
Transforming Drug Design: Innovations in Computer-Aided Discovery for Biosimilar Agents



Abstract
:1. Introduction
2. Biosimilar Agents
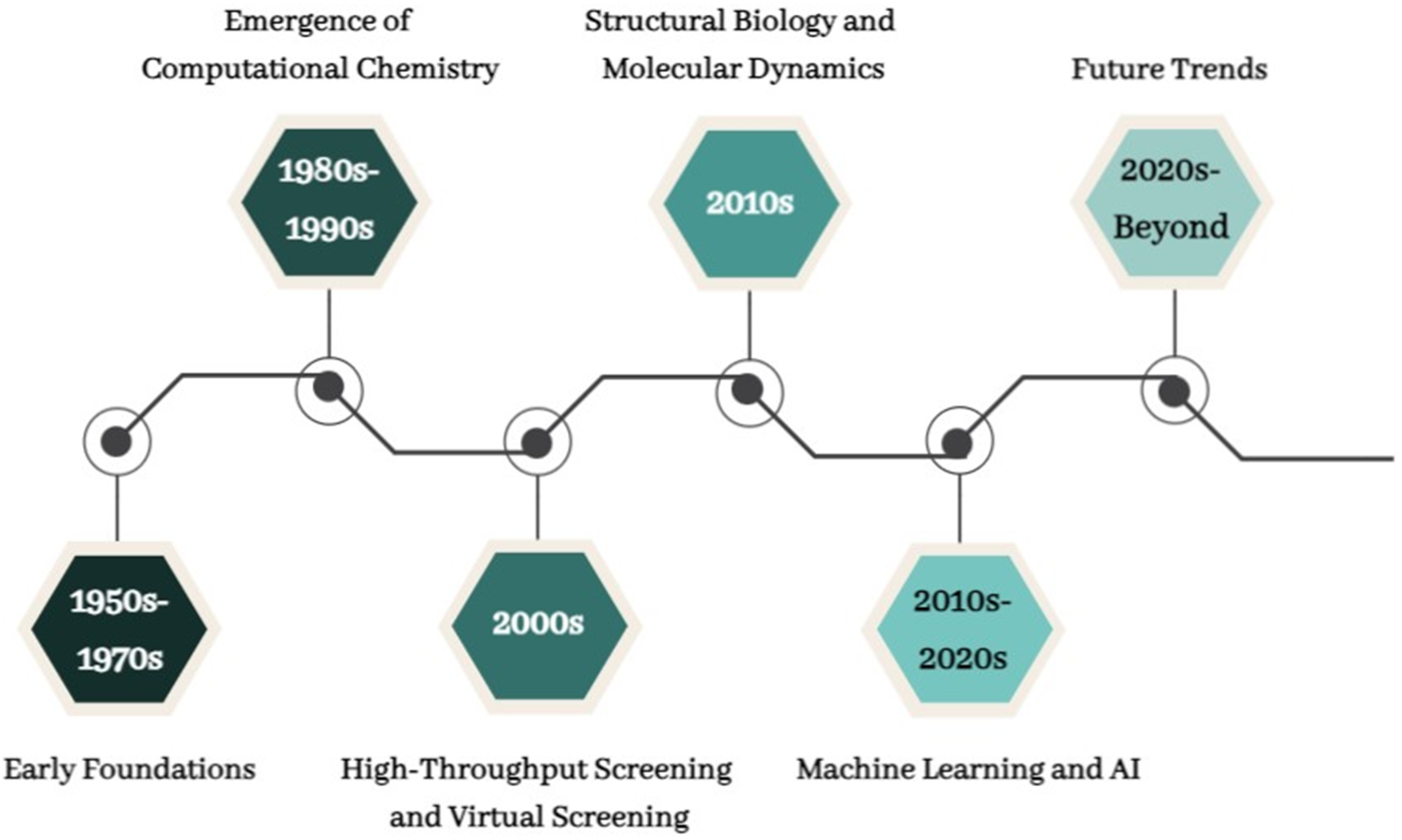
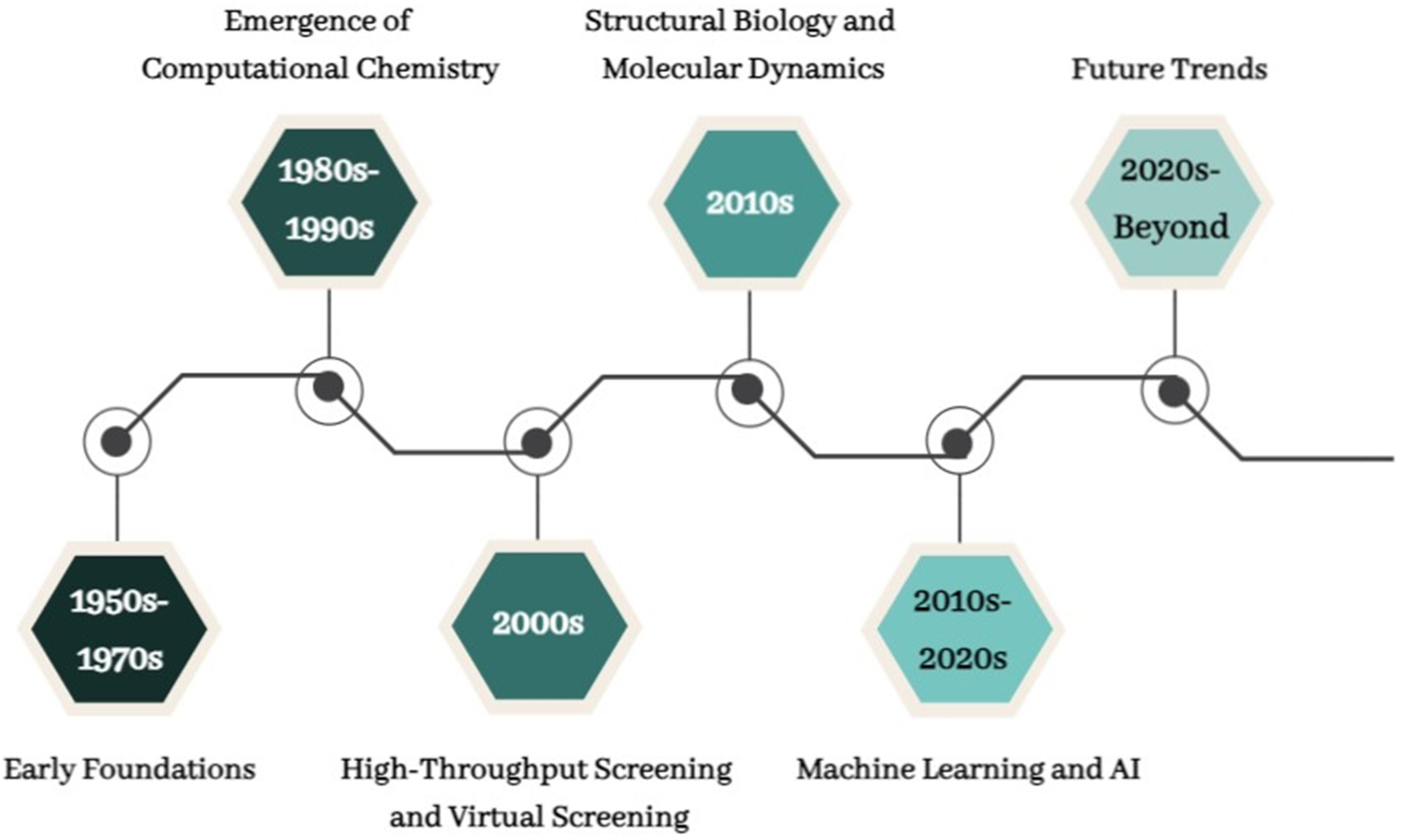
3. Evolution of CADD
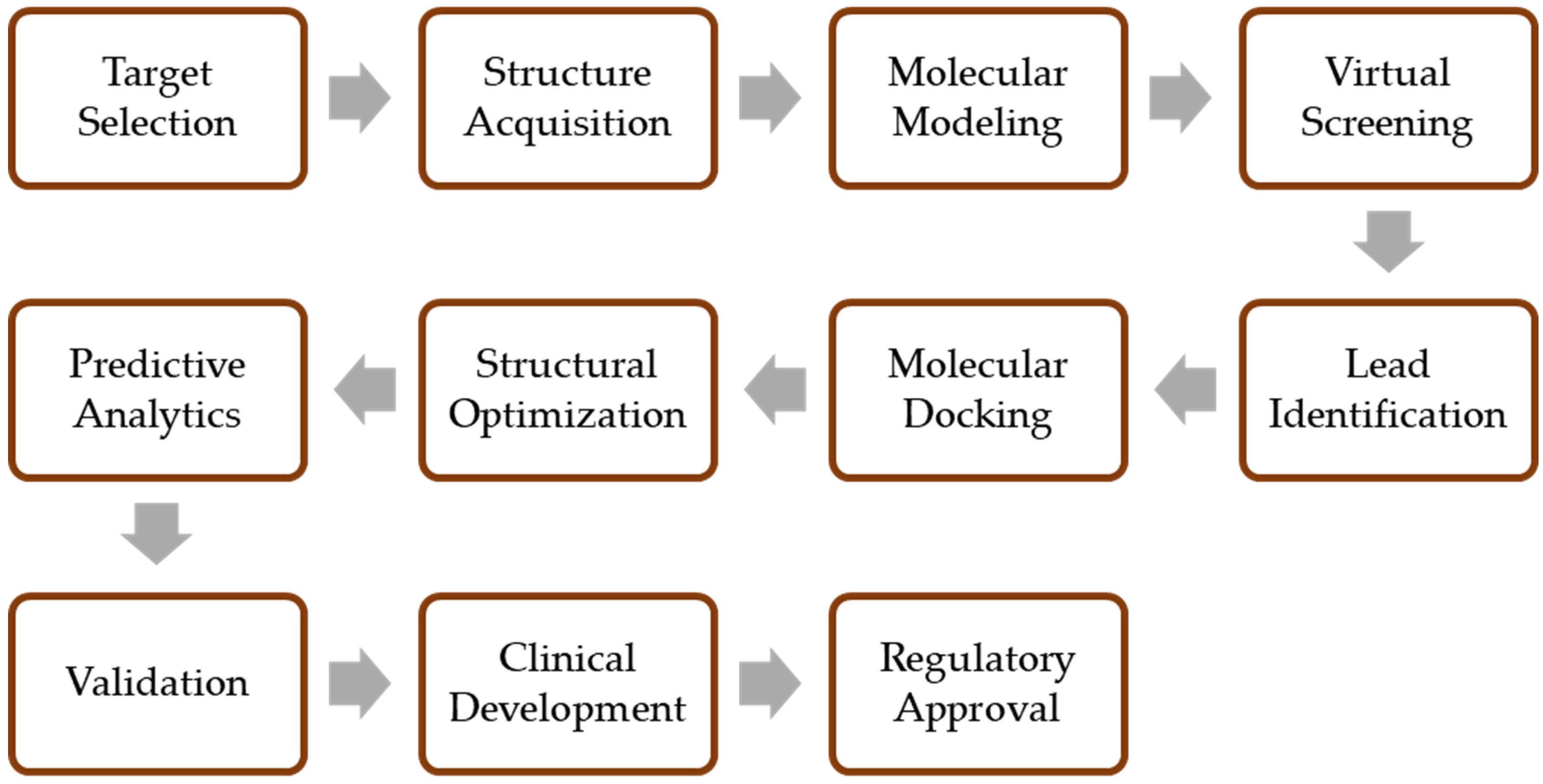
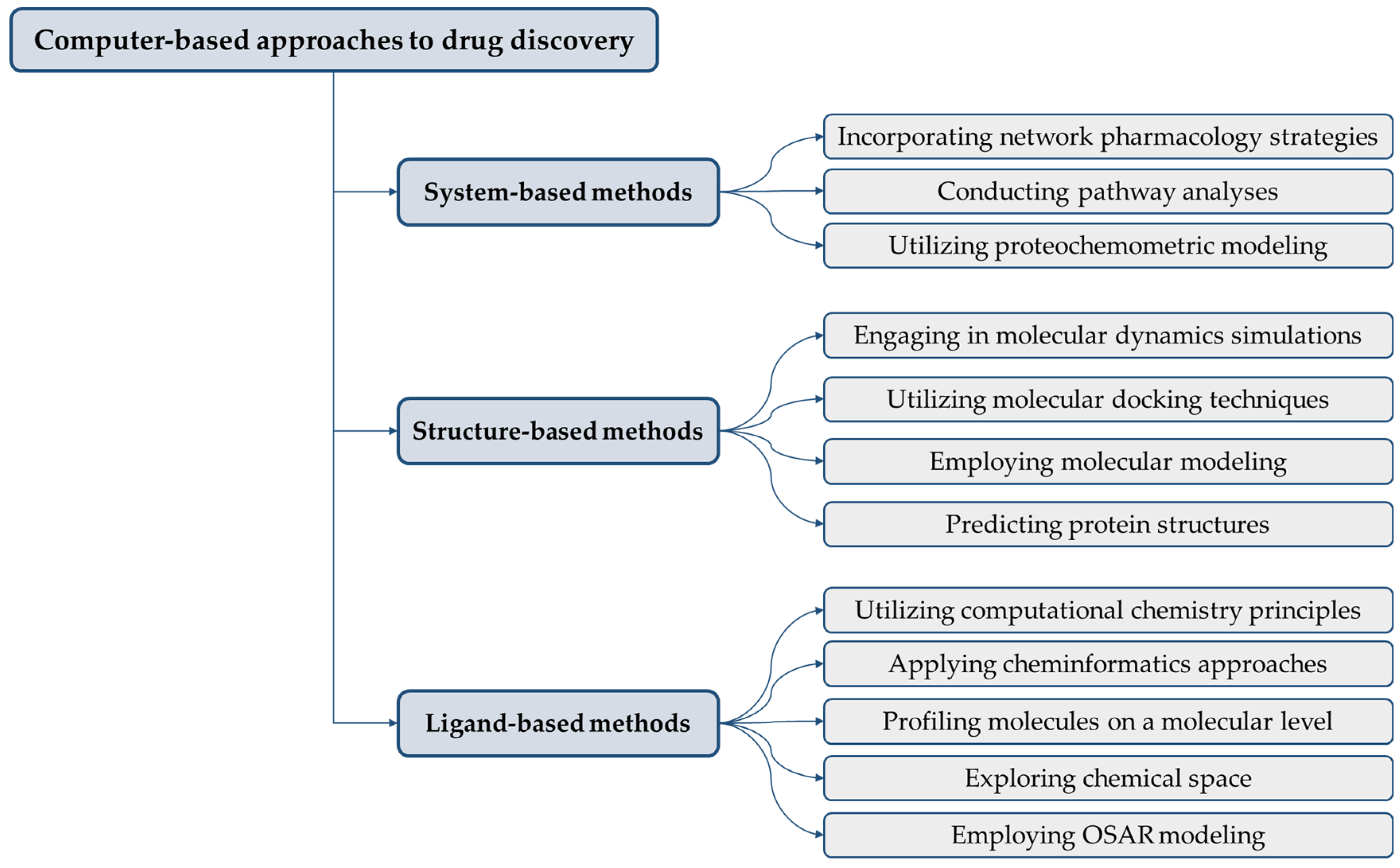
4. Applications of Computer-Aided Discovery in Biosimilar Development
4.1. Molecular Modeling and Simulation
4.1.1. Homology Modeling for Predicting Biosimilar Structure
4.1.2. Molecular Dynamics Simulations to Analyze Stability and Interactions
4.2. Virtual Screening
4.2.1. High-Throughput Virtual Screening to Identify Potential Biosimilar Candidates
4.2.2. Ligand-Based and Structure-Based Approaches for Target Identification
4.3. QSAR Modeling
4.4. Data Mining and Bioinformatics
5. Challenges and Future Directions
5.1. Validation and Accuracy of CADD Predictions
5.2. Integration of AI and Machine Learning in Biosimilar Discovery
5.3. Regulatory Considerations for CADD-Generated Biosimilars
5.4. Ethics of Using AI in Drug Creation
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- US Food and Drug Administration. Drugs@FDA Glossary of Terms; US Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Young, D.C. Computational Drug Design: A Guide for Computational and Medicinal Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Luu, K.T.; Kraynov, E.; Kuang, B.; Vicini, P.; Zhong, W.-Z. Modeling, simulation, and translation framework for the preclinical development of monoclonal antibodies. AAPS J. 2013, 15, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Gustafson, C.J.; Sandoval, L.F.; Davis, S.A.; Feldman, S.R. Cost effectiveness of biologic therapies for plaque psoriasis. Am. J. Clin. Dermatol. 2013, 14, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.C.; Gupta, R.; Brown, G.; Malakouti, M.; Koo, J. Biologic fatigue in psoriasis. J. Dermatol. Treat. 2014, 25, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Gniadecki, R.; Bang, B.; Bryld, L.; Iversen, L.; Lasthein, S.; Skov, L. Comparison of long-term drug survival and safety of biologic agents in patients with psoriasis vulgaris. Br. J. Dermatol. 2015, 172, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, M.; Covic, A. The protein science of biosimilars. Nephrol. Dial. Transplant. 2006, 21 (Suppl. S5), v4–v8. [Google Scholar] [CrossRef]
- Azevedo, V.; Hassett, B.; Fonseca, J.E.; Atsumi, T.; Coindreau, J.; Jacobs, I.; Mahgoub, E.; O’brien, J.; Singh, E.; Vicik, S.; et al. Differentiating biosimilarity and comparability in biotherapeutics. Clin. Rheumatol. 2016, 35, 2877–2886. [Google Scholar] [CrossRef]
- Ahmed, I.; Kaspar, B.; Sharma, U. Biosimilars: Impact of biologic product life cycle and European experience on the regulatory trajectory in the United States. Clin. Ther. 2012, 34, 400–419. [Google Scholar] [CrossRef]
- Blauvelt, A.; Cohen, A.; Puig, L.; Vender, R.; van der Walt, J.; Wu, J. Biosimilars for psoriasis: Preclinical analytical assessment to determine similarity. Br. J. Dermatol. 2016, 174, 282–286. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Drugs: Information for Healthcare Professionals (Biosimilars). 2015. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/information-healthcare-professionals-technetium-99m-tc-fanolesomab-marketed-neutrospec-122005 (accessed on 10 October 2023).
- Olech, E. Biosimilars: Rationale and current regulatory landscape. In Seminars in Arthritis and Rheumatism; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Weise, M.; Bielsky, M.C.; De Smet, K.; Ehmann, F.; Ekman, N.; Giezen, T.J.; Gravanis, I.; Heim, H.-K.; Heinonen, E.; Ho, K.; et al. Biosimilars: What clinicians should know. Blood J. Am. Soc. Hematol. 2012, 120, 5111–5117. [Google Scholar] [CrossRef]
- McCamish, M.; Woollett, G. The state of the art in the development of biosimilars. Clin. Pharmacol. Ther. 2012, 91, 405–417. [Google Scholar] [CrossRef]
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef]
- Gamez-Belmonte, R.; Hernández-Chirlaque, C.; Arredondo-Amador, M.; Aranda, C.J.; González, R.; Martínez-Augustin, O.; de Medina, F.S. Biosimilars: Concepts and controversies. Pharmacol. Res. 2018, 133, 251–264. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, present, and future of rituximab—The world’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Liu, Y.; Li, R.; Liang, F.; Deng, C.; Seidi, F.; Xiao, H. Fluorescent paper-based analytical devices for ultra-sensitive dual-type RNA detections and accurate gastric cancer screening. Biosens. Bioelectron. 2022, 197, 113781. [Google Scholar] [CrossRef]
- Fotis, C.; Antoranz, A.; Hatziavramidis, D.; Sakellaropoulos, T.; Alexopoulos, L.G. Network-based technologies for early drug discovery. Drug Discov. Today 2018, 23, 626–635. [Google Scholar] [CrossRef]
- Salazar, D.E.; Gormley, G. Modern drug discovery and development. In Clinical and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; pp. 719–743. [Google Scholar]
- Surabhi, S.; Singh, B. Computer aided drug design: An overview. J. Drug Deliv. Ther. 2018, 8, 504–509. [Google Scholar] [CrossRef]
- Safaei, M.; Sodano, H.A.; Anton, S.R. A review of energy harvesting using piezoelectric materials: State-of-the-art a decade later (2008–2018). Smart Mater. Struct. 2019, 28, 113001. [Google Scholar] [CrossRef]
- Massetti, M.; Jiao, F.; Ferguson, A.J.; Zhao, D.; Wijeratne, K.; Würger, A.; Blackburn, J.L.; Crispin, X.; Fabiano, S. Unconventional thermoelectric materials for energy harvesting and sensing applications. Chem. Rev. 2021, 121, 12465–12547. [Google Scholar] [CrossRef]
- Kore, P.P.; Mutha, M.M.; Antre, R.V.; Oswal, R.J.; Kshirsagar, S.S. Computer-aided drug design: An innovative tool for modeling. Open J. Med. Chem. 2012, 2, 139–148. [Google Scholar] [CrossRef]
- Veselovsky, A.; Ivanov, A. Strategy of computer-aided drug design. Curr. Drug Targets-Infect. Disord. 2003, 3, 33–40. [Google Scholar] [CrossRef]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef]
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef]
- Rader, R.A. (Re) defining biopharmaceutical. Nat. Biotechnol. 2008, 26, 743–751. [Google Scholar] [CrossRef]
- Socinski, M.A.; Curigliano, G.; Jacobs, I.; Gumbiner, B.; MacDonald, J.; Thomas, D. Clinical considerations for the development of biosimilars in oncology. In MAbs; Taylor & Francis: Abingdon, UK, 2015. [Google Scholar]
- Cherny, N.; Sullivan, R.; Torode, J.; Saar, M.; Eniu, A. ESMO European Consortium Study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann. Oncol. 2016, 27, 1423–1443. [Google Scholar] [CrossRef]
- Monk, B.J.; Lammers, P.E.; Cartwright, T.; Jacobs, I. Barriers to the access of bevacizumab in patients with solid tumors and the potential impact of biosimilars: A physician survey. Pharmaceuticals 2017, 10, 19. [Google Scholar] [CrossRef]
- Baer, W.H.; Maini, A.; Jacobs, I. Barriers to the access and use of rituximab in patients with non-Hodgkin’s lymphoma and chronic lymphocytic leukemia: A physician survey. Pharmaceuticals 2014, 7, 530–544. [Google Scholar] [CrossRef]
- Kaida-Yip, F.; Deshpande, K.; Saran, T.; Vyas, D. Biosimilars: Review of current applications, obstacles, and their future in medicine. World J. Clin. Cases 2018, 6, 161. [Google Scholar] [CrossRef]
- Lyman, G.H. Emerging opportunities and challenges of biosimilars in oncology practice. J. Oncol. Pract. 2017, 13 (Suppl. S9), 7s–9s. [Google Scholar] [CrossRef]
- Dutta, B.; Huys, I.; Vulto, A.G.; Simoens, S. Identifying key benefits in European off-patent biologics and biosimilar markets: It is not only about price! BioDrugs 2020, 34, 159–170. [Google Scholar] [CrossRef]
- Lucio, S.D.; Stevenson, J.G.; Hoffman, J.M. Biosimilars: Implications for health-system pharmacists. Am. J. Health-Syst. Pharm. 2013, 70, 2004–2017. [Google Scholar] [CrossRef]
- Zelenetz, A.D. Biosimilars in oncology. Oncol. Hematol. Rev. 2016, 12, 22–28. [Google Scholar] [CrossRef]
- Chopra, R.; Lopes, G. Improving access to cancer treatments: The role of biosimilars. J. Glob. Oncol. 2017, 3, 596–610. [Google Scholar] [CrossRef]
- Farhat, F.; Torres, A.; Park, W.; Lopes, G.d.L.; Mudad, R.; Ikpeazu, C.; Aad, S.A. The concept of biosimilars: From characterization to evolution—A narrative review. Oncologist 2018, 23, 346–352. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Lönnfors, S.; Avedano, L.; Danese, S. Changes in inflammatory bowel disease patients’ perspectives on biosimilars: A follow-up survey. United Eur. Gastroenterol. J. 2019, 7, 1345–1352. [Google Scholar] [CrossRef]
- Cohen, H.; Beydoun, D.; Chien, D.; Lessor, T.; McCabe, D.; Muenzberg, M.; Popovian, R.; Uy, J. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv. Ther. 2016, 33, 2160–2172. [Google Scholar] [CrossRef]
- Christl, L.; Lim, S. Biosimilar and Interchangeable Products in the United States: Scientific Concepts, Clinical Use, and Practical Considerations. 2020. Available online: https://www.fda.gov/media/122832/download (accessed on 10 October 2023).
- Cohen, H.P.; Lamanna, W.C.; Schiestl, M. Totality of evidence and the role of clinical studies in establishing biosimilarity. In Biosimilars: Regulatory, Clinical, and Biopharmaceutical Development; Springer: Berlin/Heidelberg, Germany, 2018; pp. 601–628. [Google Scholar]
- Krendyukov, A.; Schiestl, M. Extrapolation concept at work with biosimilar: A decade of experience in oncology. ESMO Open 2018, 3, E000319. [Google Scholar] [CrossRef]
- Cohen, H.P.; McCabe, D. The importance of countering biosimilar disparagement and misinformation. BioDrugs 2020, 34, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Considerations in Demonstrating Interchangeability with a Reference Product: Guidance for Industry. Available online: https://www.fda.gov/media/124907/download (accessed on 10 October 2023).
- EFPIA MID3 Workgroup; Marshall, S.; Cosson, V.; Cheung, S.Y.A.; Chenel, M.; DellaPasqua, O.; Frey, N.; Hamrén, B.; Harnisch, L.; Ivanow, F.; et al. Good practices in model-informed drug discovery and development: Practice, application, and documentation. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar]
- Mullard, A. Biotech R&D spend jumps by more than 15%. Nat. Rev. Drug Discov. 2016, 15, 447–448. [Google Scholar] [PubMed]
- Ghosh, B.; Choudhuri, S. Drug Design for Malaria with Artificial Intelligence (AI). In Plasmodium Species and Drug Resistance; IntechOpen: London, UK, 2021. [Google Scholar]
- Am Ende, D.J.; am Ende, M.T. Chemical engineering in the pharmaceutical industry: An introduction. In Chemical Engineering in the Pharmaceutical Industry: Drug Product Design, Development, and Modeling; Wiley: Hoboken, NJ, USA, 2019; pp. 1–17. [Google Scholar]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-aided drug design in epigenetics. Front. Chem. 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Xiang, M.; Cao, Y.; Fan, W.; Chen, L.; Mo, Y. Computer-aided drug design: Lead discovery and optimization. Comb. Chem. High Throughput Screen. 2012, 15, 328–337. [Google Scholar] [CrossRef]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D. Computer-aided drug design methods. In Antibiotics: Methods and protocols; Springer: Berlin/Heidelberg, Germany, 2017; pp. 85–106. [Google Scholar]
- Wang, A.; Durrant, J.D. Open-Source Browser-Based Tools for Structure-Based Computer-Aided Drug Discovery. Molecules 2022, 27, 4623. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminformatics 2015, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Model. 2012, 52, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Blomme, E.A.; Will, Y. Toxicology strategies for drug discovery: Present and future. Chem. Res. Toxicol. 2016, 29, 473–504. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Bajorath, J.; Medina-Franco, J.L. Informatics for chemistry, biology, and biomedical sciences. J. Chem. Inf. Model. 2020, 61, 26–35. [Google Scholar] [CrossRef]
- Gasteiger, J. Chemistry in times of artificial intelligence. ChemPhysChem 2020, 21, 2233–2242. [Google Scholar] [CrossRef]
- Bajorath, J. State-of-the-art of artificial intelligence in medicinal chemistry. Future Sci. 2021, 7, FSO702. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Deo, R.C. Machine learning in medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Martinez-Mayorga, K.; Gortari, E.F.-D.; Kirchmair, J.; Bajorath, J. Rationality over fashion and hype in drug design. F1000Research 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R. The trouble with QSAR (or how I learned to stop worrying and embrace fallacy). J. Chem. Inf. Model. 2008, 48, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Bharath, E.; Manjula, S.; Vijaychand, A. In silico drug design tool for overcoming the innovation deficit in the drug discovery process. Int. J. Pharm. Pharm. Sci. 2011, 3, 8–12. [Google Scholar]
- Chu, X.; Wang, Y.; Tian, P.; Li, W.; Mercadante, D. Advanced Sampling and Modeling in Molecular Simulations for Slow and Large-Scale Biomolecular Dynamics. Front. Mol. Biosci. 2021, 8, 795991. [Google Scholar] [CrossRef]
- Badar, M.S.; Shamsi, S.; Ahmed, J.; Alam, M.A. Molecular dynamics simulations: Concept, methods, and applications. In Transdisciplinarity; Springer: Berlin/Heidelberg, Germany, 2022; pp. 131–151. [Google Scholar]
- Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide; Springer: Berlin/Heidelberg, Germany, 2010; Volume 2. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci. 2019, 31, 595–601. [Google Scholar] [CrossRef]
- Nunes, R.R.; da Fonseca, A.L.; Pinto, A.C.d.S.; Maia, E.H.B.; da Silva, A.M.; Varotti, F.d.P.; Taranto, A.G. Brazilian malaria molecular targets (BraMMT): Selected receptors for virtual high-throughput screening experiments. Memórias Do Inst. Oswaldo Cruz 2019, 114. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Yang, J.; Yu, S.; Yang, Z.; Yan, Y.; Chen, Y.; Zeng, H.; Ma, F.; Shi, Y.; Shi, Y.; Zhang, Z.; et al. Efficacy and safety of anti-cancer biosimilars compared to reference biologics in oncology: A systematic review and meta-analysis of randomized controlled trials. BioDrugs 2019, 33, 357–371. [Google Scholar] [CrossRef]
- Bloomfield, D.; D’Andrea, E.; Nagar, S.; Kesselheim, A. Characteristics of clinical trials evaluating biosimilars in the treatment of cancer: A systematic review and meta-analysis. JAMA Oncol. 2022, 8, 537–545. [Google Scholar] [CrossRef]
- Gbeddy, G.; Egodawatta, P.; Goonetilleke, A.; Ayoko, G.; Chen, L. Application of quantitative structure-activity relationship (QSAR) model in comprehensive human health risk assessment of PAHs, and alkyl-, nitro-, carbonyl-, and hydroxyl-PAHs laden in urban road dust. J. Hazard. Mater. 2020, 383, 121154. [Google Scholar] [CrossRef] [PubMed]
- Ray, R. Understanding the Structural Importance of the Non-Binding and Binding Parts of Bedaquiline and Its Analogues with ATP Synthase Subunit C Using Molecular Docking, Molecular Dynamics Simulation and 3D-QSAR Techniques. In Proceedings of the International Conference on Drug Discovery (ICDD), Hyderabad, India, 29 February–2 March 2020. [Google Scholar]
- Padole, S.S.; Asnani, A.J.; Chaple, D.R.; Katre, S.G. A review of approaches in computer-aided drug design in drug discovery. GSC Biol. Pharm. Sci. 2022, 19, 075–083. [Google Scholar] [CrossRef]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinform. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Wang, X.S.; Zhu, H.; Tropsha, A. Predictive QSAR modeling: Methods and applications in drug discovery and chemical risk assessment. Handb. Comput. Chem. 2012, 1309–1342. [Google Scholar]
- Kausar, S.; Falcao, A.O. An automated framework for QSAR model building. J. Cheminformatics 2018, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, N. Role of data mining techniques in bioinformatics. Int. J. Appl. Res. Bioinform. 2021, 11, 51–60. [Google Scholar] [CrossRef]
- Raza, K. Application of data mining in bioinformatics. arXiv 2012, arXiv:1205.1125. [Google Scholar]
- Avila-Portillo, L.M.; Aristizabal, F.; Riveros, A.; Abba, M.C.; Correa, D. Modulation of adipose-derived mesenchymal stem/stromal cell transcriptome by G-CSF stimulation. Stem Cells Int. 2020, 2020, 5045124. [Google Scholar] [CrossRef]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Bhunia, S.S.; Saxena, M.; Saxena, A.K. Ligand-and structure-based virtual screening in drug discovery. In Biophysical and Computational Tools in Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2021; pp. 281–339. [Google Scholar]
- Florian, J.; Sun, Q.; Schrieber, S.J.; White, R.; Shubow, S.; Johnson-Williams, B.E.; Sheikhy, M.; Harrison, N.R.; Parker, V.J.; Wang, Y.; et al. Pharmacodynamic biomarkers for biosimilar development and approval: A workshop summary. Clin. Pharmacol. Ther. 2023, 113, 1030–1035. [Google Scholar] [CrossRef]
- Han, Y.; Klinger, K.; Rajpal, D.K.; Zhu, C.; Teeple, E. Empowering the discovery of novel target-disease associations via machine learning approaches in the open targets platform. BMC Bioinform. 2022, 23, 1–19. [Google Scholar] [CrossRef]
- Hosseini, M.; Hammami, B.; Kazemi, M. Identification of potential diagnostic biomarkers and therapeutic targets for endometriosis based on bioinformatics and machine learning analysis. J. Assist. Reprod. Genet. 2023, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Adinew, G.M.; Messeha, S.; Taka, E.; Ahmed, S.A.; Soliman, K.F. The Role of Apoptotic Genes and Protein-Protein Interactions in Triple-negative Breast Cancer. Cancer Genom. Proteom. 2023, 20, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Chujan, S.; Kitkumthorn, N.; Satayavivad, J. Identification of potential molecular mechanisms and prognostic markers for oral squamous cell carcinoma: A bioinformatics analysis. J. Int. Soc. Prev. Community Dent. 2023, 13, 237. [Google Scholar] [CrossRef]
- Namba, S.; Iwata, M.; Yamanishi, Y. From drug repositioning to target repositioning: Prediction of therapeutic targets using genetically perturbed transcriptomic signatures. Bioinformatics 2022, 38 (Suppl. S1), i68–i76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, F.; Yang, N.; Zhan, X.; Liao, J.; Mai, S.; Huang, Z. In silico methods for identification of potential therapeutic targets. Interdiscip. Sci. Comput. Life Sci. 2022, 14, 285–310. [Google Scholar] [CrossRef]
- Taherdoost, H.; Madanchian, M. Artificial intelligence and sentiment analysis: A review in competitive research. Computers 2023, 12, 37. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, F.; Hu, G.; Shao, S.; He, H.; Zhang, W.; Zhang, X.; Li, L. Dynamic contribution of microbial residues to soil organic matter accumulation influenced by maize straw mulching. Geoderma 2019, 333, 35–42. [Google Scholar] [CrossRef]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminformatics 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Raina, R.; Madhavan, A.; Ng, A.Y. Large-Scale Deep Unsupervised Learning Using Graphics Processors. In Proceedings of the 26th Annual International Conference on Machine Learning, Montreal, QC, Canada, 14–18 June 2009. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Smalley, E. AI-powered drug discovery captures pharma interest. Nat. Biotechnol. 2017, 35, 604–606. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, J.; Jun, J.J.; Xie, X.-Q. Deep convolutional generative adversarial network (dcGAN) models for screening and design of small molecules targeting cannabinoid receptors. Mol. Pharm. 2019, 16, 4451–4460. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative molecular design in low data regimes. Nat. Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Rahalkar, H.; Sheppard, A.; Santos, G.M.L.; Dasgupta, C.; Perez-Tapia, S.M.; Lopez-Morales, C.A.; Salek, S. Current regulatory requirements for biosimilars in six member countries of BRICS-TM: Challenges and opportunities. Front. Med. 2021, 8, 726660. [Google Scholar] [CrossRef]
- Gundersen, T.; Bærøe, K. The future ethics of artificial intelligence in medicine: Making sense of collaborative models. Sci. Eng. Ethics 2022, 28, 17. [Google Scholar] [CrossRef]
- Nupur, N.; Joshi, S.; Gulliarme, D.; Rathore, A.S. Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Front. Bioeng. Biotechnol. 2022, 10, 832059. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aspect | Biosimilars | Small Molecule Pharmaceuticals |
|---|---|---|
| Development Process | Complex, requires demonstration of similarity in safety, purity, and potency | Relatively simpler, focus on chemical synthesis |
| Regulatory Approval | Based on “totality of evidence” approach, relies on prior FDA findings | Strict approval requirements, separate Phase III studies for each indication |
| Cost | Typically 30% cheaper than reference drugs | Cost varies based on manufacturing and development |
| Advantages | More affordable, potential for business rivalry | Well-established development process, easily understood by doctors and patients |
| Disadvantages | Potential for immunogenicity, lack of awareness and acceptance | Limited structural understanding, potential for side effects |
| Challenges | Indication extrapolation, interchangeability confusion | Potential for misunderstanding due to the complex nature |
| Communication | Clear communication essential for interchangeability and clinical switching investigations | Straightforward communication due to a well-understood development process |
| Use of CADD | Necessary to simplify the complex development process | Less necessary due to the simpler development process |
| Aspect | Traditional Drug Development | CADD |
|---|---|---|
| Steps | Discovery, Target Identification, Lead Compound Identification, Preclinical Testing, Clinical Trials (Phase I, II, III), Regulatory Approval, Post-Marketing Surveillance | Bioinformatics, Molecular Modeling, Virtual Screening, In Silico Testing, Predictive Modeling, Data Analysis |
| Time Investment | Each step is time-consuming, often taking years | Significantly reduced time per step |
| Cost Investment | High costs associated with extensive laboratory work, clinical trials, and regulatory processes | Relatively lower costs due to reduced experimentation and reliance on computational methods |
| Benefits of Approach | Well-established process with proven success, suitable for novel mechanisms with limited data | Faster identification of potential compounds, reduced cost due to in silico testing, streamlined data analysis and prediction |
| Considerations for Use | Relevant for complex biological systems requiring extensive testing and validation | More suitable for situations with available data and where in silico methods can provide valuable insights |
| Overall Efficiency | Slower progress due to lengthy experimental phases | Accelerated progress due to computational speed and reduced reliance on physical experiments |
| Flexibility | Limited flexibility once experiments are initiated | Greater flexibility to adjust and optimize approaches |
| Risk Management | Higher risk due to resource-intensive nature | Lower risk due to the ability to simulate and predict outcomes |
| Data Utilization | Heavy reliance on experimental data | Leveraging available data for predictions and insights |
| Regulatory Approval | Adheres to established regulatory pathways | May require adaptation of regulatory standards for computational methods |
| Applications | Use Cases | Pros | Cons |
|---|---|---|---|
| Molecular Modeling and Simulation | Predicting 3D structure and dynamic behavior of biosimilars for similarity assessment and optimization |
|
|
| Virtual Screening | Identifying potential biosimilar candidates with a high binding affinity and biological activity |
|
|
| QSAR Modeling | Predicting the activity of biosimilar candidates against specific targets for lead compound selection |
|
|
| Data Mining and Bioinformatics | Identifying biomarkers, therapeutic targets, and optimizing biosimilar candidates from large datasets |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Askari, S.; Ghofrani, A.; Taherdoost, H. Transforming Drug Design: Innovations in Computer-Aided Discovery for Biosimilar Agents. BioMedInformatics 2023, 3, 1178-1196. https://doi.org/10.3390/biomedinformatics3040070
Askari S, Ghofrani A, Taherdoost H. Transforming Drug Design: Innovations in Computer-Aided Discovery for Biosimilar Agents. BioMedInformatics. 2023; 3(4):1178-1196. https://doi.org/10.3390/biomedinformatics3040070
Chicago/Turabian StyleAskari, Shadi, Alireza Ghofrani, and Hamed Taherdoost. 2023. "Transforming Drug Design: Innovations in Computer-Aided Discovery for Biosimilar Agents" BioMedInformatics 3, no. 4: 1178-1196. https://doi.org/10.3390/biomedinformatics3040070
APA StyleAskari, S., Ghofrani, A., & Taherdoost, H. (2023). Transforming Drug Design: Innovations in Computer-Aided Discovery for Biosimilar Agents. BioMedInformatics, 3(4), 1178-1196. https://doi.org/10.3390/biomedinformatics3040070