
Comparative Analysis and Ancestral Sequence Reconstruction of Bacterial Sortase Family Proteins Generates Functional Ancestral Mutants with Different Sequence Specificities


Abstract
:1. Introduction
2. Results
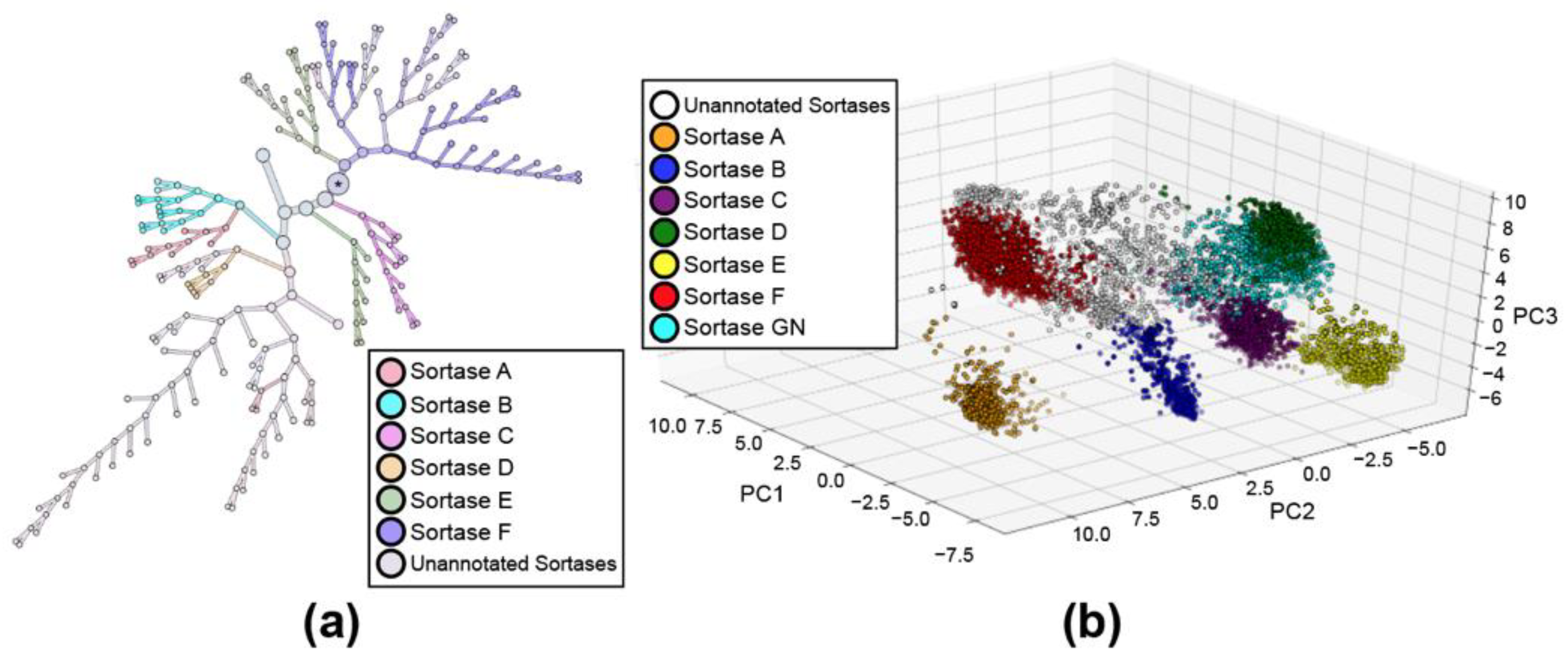
2.1. Principal Component Analysis (PCA) of Bacterial Sortases
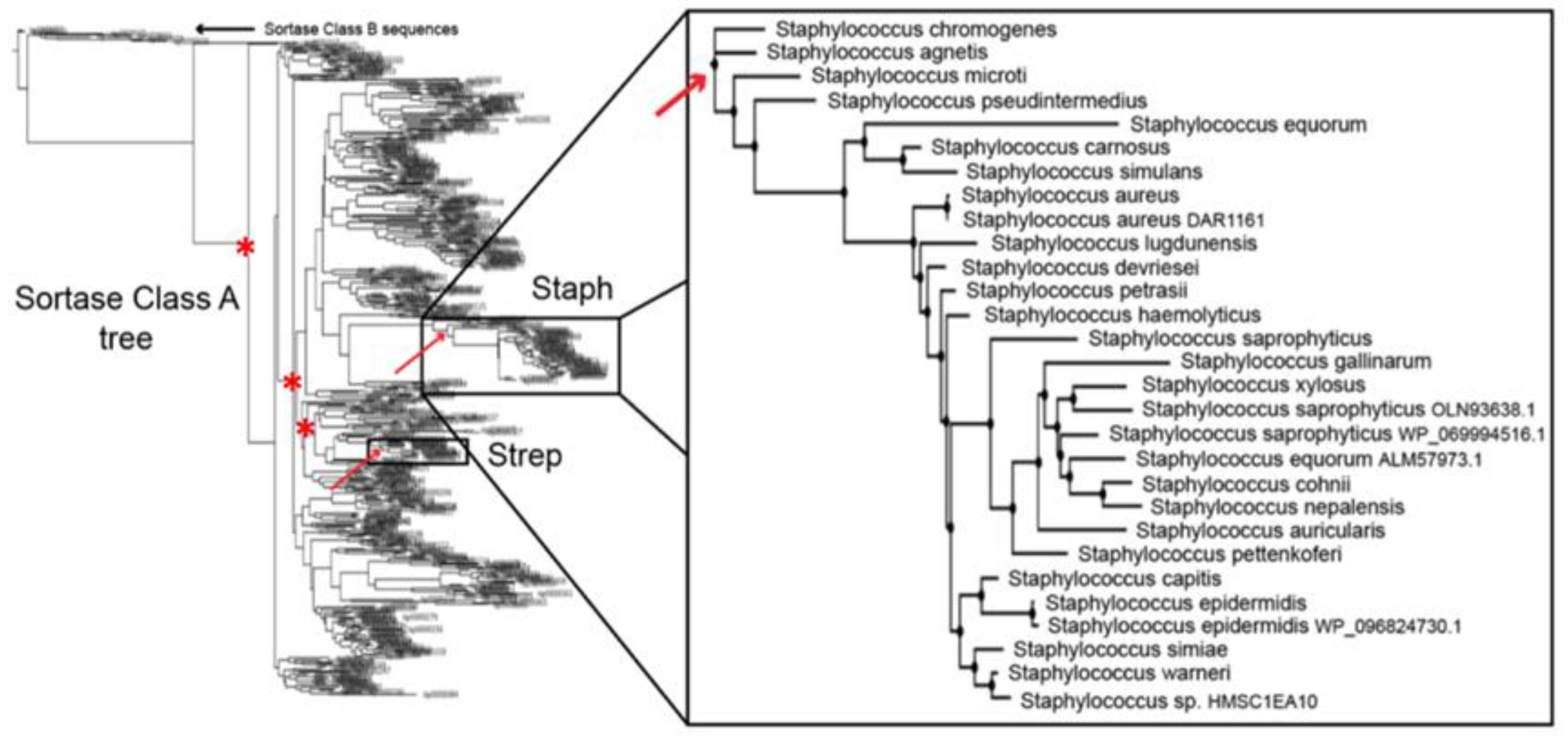
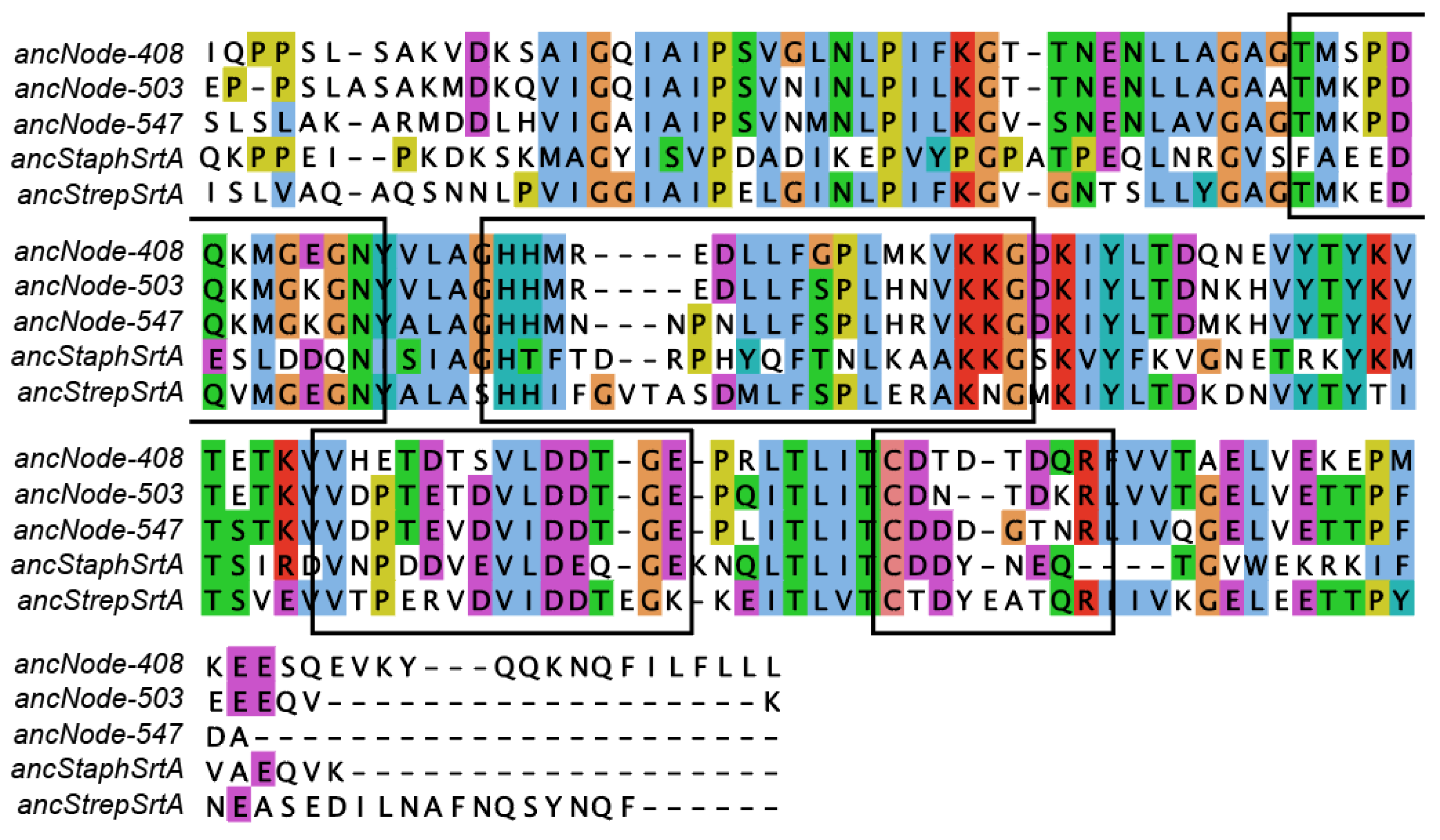
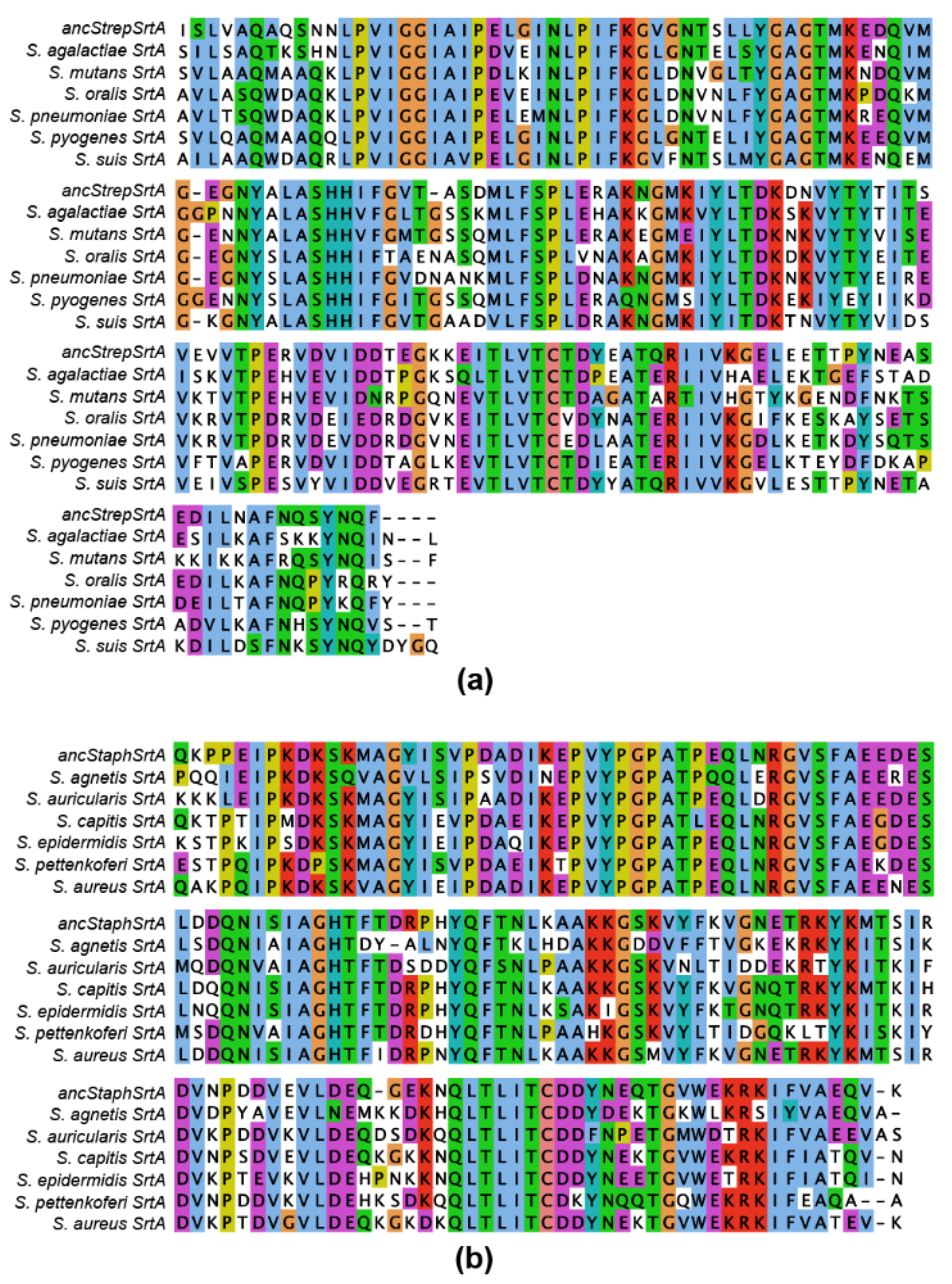
2.2. Ancestral Sequence Reconstruction of Class A Sortases
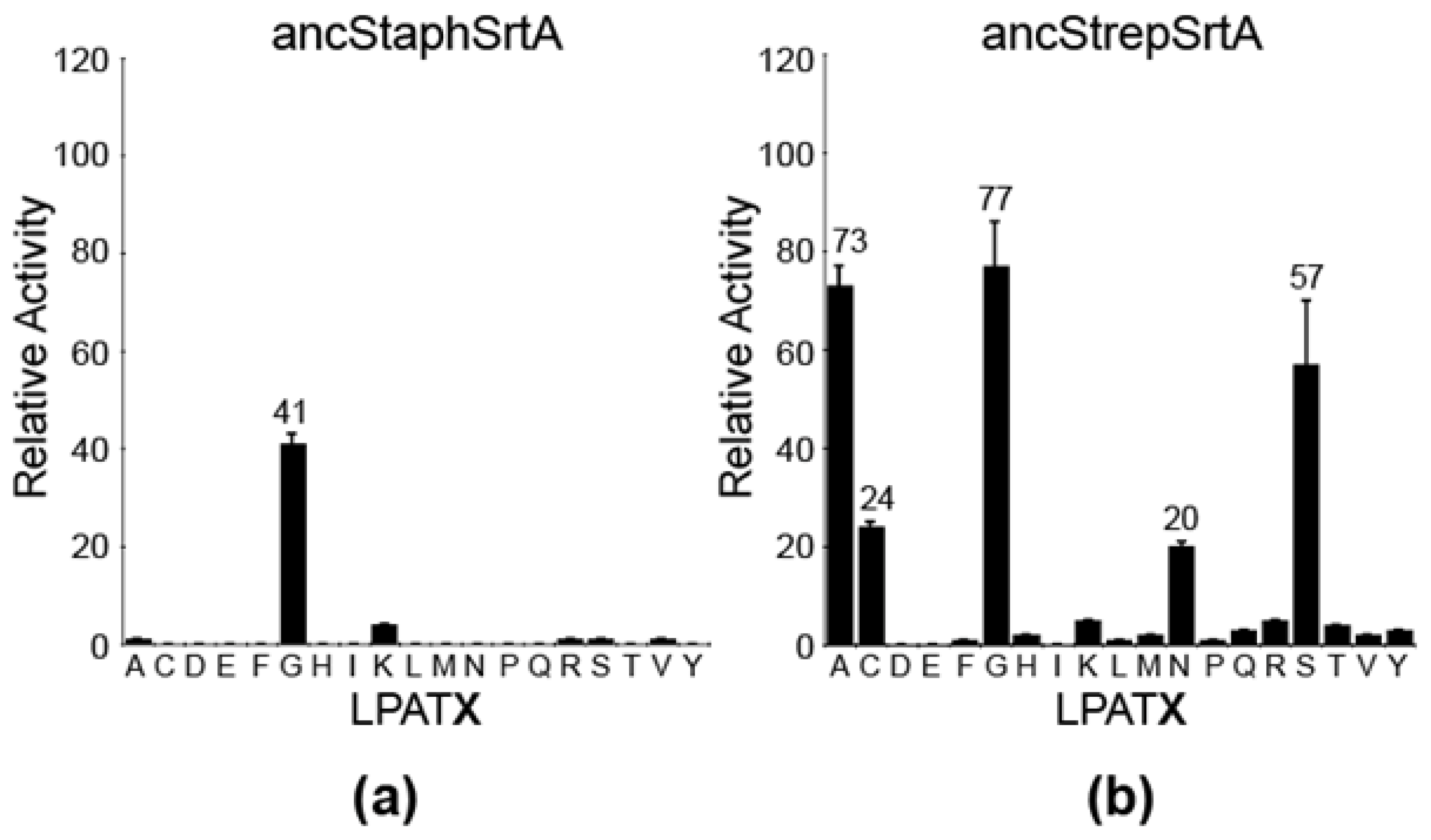
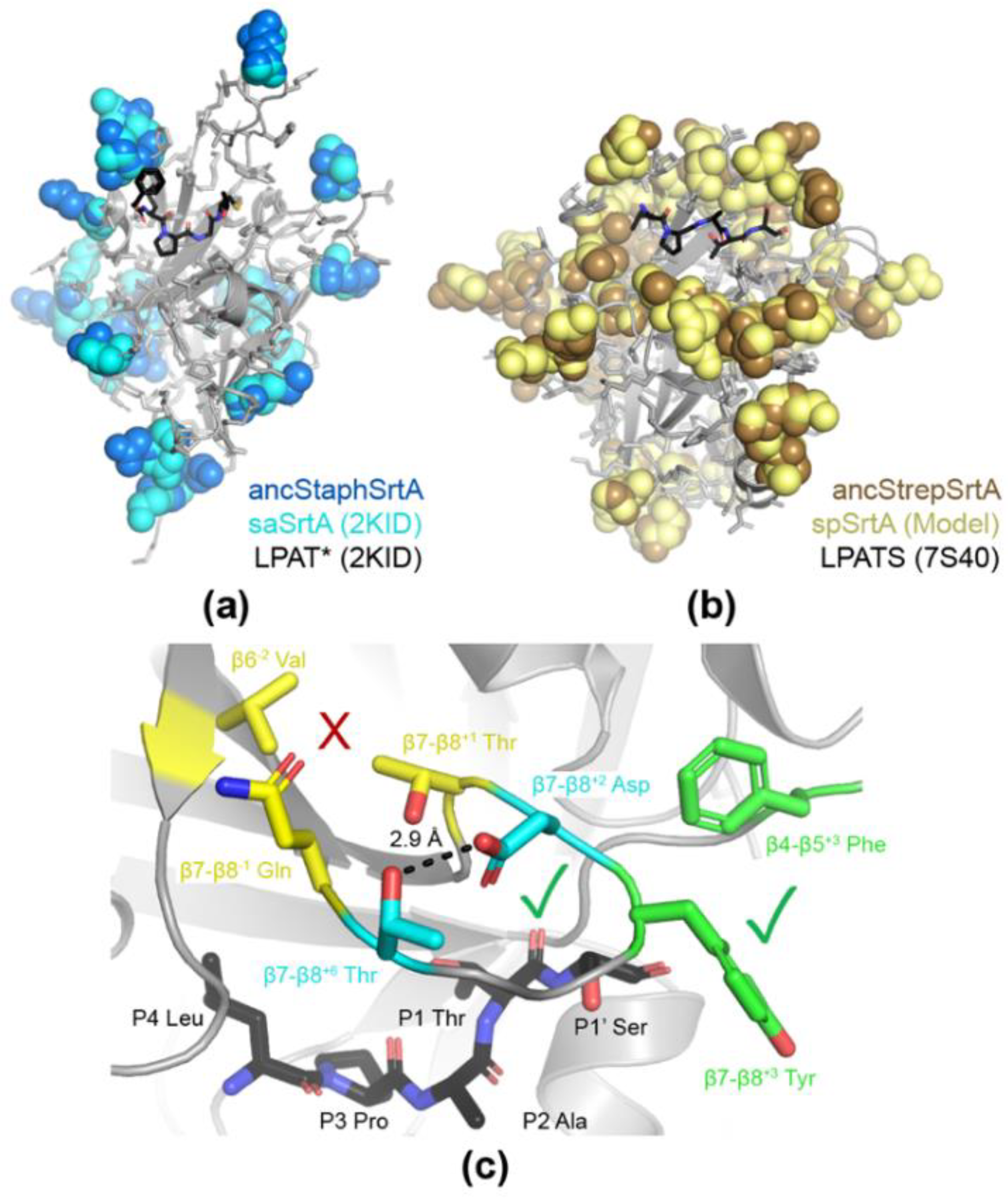
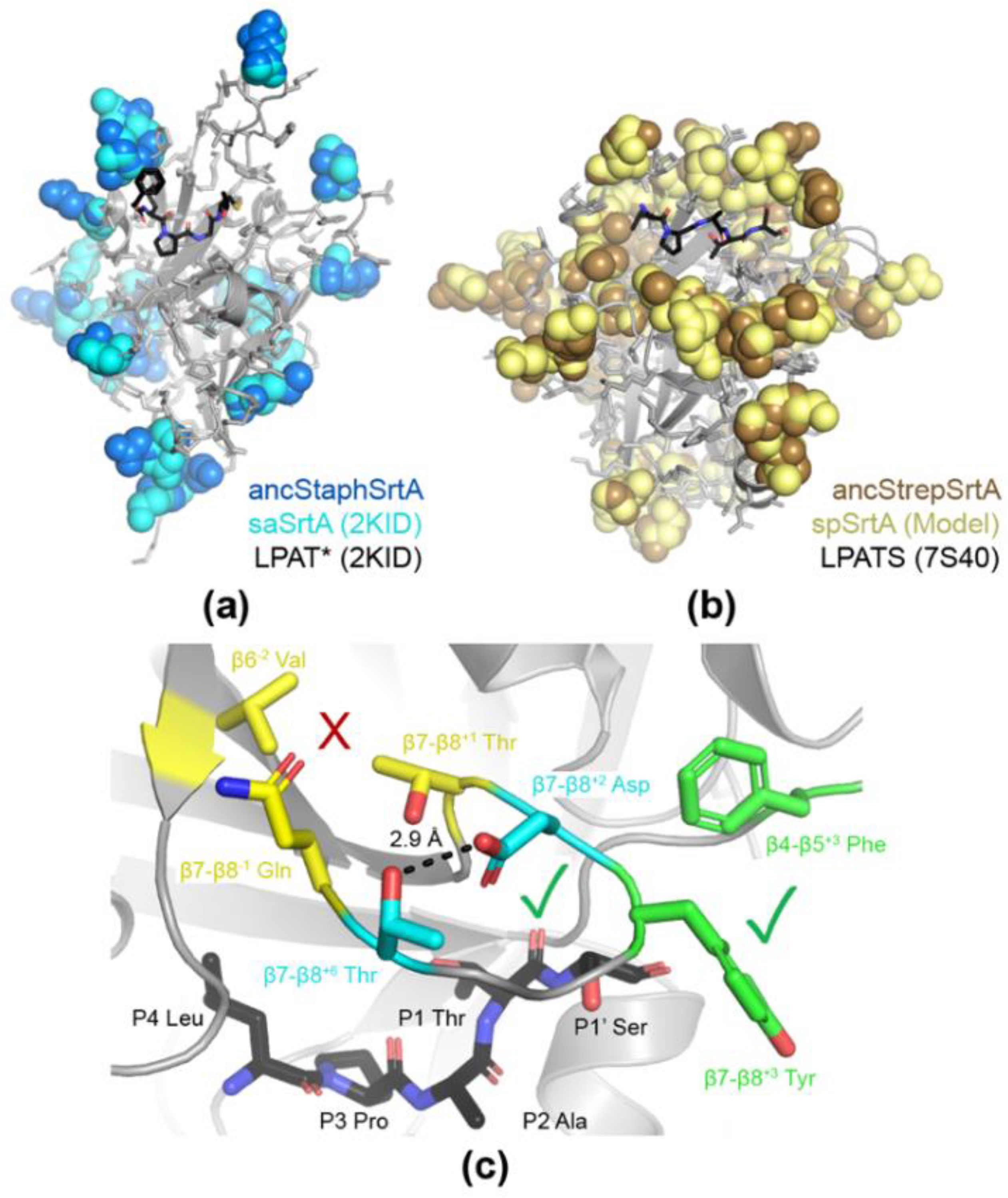
2.3. Structural Analyses of Ancestral SrtA Proteins
2.4. Investigating Ancestral Proteins at Distant Nodes
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sizar, O.; Unakal, C.G. Gram Positive Bacteria. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Diekema, D.J.; Hsueh, P.-R.; Mendes, R.E.; Pfaller, M.A.; Rolston, K.V.; Sader, H.S.; Jones, R.N. The Microbiology of Bloodstream Infection: 20-Year Trends from the SENTRY Antimicrobial Surveillance Program. Antimicrob. Agents Chemother. 2019, 63, e00355-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharath, A.; Ahmed, M.S. Bacterial Etiology of Bloodstream Infections and Antimicrobial Resistance Patterns from a Tertiary Care Hospital in Malé, Maldives. Int. J. Microbiol. 2021, 2021, 3088202. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yue, Y.; Zhu, L.; Cui, J.; Zhu, M.; Chen, L.; Yang, Z.; Liang, Z. Epidemiology and microbiology of Gram-positive bloodstream infections in a tertiary-care hospital in Beijing, China: A 6-year retrospective study. Antimicrob. Resist. Infect. Control 2018, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.F.; Cho, G.; Doolittle, R.F. Determining divergence times with a protein clock: Update and reevaluation. Proc. Natl. Acad. Sci. USA 1997, 94, 13028–13033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marraffini, L.A.; DeDent, A.C.; Schneewind, O. Sortases and the Art of Anchoring Proteins to the Envelopes of Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 192–221. [Google Scholar] [CrossRef] [Green Version]
- Ton-That, H.; Mazmanian, S.K.; Alksne, L.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Cysteine 184 and histidine 120 of sortase form a thiolate-imidazolium ion pair for catalysis. J. Biol. Chem. 2002, 277, 7447–7452. [Google Scholar] [CrossRef] [Green Version]
- Jacobitz, A.W.; Kattke, M.D.; Wereszczynski, J.; Clubb, R.T. Sortase transpeptidases: Structural biology and catalytic mechanism. Adv. Protein Chem. Struct. Biol. 2017, 109, 223–264. [Google Scholar]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef]
- Antos, J.M.; Truttmann, M.C.; Ploegh, H.L. Recent advances in sortase-catalyzed ligation methodology. Curr. Opin. Struct. Biol. 2016, 38, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.-T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, I.M.; Struyvenberg, S.A.; Valgardson, J.D.; Johnson, D.A.; Gao, M.; Johnston, K.; Svendsen, J.E.; Kodama, H.M.; Hvorecny, K.L.; Antos, J.M.; et al. Sequence variation in the β7–β8 loop of bacterial class A sortase enzymes alters substrate selectivity. J. Biol. Chem. 2021, 297, 100981. [Google Scholar] [CrossRef]
- Gao, M.; Johnson, D.A.; Piper, I.M.; Kodama, H.M.; Svendsen, J.E.; Tahti, E.; Longshore-Neate, F.; Vogel, B.; Antos, J.M.; Amacher, J.F. Structural and biochemical analyses of selectivity determinants in chimeric Streptococcus Class A sortase enzymes. Protein Sci. 2022, 31, 701–715. [Google Scholar] [CrossRef]
- Dai, X.; Böker, A.; Glebe, U. Broadening the scope of sortagging. RSC Adv. 2019, 9, 4700–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podracky, C.J.; An, C.; DeSousa, A.; Dorr, B.M.; Walsh, D.M.; Liu, D.R. Laboratory evolution of a sortase enzyme that modifies amyloid-β protein. Nat. Chem. Biol. 2021, 17, 317–325. [Google Scholar] [CrossRef]
- Bierlmeier, J.; Álvaro-Benito, M.; Scheffler, M.; Sturm, K.; Rehkopf, L.; Freund, C.; Schwarzer, D. Sortase-Mediated Multi-Fragment Assemblies by Ligation Site Switching. Angew. Chem. Int. Ed. 2022, 61, e202109032. [Google Scholar] [CrossRef]
- Kruger, R.G.; Otvos, B.; Frankel, B.A.; Bentley, M.; Dostal, P.; McCafferty, D.G. Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry 2004, 43, 1541–1551. [Google Scholar] [CrossRef]
- Freund, C.; Schwarzer, D. Engineered sortases in peptide and protein chemistry. Chembiochem 2021, 22, 1347–1356. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, H.; Ishikawa, S.; Nagamune, T. Design of Ca2+-independent Staphylococcus aureus sortase A mutants. Biotechnol. Bioeng. 2012, 109, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, M.; Vázquez Torres, S.; Quax, W.J.; Boersma, Y.L. Sortase mutants with improved protein thermostability and enzymatic activity obtained by consensus design. Protein Eng. Des. Sel. 2019, 32, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, M.; Szala, K.; van Merkerk, R.; Quax, W.J.; Boersma, Y.L. Engineering the specificity of Streptococcus pyogenes sortase A by loop grafting. Proteins 2020, 88, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Nikghalb, K.D.; Horvath, N.M.; Prelesnik, J.L.; Banks, O.G.B.; Filipov, P.A.; Row, R.D.; Roark, T.J.; Antos, J.M. Expanding the Scope of Sortase-Mediated Ligations by Using Sortase Homologues. Chembiochem 2018, 19, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, L.; Bierlmeier, J.; von Kügelgen, N.; Kurz, L.; Reis, P.; Barthels, F.; Mach, P.; Schutkowski, M.; Freund, C.; Schwarzer, D. Identification of sortase substrates by specificity profiling. Bioorg. Med. Chem. 2017, 25, 5002–5007. [Google Scholar] [CrossRef]
- Malik, A.; Kim, S.B. A comprehensive in silico analysis of sortase superfamily. J. Microbiol. 2019, 57, 431–443. [Google Scholar] [CrossRef]
- Di Girolamo, S.; Puorger, C.; Castiglione, M.; Vogel, M.; Gébleux, R.; Briendl, M.; Hell, T.; Beerli, R.R.; Grawunder, U.; Lipps, G. Characterization of the housekeeping sortase from the human pathogen Propionibacterium acnes: First investigation of a class F sortase. Biochem. J. 2019, 476, 665–682. [Google Scholar] [CrossRef]
- Harms, M.J.; Thornton, J.W. Analyzing protein structure and function using ancestral gene reconstruction. Curr. Opin. Struct. Biol. 2010, 20, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Pillai, A.S.; Chandler, S.A.; Liu, Y.; Signore, A.V.; Cortez-Romero, C.R.; Benesch, J.L.P.; Laganowsky, A.; Storz, J.F.; Hochberg, G.K.A.; Thornton, J.W. Origin of complexity in haemoglobin evolution. Nature 2020, 581, 480–485. [Google Scholar] [CrossRef]
- Wheeler, L.C.; Lim, S.A.; Marqusee, S.; Harms, M.J. The thermostability and specificity of ancient proteins. Curr. Opin. Struct. Biol. 2016, 38, 37–43. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.W.; Zhou, Y.; Petrovic, J.; Fasolino, M.; Xu, L.; Shaffer, S.M.; Pear, W.S.; Vahedi, G.; Faryabi, R.B. TooManyCells identifies and visualizes relationships of single-cell clades. Nat. Methods 2020, 17, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Kattke, M.D.; Chan, A.H.; Duong, A.; Sexton, D.L.; Sawaya, M.R.; Cascio, D.; Elliot, M.A.; Clubb, R.T. Crystal Structure of the Streptomyces coelicolor Sortase E1 Transpeptidase Provides Insight into the Binding Mode of the Novel Class E Sorting Signal. PLoS ONE 2016, 11, e0167763. [Google Scholar] [CrossRef]
- Bentley, M.L.; Gaweska, H.; Kielec, J.M.; McCafferty, D.G. Engineering the substrate specificity of Staphylococcus aureus Sortase A. The beta6/beta7 loop from SrtB confers NPQTN recognition to SrtA. J. Biol. Chem. 2007, 282, 6571–6581. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Nöth, M.; Jakob, F.; Schwaneberg, U. Designed Streptococcus pyogenes Sortase A Accepts Branched Amines as Nucleophiles in Sortagging. Bioconjug. Chem. 2020, 31, 2476–2481. [Google Scholar] [CrossRef]
- Kruger, R.G.; Dostal, P.; McCafferty, D.G. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 2004, 326, 42–48. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, W.J.; Davies, A.H.; Chambers, C.J.; Roberts, A.K.; Shone, C.C.; Acharya, K.R. Molecular features of the sortase enzyme family. FEBS J. 2015, 282, 2097–2114. [Google Scholar] [CrossRef] [Green Version]
- Comfort, D.; Clubb, R.T. A comparative genome analysis identifies distinct sorting pathways in gram-positive bacteria. Infect. Immun. 2004, 72, 2710–2722. [Google Scholar] [CrossRef] [Green Version]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
- NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 16 March 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |||||||
|---|---|---|---|---|---|---|---|
| ancStaph | S. agnetis | S. aureus | S. auricularis | S. capitis | S. epidermidis | S. pettenkoferi | |
| ancStaph | X | ||||||
| S. agnetis | 68% (97/142) | X | |||||
| S. aureus | 86% (127/147) | 76% (108/142) | X | ||||
| S. auricularis | 73% (107/146) | 62% (91/146) | 70% (101/144) | X | |||
| S. capitis | 88% (129/146) | 62% (88/141) | 84% (122/146) | 68% (96/141) | X | ||
| S. epidermidis | 81% (116/143) | 59% (84/142) | 78% (114/146) | 69% (98/142) | 84% (124/147) | X | |
| S. pettenkoferi | 74% (105/141) | 58% (81/140) | 67% (96/143) | 78% (109/140) | 73% (104/143) | 71% (101/143) | X |
| (b) | |||||||
| ancStrep | S. agalactiae | S. mutans | S. oralis | S. pneumoniae | S. pyogenes | S. suis | |
| ancStrep | X | ||||||
| S. agalactiae | 68% (113/165) | X | |||||
| S. mutans | 65% (106/163) | 64% (109/169) | X | ||||
| S. oralis | 69% (113/163) | 58% (97/166) | 65% (107/165) | X | |||
| S. pneumoniae | 68% (111/164) | 57% (95/167) | 64% (107/166) | 81% (136/167) | X | ||
| S. pyogenes | 71% (117/164) | 65% (109/168) | 70% (117/168) | 63% (104/166) | 63% (105/166) | X | |
| S. suis | 76% (125/164) | 58% (98/168) | 60% (101/168) | 61% (101/165) | 62% (103/166) | 63% (104/166) | X |
| ancStaph | ancStrep | ancNode-408 | ancNode-503 | ancNode-547 | |
|---|---|---|---|---|---|
| ancStaph | X | ||||
| ancStrep | 30% (35/117) | X | |||
| ancNode-408 | 35% (47/133) | 51% (77/151) | X | ||
| ancNode-503 | 33% (50/150) | 56% (76/136) | 78% (156/200) | X | |
| ancNode-547 | 54% (64/118) | 59% (85/145) | 64% (147/199) | 77% (168/199) | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valgardson, J.D.; Struyvenberg, S.A.; Sailer, Z.R.; Piper, I.M.; Svendsen, J.E.; Johnson, D.A.; Vogel, B.A.; Antos, J.M.; Harms, M.J.; Amacher, J.F. Comparative Analysis and Ancestral Sequence Reconstruction of Bacterial Sortase Family Proteins Generates Functional Ancestral Mutants with Different Sequence Specificities. Bacteria 2022, 1, 121-135. https://doi.org/10.3390/bacteria1020011
Valgardson JD, Struyvenberg SA, Sailer ZR, Piper IM, Svendsen JE, Johnson DA, Vogel BA, Antos JM, Harms MJ, Amacher JF. Comparative Analysis and Ancestral Sequence Reconstruction of Bacterial Sortase Family Proteins Generates Functional Ancestral Mutants with Different Sequence Specificities. Bacteria. 2022; 1(2):121-135. https://doi.org/10.3390/bacteria1020011
Chicago/Turabian StyleValgardson, Jordan D., Sarah A. Struyvenberg, Zachary R. Sailer, Isabel M. Piper, Justin E. Svendsen, D. Alex Johnson, Brandon A. Vogel, John M. Antos, Michael J. Harms, and Jeanine F. Amacher. 2022. "Comparative Analysis and Ancestral Sequence Reconstruction of Bacterial Sortase Family Proteins Generates Functional Ancestral Mutants with Different Sequence Specificities" Bacteria 1, no. 2: 121-135. https://doi.org/10.3390/bacteria1020011
APA StyleValgardson, J. D., Struyvenberg, S. A., Sailer, Z. R., Piper, I. M., Svendsen, J. E., Johnson, D. A., Vogel, B. A., Antos, J. M., Harms, M. J., & Amacher, J. F. (2022). Comparative Analysis and Ancestral Sequence Reconstruction of Bacterial Sortase Family Proteins Generates Functional Ancestral Mutants with Different Sequence Specificities. Bacteria, 1(2), 121-135. https://doi.org/10.3390/bacteria1020011