
Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles

 , ,
, ,  ,
,
Abstract
:1. Introduction
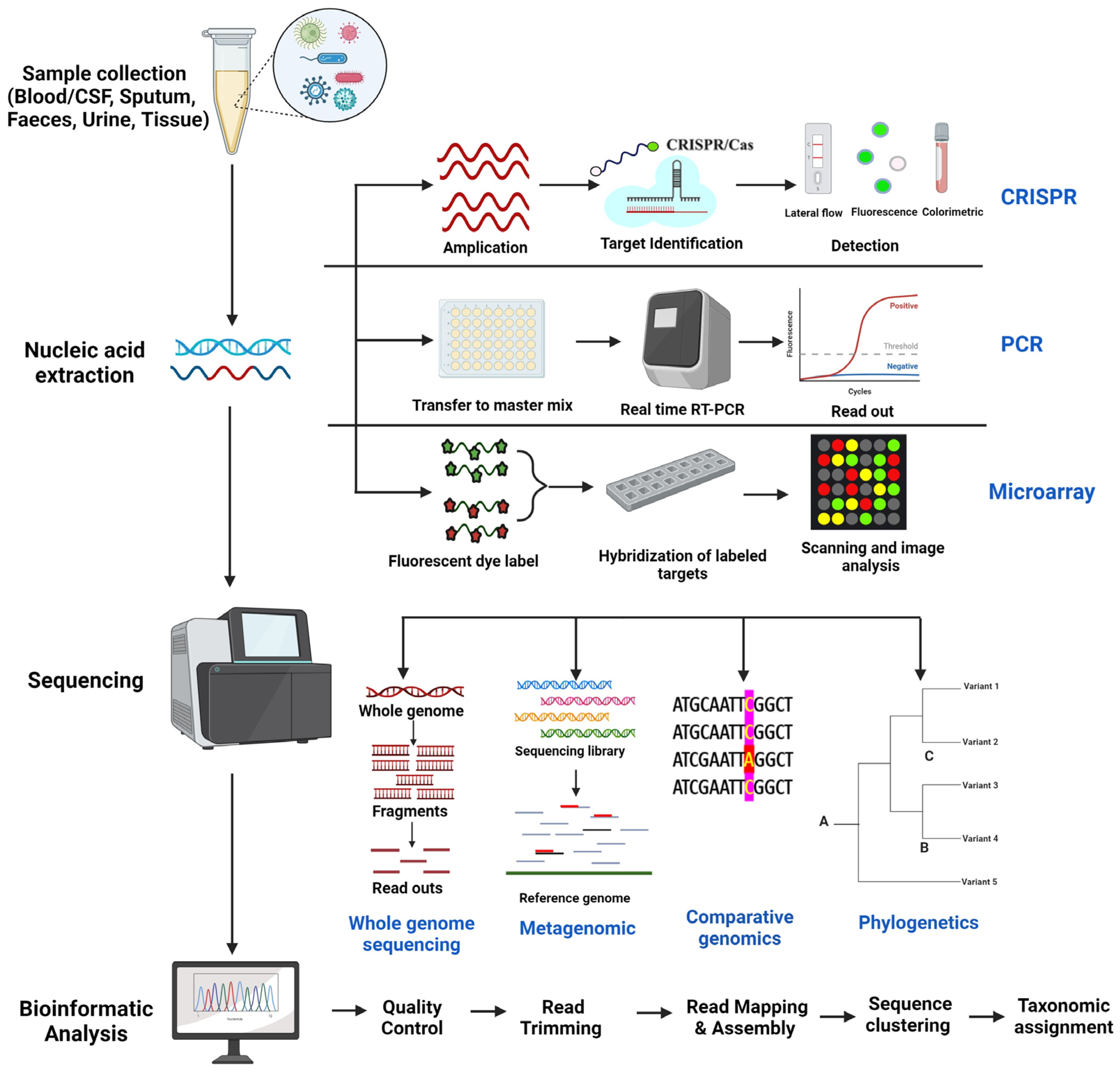
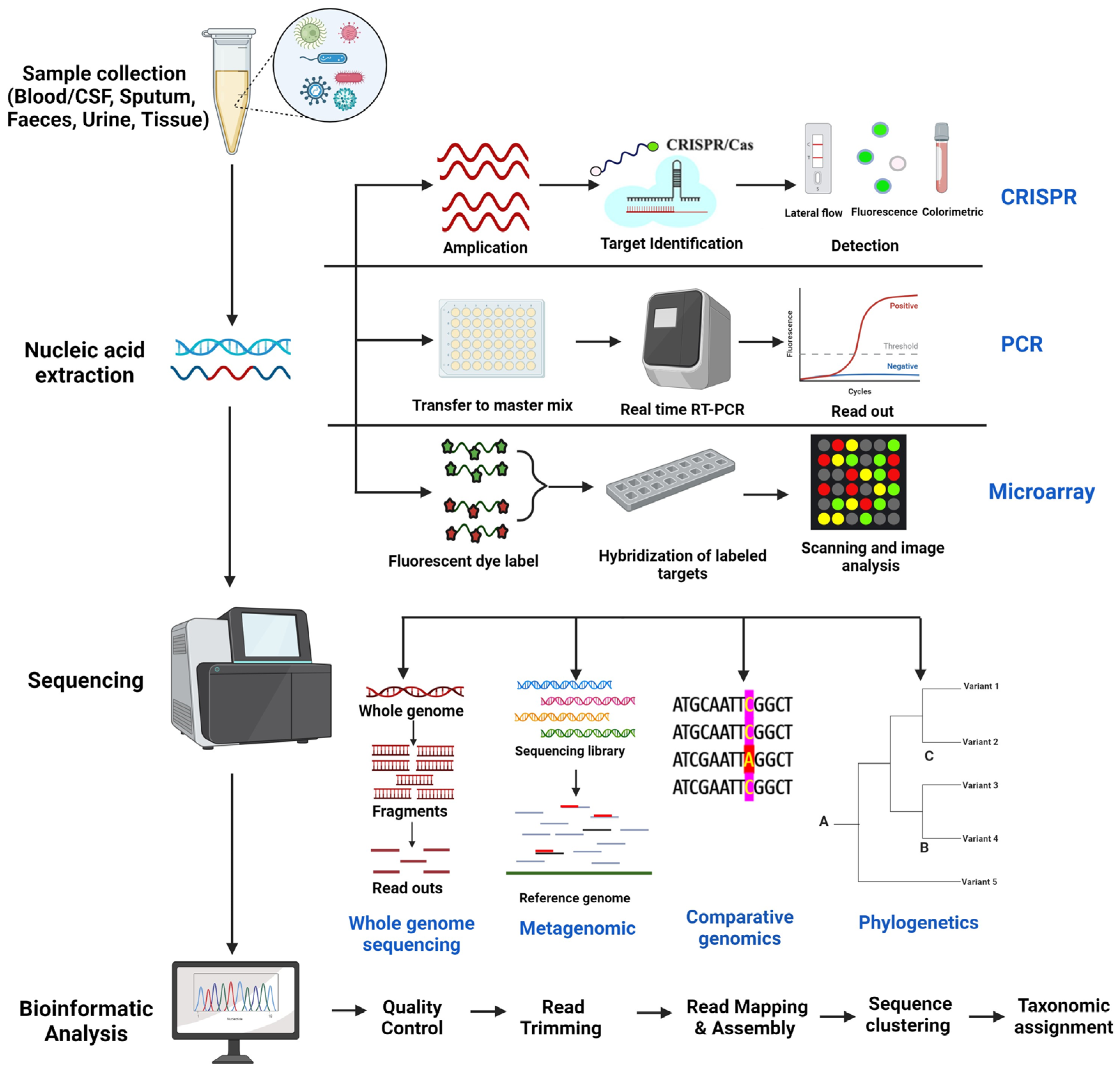
2. Genomic Techniques for Pathogen Detection and Tracking
2.1. Whole Genome Sequencing

{kind=link}
{kind=link}
| Sequencing Technique | Advantage | Disadvantage | Pathogen [Reference] |
|---|---|---|---|
| 454 pyrosequencing | First developed HTS method—preferred sequencing method for metabarcoding projects for a while but now discontinued. | Lower throughput and subsequently higher sequencing cost per base | Ebola [38] |
| Illumina | A more popular choice for both metabarcoding and shotgun metagenomics studies. Best bases/cost ratio—Short (150–300 bps) but high-quality (99.9% accuracy) paired-end (P.E.) sequences—Very high level of accuracy | Does not perform well with low-quality material | SARS-CoV-2 [39] |
| Mycobacterium tuberculosis [40] | |||
| Candida auris [41] | |||
| IonTorrent | Bidirectional amplification and greater read length (400–450 bp) | Robust species inference | Zika [42] |
| PacBio | Produces long reads of 30–100 kb—Better average contig length and a higher number of large contigs in shotgun metagenomics studies. Allows for sequencing of longer PCR fragments such as the full ITS1-5.8S-ITS2 in metabarcoding studies. | Does not perform well with low-quality material. Higher rates of sequencing errors. Higher cost. | Influenza A (H1N1) [43] |
| Oxford Nanopore Technologies | Faster than Illumina or PacBio—Enables users to detect pathogens within minutes of the start of sequencing—small size and ability to be operated from a simple laptop. | Less accuracy (around 95% consensus)—Requires more DNA—More susceptib le to library construction or sequencing inhibitors. | Zika Virus in Brazil and America [44] |
| Ebola Virus [45] |
2.2. Metagenomics
| Pathogen | Metagenomics Implications | Platform Used | References |
|---|---|---|---|
| Zika virus | Detected in Aedes mosquitoes during the epidemic. Arbovirus detection can be a useful tool for identifying epidemic-causing arboviruses. | Illumina MiSeq, Illumina HiSeq | [57] |
| Ebola virus | Broad-based pathogen detection and outbreak surveillance | Ion Torrent PGM | [58] |
| MERS-CoV | Rapid sequencing for genotype information and co-infections enables identification of genotype changes, including insertions, deletions, and minor variants, while also providing insights into the background microbiome. | Amplicon-based approach coupled to Oxford Nanopore long read length sequencing | [59] |
| SARS-CoV-2 | It successfully assembled complete or near-complete genomes and accurately classified phylogenetic lineages, including the identification of Variant of Concern (VOC) strains. The assay’s capability to distinguish between different SARS-CoV-2 variants, such as Alpha and Gamma, surpassed the standard VOC PCR method. | Nanopore-based Sequence-Independent Single Primer Amplification (SISPA) | [50] |
| Chikungunya Dengue, Zika virus | Viral metagenomics was found to be a potent method for the identification of emerging arboviruses. | Illumina NextSeq 2000 | [60] |
| Avian influenza virus (H7N9) | Viral infection surveillance in poultry farms. | Ion Torrent PGM | [61] |
| Influenza virus | Diagnostic test, insights on transmission, evolution, and drug resistance. | Oxford Nanopore | [62] |
| Shiga-toxigenic Escherichia coli (STEC) O104:H4 | Identification and characterization of bacterial strains during diarrheal disease outbreaks, including the STEC outbreak strain, as well as detection of other pathogens. | Illumina HiSeq 2500 Illumina MiSeq2500 | [63] |
2.3. Comparative Genomics
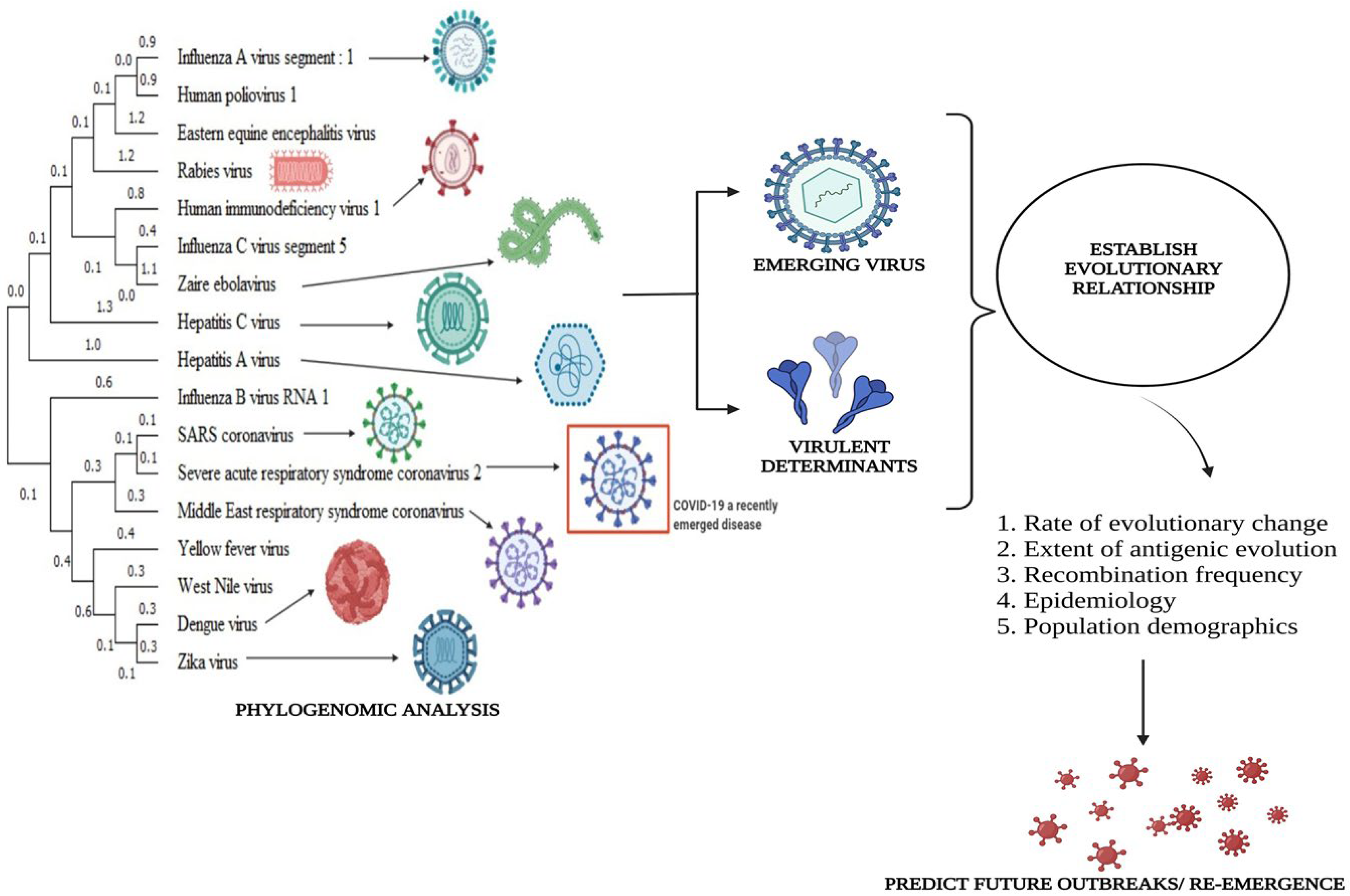
2.4. Phylogenetic Analysis
3. Other Genomic Techniques
3.1. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Based Methods
| Pathogen | Sample Type | Detection Technique | Sensitivity | Specificity | References |
|---|---|---|---|---|---|
| Helicobacter pylori | Stool | CRISPR-Cas12, a system-based method | [99] | ||
| Zika | Plasma of a viremic macaque. | NSBA CRISPR-Cas9 | [91] | ||
| SARS-CoV-2 | Nasopharyngeal swab samples | Isotachophoresis (ITP)-enhanced CRISPR-Cas12a | [100] | ||
| Pneumocystis jirovecii | Bronchial alveolar lavage fluid | Transcription-mediated amplification/ CRISPR-Cas13a/ (Fluorescence plate reader) | 78.9% | 97.7% | [101] |
| SARS-CoV-2 | Nasopharyngeal swabs | Loop-mediated Isothermal Amplification (LAMP)/SHERLOCK/(lateral flow) | - | 100% | [102] |
| Zika and Dengue | Human Serum/Saliva/Urine | RT-RPA–HUDSON/ Cas13-based SHERLOCK/ (Fluorescence/calorimetric) | - | 100% | [103] |
| Mycobacterium tuberculosis | Clinical sputum samples | LACD (loop-mediated isothermal amplification)/CRISPR-Cas12a/ (Later flow/real-time fluorescence) | ~10 copies/test | 100% | [104] |
| Mycobacterium tuberculosis | Sputum | RPA/CRISPR-Cas12a/(Fluorescent detection) | 79% | 98% | [105] |
| Hepatitis B virus | Plasma | RT-LAMP/CRISPR–Cas12a/ (Lateral flow strips or fluorescence detector) | 96% | 100% | [106] |
| Monkey pox | Synthetically produced Congo Basin clade D14L and West African clade ATI and cloned into the pUC57 vector | Loop-mediated isothermal amplification (LAMP)/CRISPR-Cas12b/ real-time fluorescence and a gold nanoparticle-based lateral flow biosensor (AuNP-LFB) | 10 copies/reaction | - | [107] |
| Ebola virus | Urine, Saliva | HUDSON/CRISPR-Cas13a-based (SHERLOCK)/ (Fluorescent and lateral flow readouts) | 91% | 100% | [108] |
| Influenza (H1N1) | Synthetic DNA strands | CRISPR/Cas13a/hybridization chain reaction (HCR)/ Colorimetric biosensor | 0.152 pM | - | [109] |
3.2. PCR-Based Methods
3.3. Microarray Analysis
3.4. Bioinformatics Tools
4. Advances in Genomic Epidemiology and Pathogen Surveillance
5. Challenges and Future Directions
5.1. Lack of Trained Workforce and Networking Infrastructure
5.1.1. Shortage of Skilled Genomic Experts
5.1.2. Training Gaps
5.1.3. Data Sharing Challenges
5.1.4. Limited Collaboration Platforms
5.2. Technical Considerations of Genomics in Pathogen Detection and Tracking
- Low sensitivity of sequencing techniques in detecting low abundance or low copy number pathogens, such as those found in cerebrospinal fluid [190].
- Genomic sequencing generates vast amounts of data, which can overwhelm computational resources and expertise. Analyzing and interpreting this data require advanced bioinformatics tools and skilled personnel. This can lead to delays in obtaining actionable results, especially in resource-limited settings.
- Data analysis and interpretation complexity, including the need for bioinformatics expertise and computational resources.
- Technical variability and errors in sequencing data, particularly in the presence of genetic polymorphisms, genomic rearrangements, or repetitive regions [191].
- The accuracy of genomics-based pathogen identification relies on the quality and representativeness of the sample collected. Biases can be introduced if the sampling process is not well designed or if the pathogen is in low quantities. Additionally, the genomic material of interest might be mixed with host DNA, affecting the quality of the sequencing data.
- Pathogens can rapidly evolve through mutation and recombination, leading to genetic diversity within a single species. This diversity can make it challenging to design universal genomic markers for identification. Furthermore, the identification of novel strains or variants might require frequent updates to reference databases.
- Genomic sequencing often requires fresh or well-preserved samples. The logistics of storing and transporting samples to sequencing facilities without compromising their integrity can be challenging, particularly in remote or disaster-affected areas.
- While the cost of genomic sequencing has decreased significantly over the years, it can still be expensive, especially for large-scale surveillance or in low-resource environments. The cost of equipment, reagents, and skilled personnel can be a significant barrier to widespread adoption. Emerging initiatives, such as the Human Heredity and Health in Africa (H3Africa), the Qatar Genome Project, and the Mexico National Institute of Genomic Medicine (INMEGEN), are playing a pivotal role in bolstering the genomic research capabilities of Low- and Middle-Income Countries (LMICs). They achieve this by providing funding for locally driven research endeavors and empowering indigenous researchers to assume leadership roles in genomics projects [192]. Noteworthy accomplishments in this domain are exemplified by projects like the African Genome Variation Project [193] and the Mexico Genomic Variation Project [194]. These initiatives are dedicated to elucidating the intricate genetic structures within diverse ethnic groups, aiming to advance genomic medicine in Africa and Mexico [194].
- Effectively translating the genome sequence into actionable medical insights presents a significant hurdle. One major challenge is accurately anticipating the functional impact of genetic variations that disrupt protein-coding sequences. These variations can manifest in various ways, such as affecting transcription factor binding sites, interfering with microRNA target sites, influencing RNA splicing and stability, or even leading to protein truncation. Moreover, the intricacy of linkage disequilibrium, where seemingly benign genetic variations are situated near disease-predisposing variants, further complicates the interpretation of recurrent risk factors. Considering these complexities, there is a growing reliance on the use of in silico tools for inferring the functional consequences of mutations. As mentioned in the aforementioned sections, computational algorithms can play a crucial role in predicting the potential pathogenicity of genetic variants.
5.3. Ethical and Legal Considerations of Genomic Data Sharing and Privacy
5.4. Integration of Genomics with Other Surveillance and Diagnostic Methods
5.5. Innovations and Opportunities for Improving Genomics-Based Pathogen Detection and Tracking
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2019, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Lipkin, W.I. The genomics of emerging pathogens. Annu. Rev. Genom. Hum. Genet. 2013, 14, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.K.W. Reasons for the increase in emerging and re-emerging viral infectious diseases. Microbes Infect. 2006, 8, 905–916. [Google Scholar] [CrossRef]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Patz, J.A.; Reisen, W.K. Immunology, climate change and vector-borne diseases. Trends Immunol. 2001, 22, 171–172. [Google Scholar] [CrossRef]
- Bengis, R.G.; Leighton, F.A.; Fischer, J.R.; Artois, M.; Morner, T.; Tate, C.M. The role of wildlife in emerging and re-emerging zoonoses. Rev. Sci. Tech.-Off. Int. Epizoot. 2004, 23, 497–512. [Google Scholar]
- Caminade, C.; McIntyre, K.M.; Jones, A.E. Impact of recent and future climate change on vector-borne diseases. Ann. N. Y. Acad. Sci. 2019, 1436, 157–173. [Google Scholar] [CrossRef]
- Kilpatrick, A.M.; Randolph, S.E. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet 2012, 380, 1946–1955. [Google Scholar] [CrossRef]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature 2004, 430, 242–249. [Google Scholar] [CrossRef]
- Cutler, S.J.; Fooks, A.R.; Van Der Poel, W.H. Public health threat of new, reemerging, and neglected zoonoses in the industrialized world. Emerg. Infect. Dis. 2010, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.T.; Gayer, M.; Connolly, M.A. Epidemics after natural disasters. Emerg. Infect. Dis. 2007, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Ross, T.M. H3N2 influenza viruses in humans: Viral mechanisms, evolution, and evaluation. Hum. Vaccines Immunother. 2018, 14, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, J.L.; Gardy, J.L. A brief primer on genomic epidemiology: Lessons learned from Mycobacterium tuberculosis. Ann. N. Y. Acad. Sci. 2017, 1388, 59–77. [Google Scholar] [CrossRef]
- Eyre, D.W. Infection prevention and control insights from a decade of pathogen whole-genome sequencing. J. Hosp. Infect. 2022, 122, 180–186. [Google Scholar] [CrossRef]
- Gardy, J.L.; Johnston, J.C.; Sui, S.J.H.; Cook, V.J.; Shah, L.; Brodkin, E.; Rempel, S.; Moore, R.; Zhao, Y.; Holt, R.; et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. J. Med. 2011, 364, 730–739. [Google Scholar] [CrossRef]
- Ko, K.K.; Chng, K.R.; Nagarajan, N. Metagenomics-enabled microbial surveillance. Nat. Microbiol. 2022, 7, 486–496. [Google Scholar] [CrossRef]
- Nwadiugwu, M.C.; Monteiro, N. Applied genomics for identification of virulent biothreats and for disease outbreak surveillance. Postgrad. Med. J. 2022, 99, 403–410. [Google Scholar] [CrossRef]
- Cameron, A.; Bohrhunter, J.L.; Taffner, S.; Malek, A.; Pecora, N.D. Clinical Pathogen Genomics. Clin. Lab. Med. 2020, 40, 447–458. [Google Scholar] [CrossRef]
- Goldberg, B.; Sichtig, H.; Geyer, C.; Ledeboer, N.; Weinstock, G.M. Making the leap from research laboratory to clinic: Challenges and opportunities for next-generation sequencing in infectious disease diagnostics. MBio 2015, 6, e01888-15. [Google Scholar] [CrossRef]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Dallman, T.J.; Grant, K.A. Impact of whole genome sequencing on the investigation of food-borne outbreaks of Shiga toxin-producing Escherichia coli serogroup O157: H7, England, 2013 to 2017. Eurosurveillance 2019, 24, 1800346. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Azevedo, R.D.S.D.S.; Kraemer, M.U.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, S.J.; Dickins, B.; Jolley, K.A. Cronobacter, the emergent bacterial pathogen Enterobacter sakazakii comes of age; MLST and whole genome sequence analysis. BMC Genom. 2014, 15, 1121. [Google Scholar] [CrossRef]
- Koser, C.U.; Holden, M.T.; Ellington, M.J.; Cartwright, E.J.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef]
- Bartley, P.B.; Ben Zakour, N.L.; Stanton-Cook, M.; Muguli, R.; Prado, L.; Garnys, V.; Taylor, K.; Barnett, T.C.; Pinna, G.; Robson, J.; et al. Hospital-wide eradication of a nosocomial Legionella pneumophila serogroup 1 outbreak. Clin. Infect. Dis. 2016, 62, 273–279. [Google Scholar] [CrossRef]
- Grad, Y.H.; Lipsitch, M.; Feldgarden, M.; Arachchi, H.M.; Cerqueira, G.C.; FitzGerald, M.; Godfrey, P.; Haas, B.J.; Murphy, C.I.; Russ, C.; et al. Genomic epidemiology of the Escherichia coli O104: H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. USA 2012, 109, 3065–3070. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B. 1.1. 7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Sharma, C.; Kumar, N.; Pandey, R.; Meis, J.F.; Chowdhary, A. Whole genome sequencing of emerging multidrug resistant Candida auris isolates in India demonstrates low genetic variation. New Microbes New Infect. 2016, 13, 77–82. [Google Scholar] [CrossRef]
- O’Donnell, M.R.; Larsen, M.H.; Brown, T.S.; Jain, P.; Munsamy, V.; Wolf, A.; Uccellini, L.; Karim, F.; de Oliveira, T.; Mathema, B.; et al. Early detection of emergent extensively drug-resistant tuberculosis by flow cytometry-based phenotyping and whole-genome sequencing. Antimicrob. Agents Chemother. 2019, 63, e01834-18. [Google Scholar] [CrossRef]
- Espenhain, L.; Funk, T.; Overvad, M.; Edslev, S.M.; Fonager, J.; Ingham, A.C.; Rasmussen, M.; Madsen, S.L.; Espersen, C.H.; Sieber, R.N.; et al. Epidemiological characterisation of the first 785 SARS-CoV-2 Omicron variant cases in Denmark, December 2021. Eurosurveillance 2021, 26, 2101146. [Google Scholar] [CrossRef]
- Fonager, J.; Bennedbæk, M.; Bager, P.; Wohlfahrt, J.; Ellegaard, K.M.; Ingham, A.C.; Edslev, S.M.; Stegger, M.; Sieber, R.N.; Lassauniere, R.; et al. Molecular epidemiology of the SARS-CoV-2 variant Omicron BA. 2 sub-lineage in Denmark, 29 November 2021 to 2 January 2022. Eurosurveillance 2022, 27, 2200181. [Google Scholar] [CrossRef] [PubMed]
- Whaley, M.J.; Joseph, S.J.; Retchless, A.C.; Kretz, C.B.; Blain, A.; Hu, F.; Chang, H.Y.; Mbaeyi, S.A.; MacNeil, J.R.; Read, T.D.; et al. Whole genome sequencing for investigations of meningococcal outbreaks in the United States: A retrospective analysis. Sci. Rep. 2018, 8, 15803. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A., Jr.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; McCallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; McCauley, J.W. The WHO global influenza surveillance and response system (GISRS)—A future perspective. Influenza Other Respir. Viruses 2018, 12, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.L.; Yu, M.A.; Sacks, J.A.; Barnadas, C.; Pereyaslov, D.; Cognat, S.; Briand, S.; Ryan, M.J.; Samaan, G. Global genomic surveillance strategy for pathogens with pandemic and epidemic potential 2022–2032. Bull. World Health Organ. 2022, 100, 239–239A. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Sealy, T.K.; Khristova, M.L.; Albariño, C.G.; Conlan, S.; Reeder, S.A.; Quan, P.L.; Lipkin, W.I.; Downing, R.; Tappero, J.W.; et al. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008, 4, e1000212. [Google Scholar] [CrossRef]
- Lam, T.T.Y.; Jia, N.; Zhang, Y.W.; Shum, M.H.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef]
- Walker, T.M.; Kohl, T.A.; Omar, S.V.; Hedge, J.; Elias, C.D.O.; Bradley, P.; Iqbal, Z.; Feuerriegel, S.; Niehaus, K.E.; Wilson, D.J.; et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 1193–1202. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin. Infect. Dis. 2017, 64, 134–140. [Google Scholar] [CrossRef]
- Sardi, S.I.; Carvalho, R.H.; Pacheco, L.G.C.; Almeida, J.P.P.d.; Belitardo, E.M.M.d.A.; Pinheiro, C.S.; Campos, G.S.; Aguiar, E.R.G.R. High-quality resolution of the outbreak-related Zika virus genome and discovery of new viruses using ion torrent-based metatranscriptomics. Viruses 2020, 12, 782. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Song, T.; Rosenfeld, R.; Lin, X.; Rogers, M.B.; Zhou, B.; Sebra, R.; Halpin, R.A.; Guan, Y.; Twaddle, A.; et al. Quantifying influenza virus diversity and transmission in humans. Nat. Genet. 2016, 48, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Quick, J.; Claro, I.M.; Theze, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef]
- Piombo, E.; Abdelfattah, A.; Droby, S.; Wisniewski, M.; Spadaro, D.; Schena, L. Metagenomics approaches for the detection and surveillance of emerging and recurrent plant pathogens. Microorganisms 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Kingry, L.; Sheldon, S.; Oatman, S.; Pritt, B.; Anacker, M.; Bjork, J.; Neitzel, D.; Strain, A.; Berry, J.; Sloan, L.; et al. Targeted metagenomics for clinical detection and discovery of bacterial tick-borne pathogens. J. Clin. Microbiol. 2020, 58, e00147-20. [Google Scholar] [CrossRef]
- Balière, C.; Hourdel, V.; Kwasiborski, A.; Grassin, Q.; Feher, M.; Hoinard, D.; Vanhomwegen, J.; Taieb, F.; Consigny, P.H.; Manuguerra, J.C.; et al. Complete genome sequences of monkeypox virus from a French clinical sample and the corresponding isolated strain, obtained using nanopore sequencing. Microbiol. Resour. Announc. 2023, 12, e0000923. [Google Scholar] [CrossRef]
- Gauthier, N.P.; Nelson, C.; Bonsall, M.B.; Locher, K.; Charles, M.; MacDonald, C.; Krajden, M.; Chorlton, S.D.; Manges, A.R. Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples. PLoS ONE 2021, 16, e0259712. [Google Scholar] [CrossRef]
- Kugelman, J.R.; Wiley, M.R.; Mate, S.; Ladner, J.T.; Beitzel, B.; Fakoli, L.; Taweh, F.; Prieto, K.; Diclaro, J.W.; Minogue, T.; et al. Monitoring of Ebola virus Makona evolution through establishment of advanced genomic capability in Liberia. Emerg. Infect. Dis. 2015, 21, 1135. [Google Scholar] [CrossRef] [PubMed]
- Claro, I.M.; Romano, C.M.; Candido, D.D.S.; Lima, E.L.D.; Lindoso, J.A.L.; Ramundo, M.S.; Moreira, F.R.R.; Barra, L.A.C.; Borges, L.M.S.; Medeiros, L.A.; et al. Shotgun metagenomic sequencing of the first case of monkeypox virus in Brazil, 2022. Rev. Inst. Med. Trop. São Paulo 2022, 64. [Google Scholar] [CrossRef]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE 2010, 5, e13381. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Watson, S.J.; Kellam, P.; Al-Rabeeah, A.A.; Makhdoom, H.Q.; Assiri, A.; Al-Tawfiq, J.A.; Alhakeem, R.F.; Madani, H.; AlRabiah, F.A.; et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet 2014, 382, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Seekatz, A.M.; Aas, J.; Gessert, C.E.; Rubin, T.A.; Saman, D.M.; Bakken, J.S.; Young, V.B. Recovery of the gut microbiome following fecal microbiota transplantation. mBio 2014, 5, e00893-14. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Hakstol, R.; Kailasam, S.; Glaister, G.D.; Hudson, A.J.; Wieden, H.J. Rapid metagenomics analysis of EMS vehicles for monitoring pathogen load using nanopore DNA sequencing. PLoS ONE 2019, 14, e0219961. [Google Scholar] [CrossRef] [PubMed]
- Thannesberger, J.; Rascovan, N.; Eisenmann, A.; Klymiuk, I.; Zittra, C.; Fuehrer, H.P.; Scantlebury-Manning, T.; Gittens-St Hilaire, M.; Austin, S.; Landis, R.C.; et al. Viral metagenomics reveals the presence of novel Zika virus variants in Aedes mosquitoes from Barbados. Parasites Vectors 2021, 14, 343. [Google Scholar] [CrossRef]
- Li, T.; Mbala-Kingebeni, P.; Naccache, S.N.; Thézé, J.; Bouquet, J.; Federman, S.; Somasekar, S.; Yu, G.; Sanchez-San Martin, C.; Achari, A.; et al. Metagenomic next-generation sequencing of the 2014 Ebola virus disease outbreak in the Democratic Republic of the Congo. J. Clin. Microbiol. 2019, 57, e00827-19. [Google Scholar] [CrossRef]
- Aljabr, W.; Alruwaili, M.; Penrice-Randal, R.; Alrezaihi, A.; Harrison, A.J.; Ryan, Y.; Bentley, E.; Jones, B.; Alhatlani, B.Y.; AlShahrani, D.; et al. Amplicon and metagenomic analysis of middle east respiratory syndrome (MERS) coronavirus and the microbiome in patients with severe MERS. Msphere 2021, 6, e0021921. [Google Scholar] [CrossRef]
- Souza, J.V.C.; Santos, H.D.O.; Leite, A.B.; Giovanetti, M.; Bezerra, R.D.S.; Carvalho, E.D.; Bernardino, J.D.S.T.; Viala, V.L.; Haddad, R.; Ciccozzi, M.; et al. Viral metagenomics for the identification of emerging infections in clinical samples with inconclusive Dengue, Zika, and Chikungunya viral amplification. Viruses 2022, 14, 1933. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, S.; Huang, B.; Zhong, H.; Pan, Z.; Zhuang, Q.; Peng, C.; Hou, G.; Wang, K. Viral infection detection using metagenomics technology in six poultry farms of eastern China. PLoS ONE 2019, 14, e0211553. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic nanopore sequencing of influenza virus direct from clinical respiratory samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.M.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104: H4. JAMA 2013, 309, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Chain, P.; Kurtz, S.; Ohlebusch, E.; Slezak, T. An applications-focused review of comparative genomics tools: Capabilities, limitations and future challenges. Brief. Bioinform. 2003, 4, 105–123. [Google Scholar] [CrossRef]
- Miller, W.; Makova, K.D.; Nekrutenko, A.; Hardison, R.C. Comparative genomics. Annu. Rev. Genom. Hum. Genet. 2004, 5, 15–56. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Genetic variation, comparative genomics, and the diagnosis of disease. N. Engl. J. Med. 2019, 381, 64–74. [Google Scholar] [CrossRef]
- Zhang, Q.; Jing, S.; Cheng, Z.; Yu, Z.; Dehghan, S.; Shamsaddini, A.; Yan, Y.; Li, M.; Seto, D. Comparative genomic analysis of two emergent human adenovirus type 14 respiratory pathogen isolates in China reveals similar yet divergent genomes. Emerg. Microbes Infect. 2017, 6, e92. [Google Scholar] [CrossRef]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar] [CrossRef]
- Ahammad, I.; Hossain, M.U.; Rahman, A.; Chowdhury, Z.M.; Bhattacharjee, A.; Das, K.C.; Keya, C.A.; Salimullah, M. Wave-wise comparative genomic study for revealing the complete scenario and dynamic nature of COVID-19 pandemic in Bangladesh. PLoS ONE 2021, 16, e0258019. [Google Scholar] [CrossRef]
- Asrani, P.; Hasan, G.M.; Sohal, S.S.; Hassan, M.I. Molecular basis of pathogenesis of coronaviruses: A comparative genomics approach to planetary health to prevent zoonotic outbreaks in the 21st century. Omics J. Integr. Biol. 2020, 24, 634–644. [Google Scholar] [CrossRef]
- Khan, M.I.; Khan, Z.A.; Baig, M.H.; Ahmad, I.; Farouk, A.E.; Song, Y.G.; Dong, J.J. Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: An in silico insight. PLoS ONE 2020, 15, e0238344. [Google Scholar] [CrossRef]
- Cheng, Z.; Yan, Y.; Jing, S.; Li, W.G.; Chen, W.W.; Zhang, J.; Li, M.; Zhao, S.; Cao, N.; Ou, J.; et al. Comparative genomic analysis of re-emergent human adenovirus type 55 pathogens associated with adult severe community-acquired pneumonia reveals conserved genomes and capsid proteins. Front. Microbiol. 2018, 9, 1180. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.R.; Leuze, M.R.; Nookaew, I.; Uberbacher, E.C.; Land, M.; Zhang, Q.; Wanchai, V.; Chai, J.; Nielsen, M.; Trolle, T.; et al. Ebolavirus comparative genomics. FEMS Microbiol. Rev. 2015, 39, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, F.S.; Leipe, D.D. Phylogenetic Analysis. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins, 2nd ed.; John Wiley & Sons, Inc: New York, NY, USA, 2001; Volume 2, p. 349. [Google Scholar]
- Behl, A.; Nair, A.; Mohagaonkar, S.; Yadav, P.; Gambhir, K.; Tyagi, N.; Sharma, R.K.; Butola, B.S.; Sharma, N. Threat, challenges, and preparedness for future pandemics: A descriptive review of phylogenetic analysis based predictions. Infect. Genet. Evol. 2022, 98, 105217. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 9241–9243. [Google Scholar] [CrossRef] [PubMed]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. eLife 2017, 6, e31257. [Google Scholar]
- Yebra, G.; Ragonnet-Cronin, M.; Ssemwanga, D.; Parry, C.M.; Logue, C.H.; Cane, P.A.; Kaleebu, P.; Brown, A.J.L. Analysis of the history and spread of HIV-1 in Uganda using phylodynamics. J. Gen. Virol. 2015, 96, 1890. [Google Scholar] [CrossRef]
- Benvenuto, D.; Cella, E.; Fogolari, M.; De Florio, L.; Borsetti, A.; Donati, D.; Garilli, F.; Spoto, S.; Ceccarelli, G.; Angeletti, S.; et al. The transmission dynamic of Madariaga Virus by bayesian phylogenetic analysis: Molecular surveillance of an emergent pathogen. Microb. Pathog. 2019, 132, 80–86. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, B.M.; Tsai, H.C.; Chen, Y.P.; Huang, T.Y.; Li, K.Y.; Ji, D.D.; Lee, H.S. Molecular surveillance of Vittaforma-like microsporidia by a small-volume procedure in drinking water source in Taiwan: Evidence for diverse and emergent pathogens. Environ. Sci. Pollut. Res. 2018, 25, 18823–18837. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.R.; Driebe, E.M.; Albrecht, V.; McDougal, L.K.; Granade, M.; Roe, C.C.; Lemmer, D.; Rasheed, J.K.; Engelthaler, D.M.; Keim, P.; et al. Improved subtyping of Staphylococcus aureus clonal complex 8 strains based on whole-genome phylogenetic analysis. Msphere 2018, 3, e00464-17. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.Y.; Hon, C.C.; Tang, J.W. Use of phylogenetics in the molecular epidemiology and evolutionary studies of viral infections. Crit. Rev. Clin. Lab. Sci. 2010, 47, 5–49. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Vakulenko, Y.A.; Turbabina, N.A.; Deviatkin, A.A.; Drexler, J.F. Molecular epidemiology and phylogenetics of human enteroviruses: Is there a forest behind the trees? Rev. Med. Virol. 2018, 28, e2002. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Y.; Teng, X.; Hou, H.; Deng, R.; Li, J. CRISPR-based nucleic acid diagnostics for pathogens. TrAC Trends Anal. Chem. 2023, 160, 116980. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.I.; Makhawi, A.M. SHERLOCK and DETECTR: CRISPR-Cas systems as potential rapid diagnostic tools for emerging infectious diseases. J. Clin. Microbiol. 2021, 59, e00745-20. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. Crispr-Cas12a Target Binding Unleashes Indiscriminate Single-Stranded Dnase Activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, L.; Liu, C.; Ye, S.; Chen, W.; Li, D.; Huang, W. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J. Transl. Med. 2021, 19, 1–10. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical Validation of a Cas13-Based Assay for the Detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Agrawal, S.; Fanton, A.; Chandrasekaran, S.S.; Charrez, B.; Escajeda, A.M.; Son, S.; Mcintosh, R.; Bhuiya, A.; de León Derby, M.D.; Switz, N.A.; et al. Rapid, point-of-care molecular diagnostics with Cas13. MedRxiv 2021. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, E.; Tian, T.; Cheng, M.; Lin, W.; Wang, H.; Zhang, G.; Sun, J.; Zhou, X. Clustered regularly interspaced short palindromic repeats/Cas9-mediated lateral flow nucleic acid assay. ACS Nano 2020, 14, 2497–2508. [Google Scholar] [CrossRef]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Smith, B.M.; Jain, P.K. Enhancement of Trans-Cleavage Activity of Cas12a with Engineered Crrna Enables Amplified Nucleic Acid Detection. Nat. Commun. 2020, 11, 4906. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Zuo, L.; He, D.; Fang, Z.; Berthet, N.; Yu, C.; Wong, G. Rapid detection of Nipah virus using the one-pot RPA-CRISPR/Cas13a assay. Virus Res. 2023, 332, 199130. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, U.; Liu, C. CRISPR-based COVID-19 testing: Toward next-generation point-of-care diagnostics. Front. Cell. Infect. Microbiol. 2021, 11, 663949. [Google Scholar] [CrossRef]
- Qiu, E.; Jin, S.; Xiao, Z.; Chen, Q.; Wang, Q.; Liu, H.; Xie, C.; Chen, C.; Li, Z.; Han, S. CRISPR-based Detection of Helicobacter pylori in Stool Samples. Helicobacter 2021, 26, e12828. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Huyke, D.A.; Sharma, E.; Sahoo, M.K.; Huang, C.; Banaei, N.; Pinsky, B.A.; Santiago, J.G. Electric field-driven microfluidics for rapid CRISPR-based diagnostics and its application to detection of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 29518–29525. [Google Scholar] [CrossRef]
- Zhan, Y.; Gao, X.; Li, S.; Si, Y.; Li, Y.; Han, X.; Sun, W.; Li, Z.; Ye, F. Development and evaluation of rapid and accurate CRISPR/Cas13-based RNA diagnostics for Pneumocystis jirovecii pneumonia. Front. Cell. Infect. Microbiol. 2022, 12, 765. [Google Scholar] [CrossRef]
- Joung, J.; Ladha, A.; Saito, M.; Segel, M.; Bruneau, R.; Mee-li, W.H.; Kim, N.G.; Yu, X.; Li, J.; Walker, B.D.; et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. MedRxiv 2020. [Google Scholar] [CrossRef]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Li, S.; Zhu, X.; Wang, X.; Huang, J.; Yang, X.; Tai, J. LAMP-CRISPR-Cas12-based diagnostic platform for detection of Mycobacterium tuberculosis complex using real-time fluorescence or lateral flow test. Microchim. Acta 2021, 188, 1–9. [Google Scholar] [CrossRef]
- Ai, J.W.; Zhou, X.; Xu, T.; Yang, M.; Chen, Y.; He, G.Q.; Pan, N.; Cai, Y.; Li, Y.; Wang, X.; et al. CRISPR-based rapid and ultra-sensitive diagnostic test for Mycobacterium tuberculosis. Emerg. Microbes Infect. 2019, 8, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Kham-Kjing, N.; Ngo-Giang-Huong, N.; Tragoolpua, K.; Khamduang, W.; Hongjaisee, S. Highly Specific and Rapid Detection of hepatitis C virus using RT-LAMP-coupled CRISPR–Cas12 assay. Diagnostics 2022, 12, 1524. [Google Scholar] [CrossRef]
- Chen, Q.; Gul, I.; Liu, C.; Lei, Z.; Li, X.; Raheem, M.A.; He, Q.; Haihui, Z.; Leeansyah, E.; Zhang, C.Y.; et al. CRISPR–Cas12-based field-deployable system for rapid detection of synthetic DNA sequence of the monkeypox virus genome. J. Med. Virol. 2023, 95, e28385. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.G.; Lachenauer, A.E.; Nitido, A.; Siddiqui, S.; Gross, R.; Beitzel, B.; Siddle, K.J.; Freije, C.A.; Dighero-Kemp, B.; Mehta, S.B.; et al. Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat. Commun. 2020, 11, 4131. [Google Scholar] [CrossRef]
- Zhou, H.; Bu, S.; Xu, Y.; Xue, L.; Li, Z.; Hao, Z.; Wan, J.; Tang, F. CRISPR/Cas13a combined with hybridization chain reaction for visual detection of influenza A (H1N1) virus. Anal. Bioanal. Chem. 2022, 414, 8437–8445. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Boyle, D.S.; Lehman, D.A.; Lillis, L.; Peterson, D.; Singhal, M.; Armes, N.; Parker, M.; Piepenburg, O.; Overbaugh, J. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. MBio 2013, 4, e00135-13. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. CDC Real Time RT-PCR (rRT-PCR) Protocol for Detection and Characterization of Influenza 2009 A (H1N1) Pdm Virus—RUO International; CDC: Atlanta, GA, USA, 2010.
- Tahamtan, A.; Ardebili, A. Real-time RT-PCR in COVID-19 detection: Issues affecting the results. Expert Rev. Mol. Diagn. 2020, 20, 453–454. [Google Scholar] [CrossRef]
- Vogels, C.B.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 qRT-PCR primer-probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef]
- Faye, O.; Faye, O.; Soropogui, B.; Patel, P.; El Wahed, A.A.; Loucoubar, C.; Fall, G.; Kiory, D.; Magassouba, N.F.; Keita, S.; et al. Development and deployment of a rapid recombinase polymerase amplification Ebola virus detection assay in Guinea in 2015. Eurosurveillance 2015, 20, 30053. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, J.J.; Pinsky, B.A. Zika virus: Diagnostics for an emerging pandemic threat. J. Clin. Microbiol. 2016, 54, 860–867. [Google Scholar] [CrossRef]
- Huang, Y.; Wei, H.; Wang, Y.; Shi, Z.; Raoul, H.; Yuan, Z. Rapid detection of filoviruses by real-time TaqMan polymerase chain reaction assays. Virol. Sin. 2012, 27, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Takahashi, H.; Yamamoto, N.; Hamaguchi, S.; Ojima, M.; Hirose, T.; Yoshiya, K.; Ogura, H.; Shimazu, T.; Tomono, K. Polymerase chain reaction-based active surveillance of MRSA in emergency department patients. Infect. Drug Resist. 2015, 8, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, E.S. Multiplexed diagnosis of four serotypes of dengue virus by real-time RT-PCR. BioChip J. 2020, 14, 421–428. [Google Scholar] [CrossRef]
- Tokarz, R.; Tagliafierro, T.; Cucura, D.M.; Rochlin, I.; Sameroff, S.; Lipkin, W.I. Detection of Anaplasma phagocytophilum, Babesia microti, Borrelia burgdorferi, Borrelia miyamotoi, and Powassan virus in ticks by a multiplex real-time reverse transcription-PCR assay. MSphere 2017, 2, e00151-17. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Wang, W. Multiplex real-time PCR assay for detection of Escherichia coli O157: H7 and screening for non-O157 Shiga toxin-producing E. coli. BMC Microbiol. 2017, 17, 215. [Google Scholar] [CrossRef]
- Wei, C.; Zhong, J.; Hu, T.; Zhao, X. Simultaneous detection of Escherichia coli O157: H7, Staphylococcus aureus and Salmonella by multiplex PCR in milk. 3 Biotech 2018, 8, 76. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sahay, R.R.; Balakrishnan, A.; Mohandas, S.; Radhakrishnan, C.; Gokhale, M.D.; Balasubramanian, R.; Abraham, P.; Gupta, N.; Sugunan, A.P.; et al. Nipah virus outbreak in Kerala State, India amidst of COVID-19 pandemic. Front. Public Health 2022, 10, 818545. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- L’Huillier, A.G.; Lombos, E.; Tang, E.; Perusini, S.; Eshaghi, A.; Nagra, S.; Frantz, C.; Olsha, R.; Kristjanson, E.; Dimitrova, K.; et al. Evaluation of Altona Diagnostics RealStar Zika virus reverse transcription-PCR test kit for Zika virus PCR testing. J. Clin. Microbiol. 2017, 55, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Uhteg, K.; Mostafa, H.H. Validation and implementation of an orthopoxvirus qualitative real-time PCR for the diagnosis of monkeypox in the clinical laboratory. J. Clin. Virol. 2023, 158, 105327. [Google Scholar] [CrossRef]
- Georgacopoulos, O.; Nunnally, N.S.; Le, N.; Lysen, C.; Welsh, R.M.; Kordalewska, M.; Perlin, D.S.; Berkow, E.L.; Sexton, D.J. Performance evaluation of culture-independent SYBR green Candida auris quantitative PCR diagnostics on anterior nares surveillance swabs. J. Clin. Microbiol. 2020, 58, e00690-20. [Google Scholar] [CrossRef] [PubMed]
- Antón, A.; Marcos, M.A.; Codoñer, F.M.; de Molina, P.; Martínez, A.; Cardeñosa, N.; Godoy, P.; Torner, N.; Martínez, M.J.; Ramón, S.; et al. Influenza C virus surveillance during the first influenza A (H1N1) 2009 pandemic wave in Catalonia, Spain. Diagn. Microbiol. Infect. Dis. 2011, 69, 419–427. [Google Scholar] [CrossRef]
- Jothikumar, N.; Lowther, J.A.; Henshilwood, K.; Lees, D.N.; Hill, V.R.; Vinjé, J. Rapid and sensitive detection of noroviruses by using TaqMan-based one-step reverse transcription-PCR assays and application to naturally contaminated shellfish samples. Appl. Environ. Microbiol. 2005, 71, 1870–1875. [Google Scholar] [CrossRef]
- Fernández-Pinero, J.; Gallardo, C.; Elizalde, M.; Robles, A.; Gómez, C.; Bishop, R.; Heath, L.; Couacy-Hymann, E.; Fasina, F.O.; Pelayo, V.; et al. Molecular diagnosis of African swine fever by a new real-time PCR using universal probe library. Transbound. Emerg. Dis. 2013, 60, 48–58. [Google Scholar] [CrossRef]
- Kim, S.H.; Chang, S.Y.; Sung, M.; Park, J.H.; Bin Kim, H.; Lee, H.; Choi, J.P.; Choi, W.S.; Min, J.Y. Extensive viable Middle East respiratory syndrome (MERS) coronavirus contamination in air and surrounding environment in MERS isolation wards. Rev. Infect. Dis. 2016, 63, 363–369. [Google Scholar] [CrossRef]
- Bartlow, A.W.; Stromberg, Z.R.; Gleasner, C.D.; Hu, B.; Davenport, K.W.; Jakhar, S.; Li, P.E.; Vosburg, M.; Garimella, M.; Chain, P.S.; et al. Comparing variability in diagnosis of upper respiratory tract infections in patients using syndromic, next generation sequencing, and PCR-based methods. PLoS Glob. Public Health 2022, 2, e0000811. [Google Scholar] [CrossRef]
- Anderson, E.M.; Maldarelli, F. Quantification of HIV DNA using droplet digital PCR techniques. Curr. Protoc. Microbiol. 2018, 51, e62. [Google Scholar] [CrossRef]
- Fowler, V.L.; Armson, B.; Gonzales, J.L.; Wise, E.L.; Howson, E.L.; Vincent-Mistiaen, Z.; Fouch, S.; Maltby, C.J.; Grippon, S.; Munro, S.; et al. A highly effective reverse-transcription loop-mediated isothermal amplification (RT-LAMP) assay for the rapid detection of SARS-CoV-2 infection. J. Infect. 2021, 82, 117–125. [Google Scholar] [CrossRef]
- Donatin, E.; Drancourt, M. DNA microarrays for the diagnosis of infectious diseases. Médecine Mal. Infect. 2012, 42, 453–459. [Google Scholar] [CrossRef]
- Liu, H.; Bebu, I.; Li, X. Microarray probes and probe sets. Front. Biosci. (Elite Ed.) 2010, 2, 325. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Coscoy, L.; Zylberberg, M.; Avila, P.C.; Boushey, H.A.; Ganem, D.; DeRisi, J.L. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. USA 2002, 99, 15687–15692. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Urisman, A.; Liu, Y.T.; Springer, M.; Ksiazek, T.G.; Erdman, D.D.; Mardis, E.R.; Hickenbotham, M.; Magrini, V.; Eldred, J.; et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003, 1, e2. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Wang, Z.; Vora, G.J.; Thornton, J.A.; Schnur, J.M.; Thach, D.C.; Blaney, K.M.; Ligler, A.G.; Malanoski, A.P.; Santiago, J.; et al. Broad-spectrum respiratory tract pathogen identification using resequencing DNA microarrays. Genome Res. 2006, 16, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Malanoski, A.P.; Lin, B.; Wang, Z.; Schnur, J.M.; Stenger, D.A. Automated identification of multiple micro-organisms from resequencing DNA microarrays. Nucleic Acids Res. 2006, 34, 5300–5311. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Leski, T.A.; Lin, B.; Malanoski, A.P.; Wang, Z.; Long, N.C.; Meador, C.E.; Barrows, B.; Ibrahim, S.; Hardick, J.P.; Aitichou, M.; et al. Testing and validation of high density resequencing microarray for broad range biothreat agents detection. PLoS ONE 2009, 4, e6569. [Google Scholar] [CrossRef]
- Palacios, G.; Quan, P.L.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H.; et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 2007, 13, 73. [Google Scholar] [CrossRef]
- Quan, P.L.; Palacios, G.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Pozo, F.; Jack, P.J.; Cisterna, D.; Renwick, N.; Hui, J.; et al. Detection of respiratory viruses and subtype identification of influenza A viruses by GreeneChipResp oligonucleotide microarray. J. Clin. Microbiol. 2007, 45, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, H.; Kawasaki, J.; Kubo, K.; Fukunaga, T.; Hokao, K.; Yokoyama, G.; Ichinose, A.; Suga, K.; Hamada, M. Bioinformatics approaches for unveiling virus—Host interactions. Comput. Struct. Biotechnol. J. 2023, 21, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Saeb, A.T.; Abouelhoda, M.; Selvaraju, M.; Althawadi, S.I.; Mutabagani, M.; Adil, M.; Al Hokail, A.; Tayeb, H.T. The use of next-generation sequencing in the identification of a fastidious pathogen: A lesson from a clinical setup. Evol. Bioinform. 2017, 13, 1176934316686072. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, G.M. Genomic approaches to studying the human microbiota. Nature 2012, 489, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Skwor, T. The Use of DNASTAR Lasergene Educational Software with Molecular Techniques to Support Bacterial Identification. Proc. Assoc. Biol. Lab. Educ. 2012, 33, 327–334. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Darling, A.E.; Treangen, T.J.; Messeguer, X.; Perna, N.T. Analyzing patterns of microbial evolution using the mauve genome alignment system. Comp. Genom. 2007, 396, 135–152. [Google Scholar]
- Bragin, E.; Chatzimichali, E.A.; Wright, C.F.; Hurles, M.E.; Firth, H.V.; Bevan, A.P.; Swaminathan, G.J. DECIPHER: Database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014, 42, D993–D1000. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pathogen Detection Beta [Internet]; National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA, 2004. Available online: https://www.ncbi.nlm.nih.gov/pathogens/ (accessed on 15 August 2023).
- Snyder, E.E.; Kampanya, N.; Lu, J.; Nordberg, E.K.; Karur, H.R.; Shukla, M.; Soneja, J.; Tian, Y.; Xue, T.; Yoo, H.; et al. PATRIC: The VBI pathosystems resource integration center. Nucleic Acids Res. 2007, 35 (Suppl. S1), D401–D406. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder-distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Deneke, C.; Rentzsch, R.; Renard, B.Y. PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data. Sci. Rep. 2017, 7, 39194. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.A.; Doyle, C.J.; Jennison, A.V. Real-time investigation of a Legionella pneumophila outbreak using whole genome sequencing. Epidemiol. Infect. 2014, 142, 2347–2351. [Google Scholar] [CrossRef]
- Inns, T.; Lane, C.; Peters, T.; Dallman, T.; Chatt, C.; McFarland, N.; Crook, P.; Bishop, T.; Edge, J.; Hawker, J.; et al. A multi-country Salmonella Enteritidis phage type 14b outbreak associated with eggs from a German producer: ‘near real-time’ application of whole genome sequencing and food chain investigations, United Kingdom, May to September 2014. Eurosurveillance 2015, 20, 21098. [Google Scholar] [CrossRef]
- Quick, J.; Ashton, P.; Calus, S.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015, 16, 114. [Google Scholar] [CrossRef]
- Didelot, X.; Fraser, C.; Gardy, J.; Colijn, C. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks. Mol. Biol. Evol. 2017, 34, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.; Gaunt, E. Ecological origins of novel human pathogens. Crit. Rev. Microbiol. 2007, 33, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.A.; Preston, N.; Allen, T.; Zambrana-Torrelio, C.; Hosseini, P.R.; Daszak, P. Global biogeography of human infectious diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 12746–12751. [Google Scholar] [CrossRef]
- Kelly, T.R.; Karesh, W.B.; Johnson, C.K.; Gilardi, K.V.; Anthony, S.J.; Goldstein, T.; Olson, S.H.; Machalaba, C.; Predict Consortium; Mazet, J.A. One Health proof of concept: Bringing a transdisciplinary approach to surveillance for zoonotic viruses at the human-wild animal interface. Prev. Vet. Med. 2017, 137, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Smiley Evans, T.; Barry, P.A.; Gilardi, K.V.; Goldstein, T.; Deere, J.D.; Fike, J.; Yee, J.; Ssebide, B.J.; Karmacharya, D.; Cranfield, M.R.; et al. Optimization of a novel non-invasive oral sampling technique for zoonotic pathogen surveillance in nonhuman primates. PLoS Neglected Trop. Dis. 2015, 9, e0003813. [Google Scholar] [CrossRef]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Ali Khan, S.; et al. A strategy to estimate unknown viral diversity in mammals. MBio 2013, 4, e00598-13. [Google Scholar] [CrossRef]
- Anthony, S.J.; Islam, A.; Johnson, C.; Navarrete-Macias, I.; Liang, E.; Jain, K.; Hitchens, P.L.; Che, X.; Soloyvov, A.; Hicks, A.L.; et al. Non-random patterns in viral diversity. Nat. Commun. 2015, 6, 8147. [Google Scholar] [CrossRef]
- GHRF Commission (Commission on a Global Health Risk Framework for the Future). The Neglected Dimension of Global Security a Framework to Counter Infectious Disease Crises; Commission on Global Health Risk Framework for the Future; National Academies Press: Washington, DC, USA, 2016. [Google Scholar]
- Mandl, K.D.; Overhage, J.M.; Wagner, M.M.; Lober, W.B.; Sebastiani, P.; Mostashari, F.; Pavlin, J.A.; Gesteland, P.H.; Treadwell, T.; Koski, E.; et al. Implementing syndromic surveillance: A practical guide informed by the early experience. J. Am. Med. Inform. Assoc. 2004, 11, 141–150. [Google Scholar] [CrossRef]
- Henning, K.J. What is syndromic surveillance? Morb. Mortal. Wkly. Rep. 2004, 53, 7–11. [Google Scholar]
- Brownstein, J.S.; Freifeld, C.C.; Madoff, L.C. Digital disease detection—Harnessing the Web for public health surveillance. N. Engl. J. Med. 2009, 360, 2153. [Google Scholar] [CrossRef]
- Smolinski, M.S.; Crawley, A.W.; Baltrusaitis, K.; Chunara, R.; Olsen, J.M.; Wójcik, O.; Santillana, M.; Nguyen, A.; Brownstein, J.S. Flu near you: Crowdsourced symptom reporting spanning 2 influenza seasons. Am. J. Public Health 2015, 105, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Hulth, A.; Rydevik, G.; Linde, A. Web queries as a source for syndromic surveillance. PLoS ONE 2009, 4, e4378. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, H.A.; Mylonakis, E. Google trends: A web-based tool for real-time surveillance of disease outbreaks. Clin. Infect. Dis. 2009, 49, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J. Digital disease detection: A systematic review of event-based internet biosurveillance systems. Int. J. Med. Inform. 2017, 101, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, J.S.; Freifeld, C.C. HealthMap: The development of automated real-time internet surveillance for epidemic intelligence. Eurosurveillance 2007, 12, 3322. [Google Scholar] [CrossRef] [PubMed]
- Neher, R.A.; Bedford, T. Real-time analysis and visualization of pathogen sequence data. J. Clin. Microbiol. 2018, 56, e00480-18. [Google Scholar] [CrossRef] [PubMed]
- Bagabir, S.A.; Ibrahim, N.K.; Bagabir, H.A.; Ateeq, R. Covid-19 and Artificial Intelligence: Genome sequencing, drug development and vaccine discovery. J. Infect. Public Health 2022, 15, 289–296. [Google Scholar] [CrossRef]
- Sundermann, A.; Chen, J.; Kumar, P.; Ayres, A.; Cho, S.; Ezeonwuka, C.; Griffith, M.P.; Miller, J.Z.; Mustapha, M.M.; Pasculle, A.W.; et al. Whole-Genome sequencing surveillance and machine learning of the Electronic Health Record for enhanced healthcare outbreak detection. Clin. Infect. Dis. 2021, 75, 476–482. [Google Scholar] [CrossRef]
- Yu, K.; Beam, A.L.; Kohane, I.S. Artificial intelligence in healthcare. Nat. Biomed. Eng. 2018, 2, 719–731. [Google Scholar] [CrossRef]
- Ross, E. Perspectives on Data Sharing in Disease Surveillance; Centre on Global Health Security; The Royal Institute of International Affairs Chatham House: London, UK, 2014. [Google Scholar]
- Leber, A.L.; Peterson, E.; Bard, J.D. The hidden crisis in the times of COVID-19: Critical shortages of medical laboratory professionals in clinical microbiology. J. Clin. Microbiol. 2022, 60, e0024122. [Google Scholar] [CrossRef]
- Cornish, N.E.; Bachmann, L.H.; Diekema, D.J.; McDonald, L.C.; McNult, P.; Stevens-Garcia, J.; Raphael, B.H.; Miller, M.B. Pandemic Demand for SARS-CoV-2 Testing Led to Critical Supply and Workforce Shortages in U.S. Clinical and Public Health Laboratories. J. Clin. Microbiol. 2023, 61, e0318920. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, F.M.; Koopmans, M.G. Sharing data for global infectious disease surveillance and outbreak detection. Trends Microbiol. 2016, 24, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Sane, J.; Edelstein, M. Overcoming Barriers to Data Sharing in Public Health. A Global Perspective; Chatham House: London, UK, 2015. [Google Scholar]
- Gans, J.; Wolinsky, M.; Dunbar, J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005, 309, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 1–8. [Google Scholar] [CrossRef]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, C.N.; Abayomi, A.; Abimiku, A.; Adabayeri, V.; Adebamowo, C.; Adebiyi, E.; Ademola, A.D.; Adeyemo, A.; Adu, D.; Affolabi, D.; et al. Enabling the genomic revolution in Africa. Science 2014, 344, 1346–1348. [Google Scholar] [CrossRef]
- Gurdasani, D.; Carstensen, T.; Tekola-Ayele, F.; Pagani, L.; Tachmazidou, I.; Hatzikotoulas, K.; Karthikeyan, S.; Iles, L.; Pollard, M.; Choudhury, A.; et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2014, 517, 327–332. [Google Scholar] [CrossRef]
- Moreno-Estrada, A.; Gignoux, C.R.; Fernández-López, J.C.; Zakharia, F.; Sikora, M.; Contreras, A.; Acuña-Alonzo, V.; Sandoval, K.; Eng, C.; Romero-Hidalgo, S.; et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science 2014, 344, 1280–1285. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Langmead, B.; Nellore, A. Cloud computing for genomic data analysis and collaboration. Nat. Rev. Genet. 2018, 19, 208–219. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Magnus, D. Privacy and ethical challenges in next-generation sequencing. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 95–104. [Google Scholar] [CrossRef]
- Coltart, C.E.M.; Hoppé, A.; Parker, M.; Dawson, L.; Amon, J.J.; Simwinga, M.; Geller, G.; Henderson, G.E.; Laeyendecker, O.; Tucker, J.D.; et al. Ethical considerations in global HIV phylogenetic research. Lancet HIV 2018, 5, e656–e666. [Google Scholar] [CrossRef]
- Mutenherwa, F.; Wassenaar, D.; De Oliveira, T. Experts’ perspectives on key ethical issues associated with HIV phylogenetics as applied in HIV Transmission dynamics research. J. Empir. Res. Hum. Res. Ethics 2018, 14, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Degeling, C.; Johnson, J.; Gilbert, G.L. Perspectives of Australian policy-makers on the potential benefits and risks of technologically enhanced communicable disease surveillance—A modified Delphi survey. Health Res. Policy Syst. 2019, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Rump, B.; Cornelis, C.; Woonink, F.; Verweij, M. The need for ethical reflection on the use of molecular microbial characterisation in outbreak management. Eurosurveillance 2013, 18, 20384. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.D.S.; Van Roode, M.Y.; Haringhuizen, G.B.; Koopmans, M.; Claassen, E.; Van De Burgwal, L. How ownership rights over microorganisms affect infectious disease control and innovation: A root-cause analysis of barriers to data sharing as experienced by key stakeholders. PLoS ONE 2018, 13, e0195885. [Google Scholar] [CrossRef] [PubMed]
- Knoppers, B.M.; Harris, J.R.; Budin-Ljøsne, I.; Dove, E.S. A framework for responsible sharing of genomic and health-related data. Lancet Oncol. 2014, 15, e224–e231. [Google Scholar] [CrossRef]
- Kiermer, V.; Bourne, P.E.; Fullerton, S.M.; Chambers, C. The European General Data Protection Regulation: Challenges and considerations for research. eLife 2018, 7, e34473. [Google Scholar] [CrossRef]
- Cremers, A.J.; Coolen, J.P.; Bleeker-Rovers, C.P.; van der Geest-Blankert, A.D.; Haverkate, D.; Hendriks, H.; Henriet, S.S.; Huynen, M.A.; Kolwijck, E.; Liem, D.; et al. Surveillance-embedded genomic outbreak resolution of methicillin-susceptible Staphylococcus aureus in a neonatal intensive care unit. Sci. Rep. 2020, 10, 2619. [Google Scholar] [CrossRef] [PubMed]
- Buragohain, L.; Ghosh, M.; Kumar, R.; Dahiya, S.; Malik, Y.S.; Prasad, M. Application of Proteomics and Metabolomics in Disease Diagnosis. In Advances in Animal Disease Diagnosis; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2021; pp. 79–102. [Google Scholar]
- Sahajpal, N.S.; Mondal, A.K.; Njau, A.; Petty, Z.; Chen, J.; Ananth, S.; Ahluwalia, P.; Williams, C.; Ross, T.M.; Chaubey, A.; et al. High-throughput next-generation sequencing respiratory viral panel: A diagnostic and epidemiologic tool for SARS-CoV-2 and other viruses. Viruses 2021, 13, 2063. [Google Scholar] [CrossRef]

| Pathogen | Sample Type | Method | Sensitivity | Specificity | References |
|---|---|---|---|---|---|
| SARS-CoV-2 | Sputum as well as nose and throat swabs | Real-time RT-PCR | 95% | - | [124] |
| Zika | Serum | RealStar ZIKV rRT-PCR test kit | 91% | 97% | [125] |
| Zaire Ebolavirus (ZEBOV) | Cell lines | TaqMan RT-PCR | 109 copies to 103 copies/reaction | - | [117] |
| Monkeypox | Lesion swabs | Non-variola orthopoxvirus PCR test | 100 copies/mL and 100% agreement. | 100% | [126] |
| Candida auris | Axilla-groin composite surveillance swabs | SYBR green qPCR | 0.93 | 0.96 | [127] |
| Influenza C virus (FLUCV) | Nasopharyngeal samples (nasal and oropharyngeal swabs) | Multiplex RT-PCR | - | - | [128] |
| Noroviruses (NoV) | Stool specimens | TaqMan RT-PCR assay | <10 copies of viral genome per reaction. | - | [129] |
| African swine fever virus (ASFV) | EDTA blood and serum samples from pig | Real-time PCR/UPL PCR | 4–8 DNA copies | 10-fold high for the different ASFV isolates tested, representing p72 genotypes I (the one mostly distributed in Sardinia and West Africa), VIII (the most divergent p72 genotype) and IX (representative of East Africa), in comparison with the OIE reference TaqMan PCR | [130] |
| MERS CoV | Environmental samples (air and surface swab) | RT-PCR | - | - | [131] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vashisht, V.; Vashisht, A.; Mondal, A.K.; Farmaha, J.; Alptekin, A.; Singh, H.; Ahluwalia, P.; Srinivas, A.; Kolhe, R. Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles. BioMedInformatics 2023, 3, 1145-1177. https://doi.org/10.3390/biomedinformatics3040069
Vashisht V, Vashisht A, Mondal AK, Farmaha J, Alptekin A, Singh H, Ahluwalia P, Srinivas A, Kolhe R. Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles. BioMedInformatics. 2023; 3(4):1145-1177. https://doi.org/10.3390/biomedinformatics3040069
Chicago/Turabian StyleVashisht, Vishakha, Ashutosh Vashisht, Ashis K. Mondal, Jaspreet Farmaha, Ahmet Alptekin, Harmanpreet Singh, Pankaj Ahluwalia, Anaka Srinivas, and Ravindra Kolhe. 2023. "Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles" BioMedInformatics 3, no. 4: 1145-1177. https://doi.org/10.3390/biomedinformatics3040069
APA StyleVashisht, V., Vashisht, A., Mondal, A. K., Farmaha, J., Alptekin, A., Singh, H., Ahluwalia, P., Srinivas, A., & Kolhe, R. (2023). Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles. BioMedInformatics, 3(4), 1145-1177. https://doi.org/10.3390/biomedinformatics3040069