral Pathogenesis and Interventions: New Understandings and Developments. Acta Sci. Microbiol. 2023, 6, 2–14. [Google Scholar] [CrossRef]

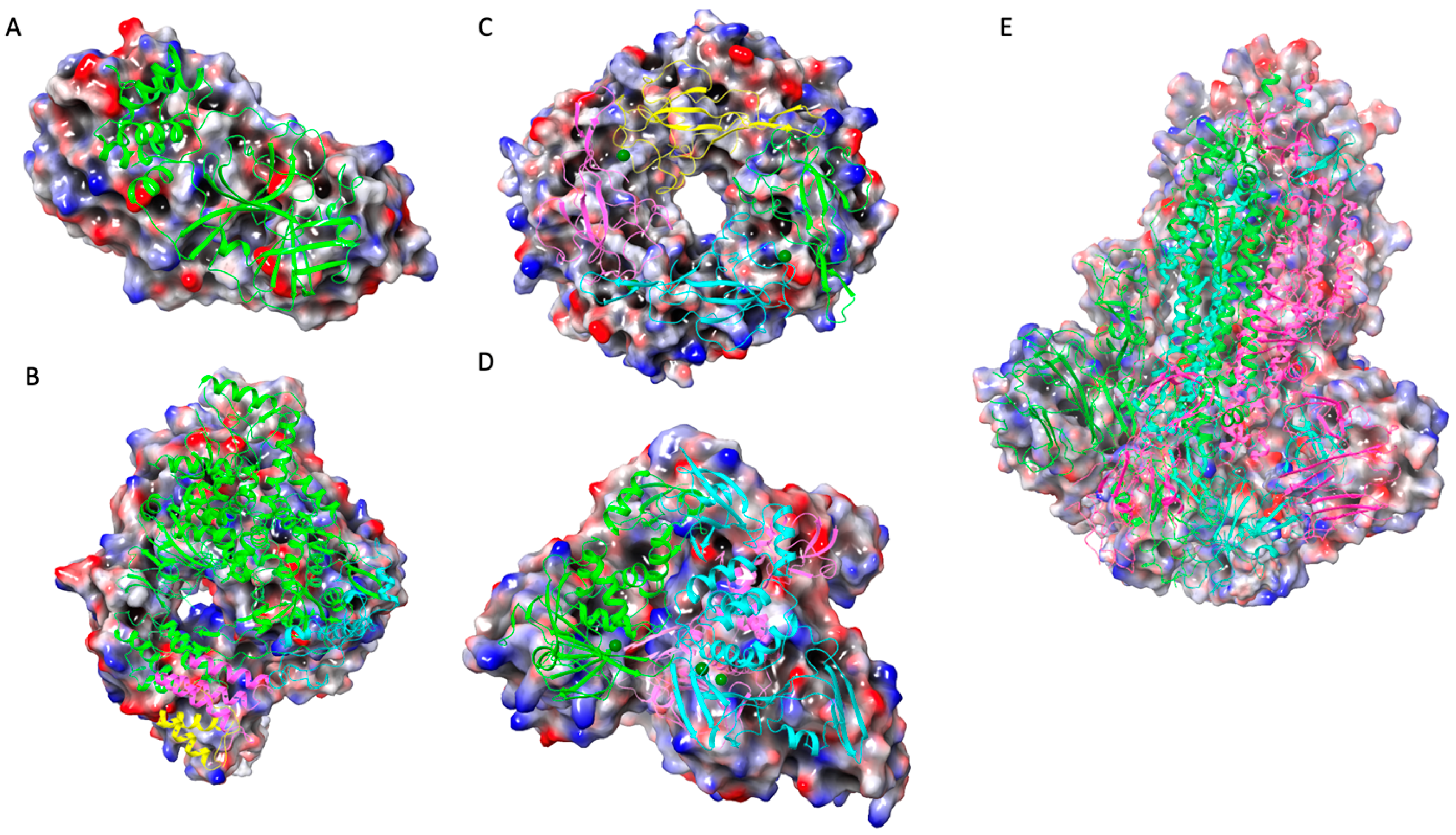



{kind=link}
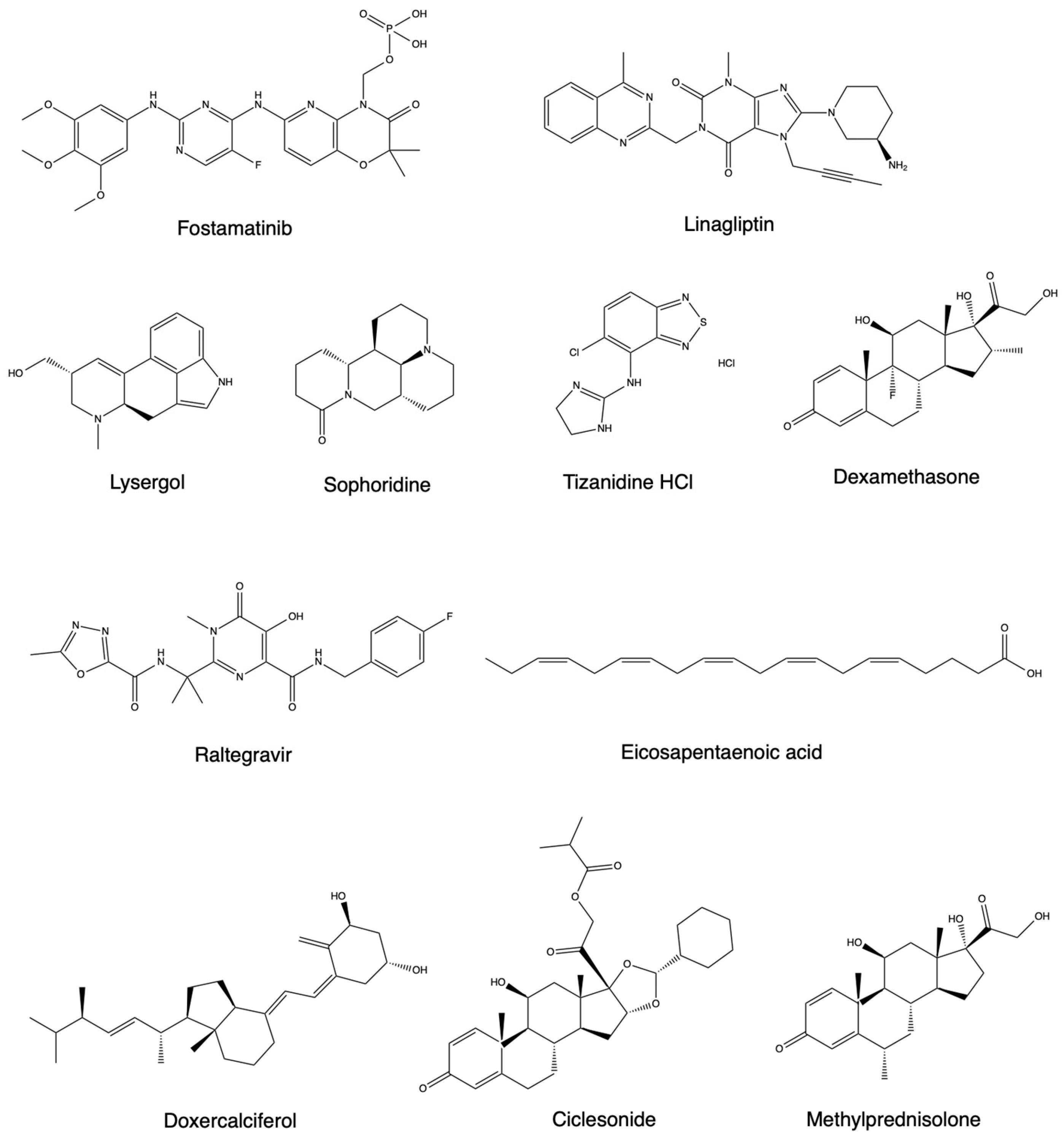
{kind=link}
{kind=link}
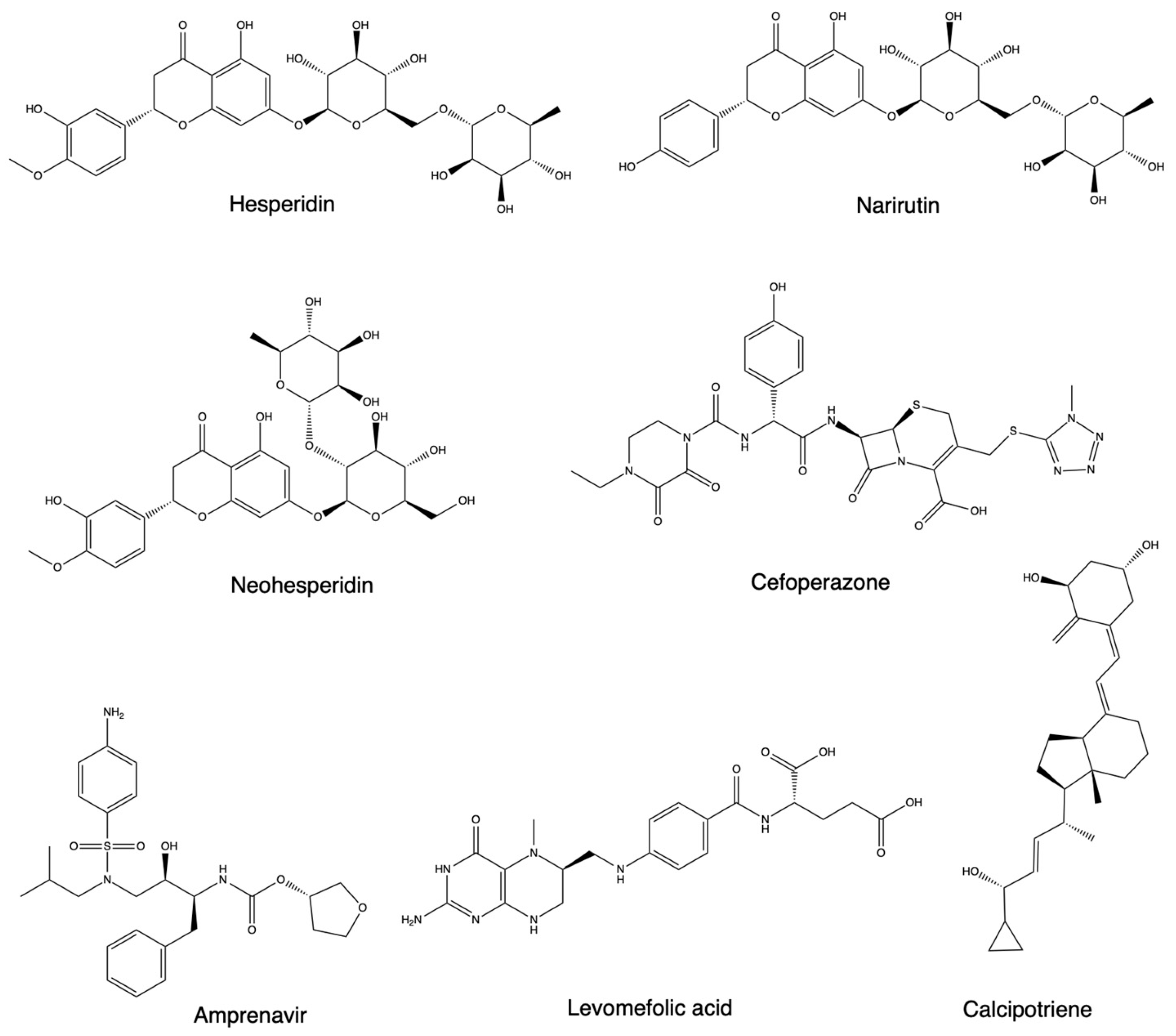{kind=link}
{kind=link}
| Serial No. | WHO Label | PANGO Lineage | Date of Designation |
|---|---|---|---|
| 1 | Alpha | B.1.1.7 and Q lineages | VOC: 29 December 2020 VBM: 21 September 2021 |
| 2 | Beta | B.1.351 and descendent lineages | VOC: 29 December 2020 VBM: 21 September 2021 |
| 3 | Gamma | P.1 and descendent lineages | VOC: 29 December 2020 VBM: 21 September 2021 |
| 4 | Delta | B.1.617.2 and descendant lineages | VOC: 15 June 2021 VBM: 14 April 2022 |
| 5 | Epsilon | B.1.427 and B.1.429 | VOC: 19 March 2021 VOI: 26 February 2021 VOI: 29 June 2021 VBM: 21 September 2021 |
| 6 | Omicron | B.1.1.529 and descendant lineages | VOC: 26 November 2021 VBM: 1 September 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, C.R.; Basharat, Z.; Lang’at, G.C. In Silico Therapeutic Study: The Next Frontier in the Fight against SARS-CoV-2 and Its Variants. Drugs Drug Candidates 2024, 3, 54-69. https://doi.org/10.3390/ddc3010005
Wei CR, Basharat Z, Lang’at GC. In Silico Therapeutic Study: The Next Frontier in the Fight against SARS-CoV-2 and Its Variants. Drugs and Drug Candidates. 2024; 3(1):54-69. https://doi.org/10.3390/ddc3010005
Chicago/Turabian StyleWei, Calvin R., Zarrin Basharat, and Godwin C. Lang’at. 2024. "In Silico Therapeutic Study: The Next Frontier in the Fight against SARS-CoV-2 and Its Variants" Drugs and Drug Candidates 3, no. 1: 54-69. https://doi.org/10.3390/ddc3010005
APA StyleWei, C. R., Basharat, Z., & Lang’at, G. C. (2024). In Silico Therapeutic Study: The Next Frontier in the Fight against SARS-CoV-2 and Its Variants. Drugs and Drug Candidates, 3(1), 54-69. https://doi.org/10.3390/ddc3010005