

Integrative Meta-Analysis during Induced Pluripotent Stem Cell Reprogramming Reveals Conserved Networks and Chromatin Accessibility Signatures in Human and Mouse


Abstract
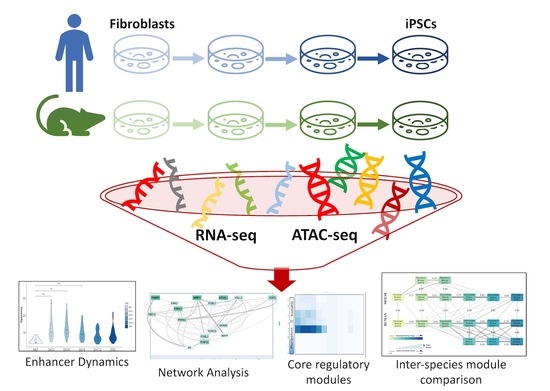
:
1. Introduction
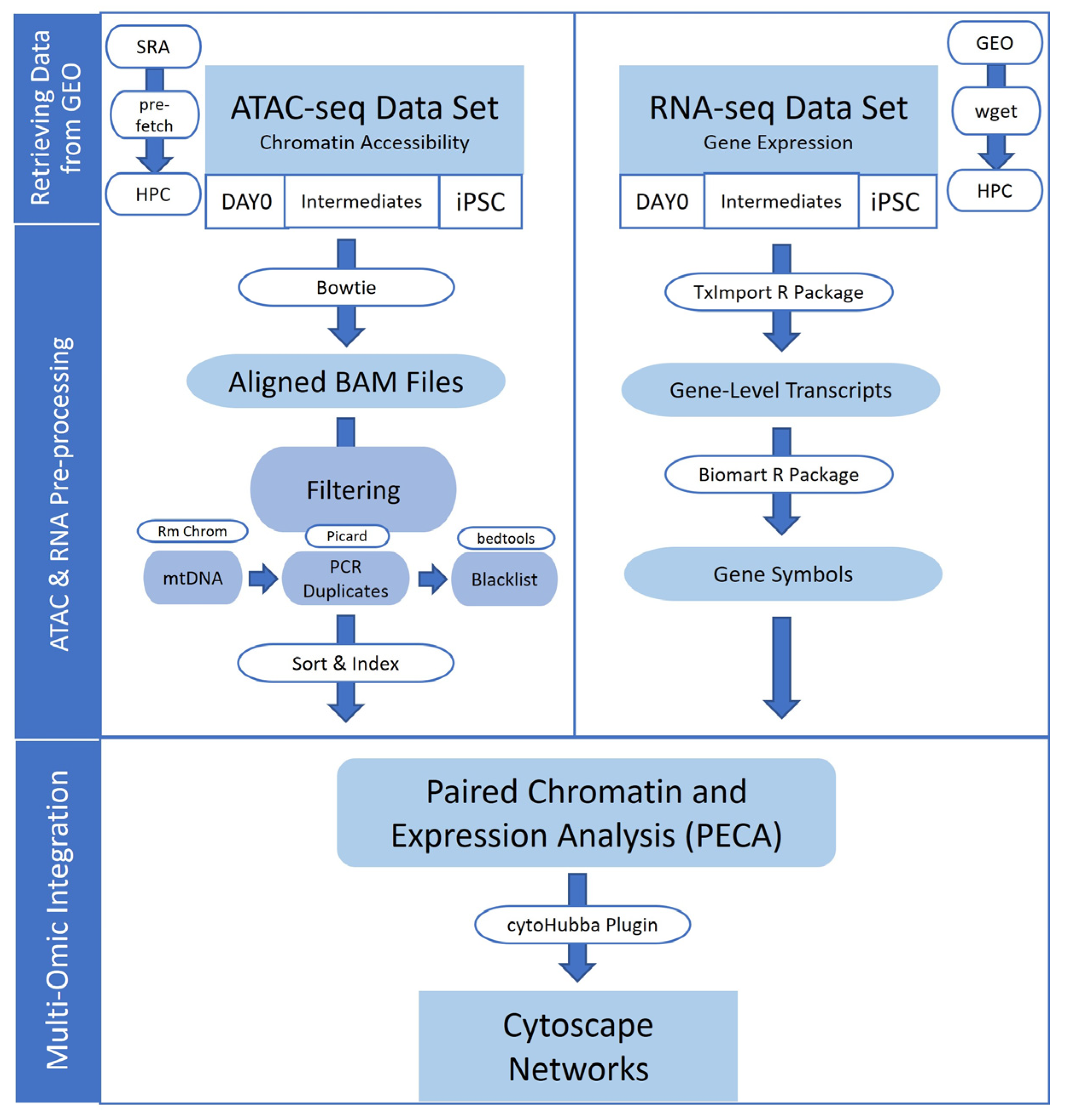
2. Materials and Methods
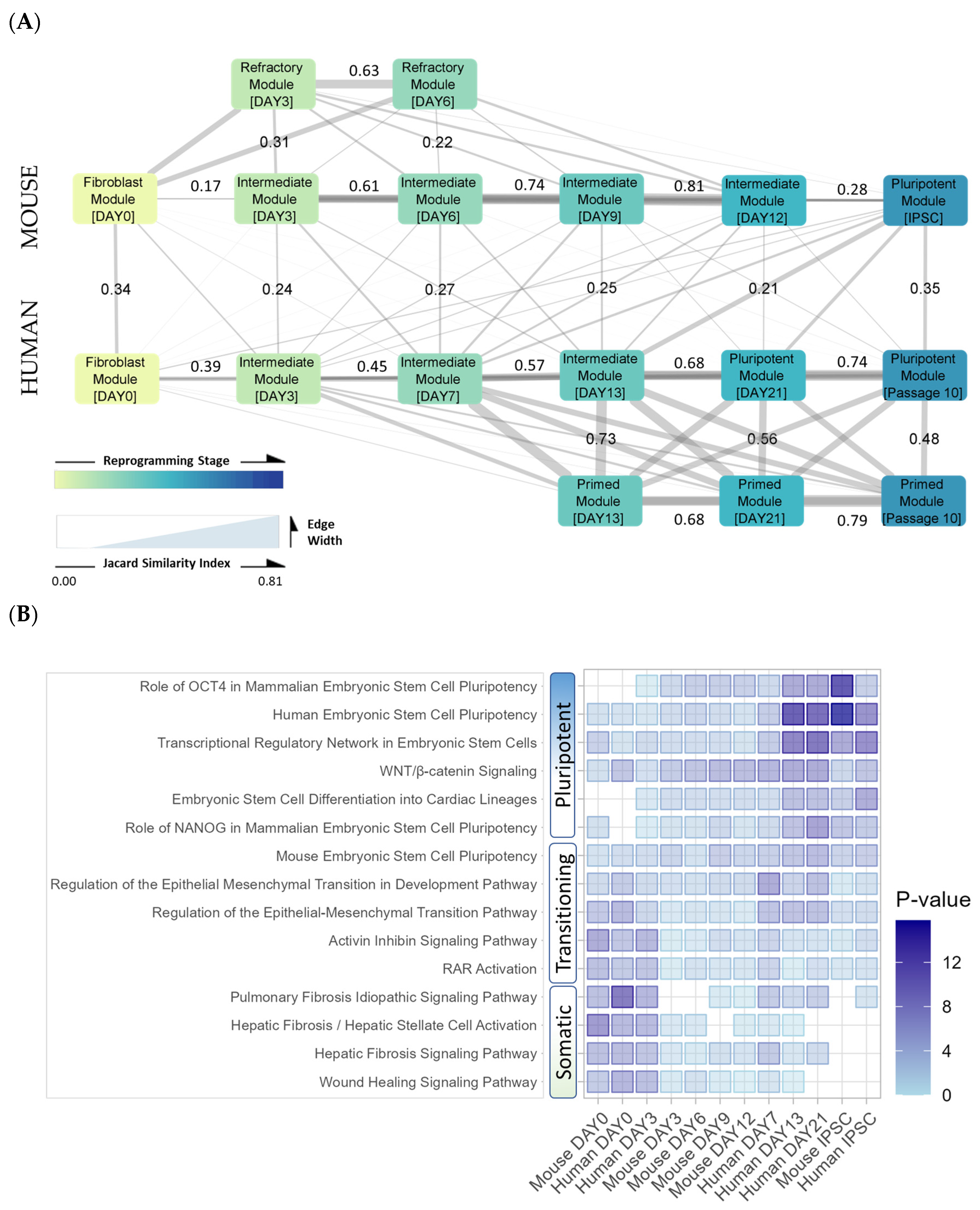
3. Results
3.1. Differential Chromatin Accessibility and Motif Enrichment Analysis
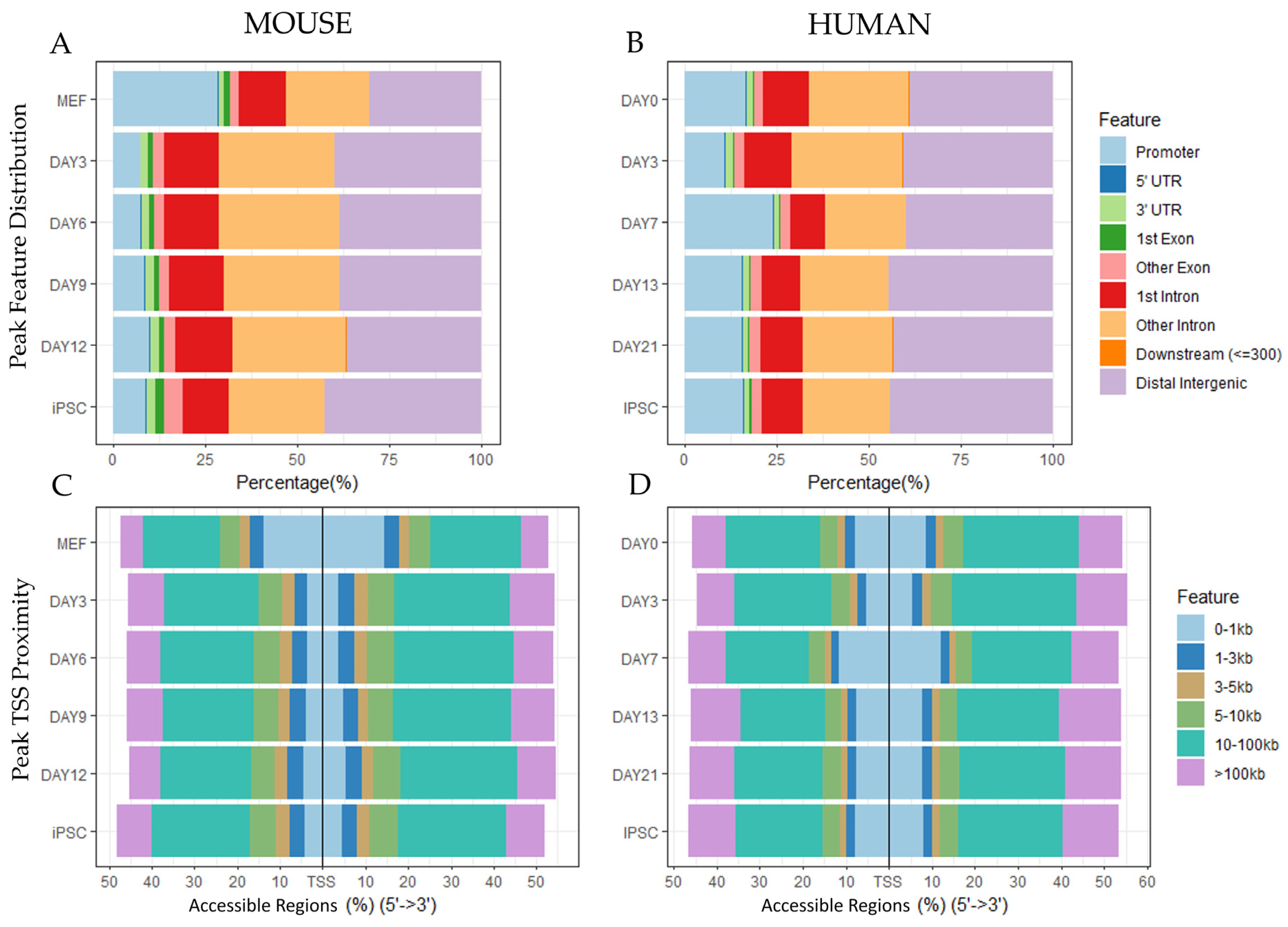
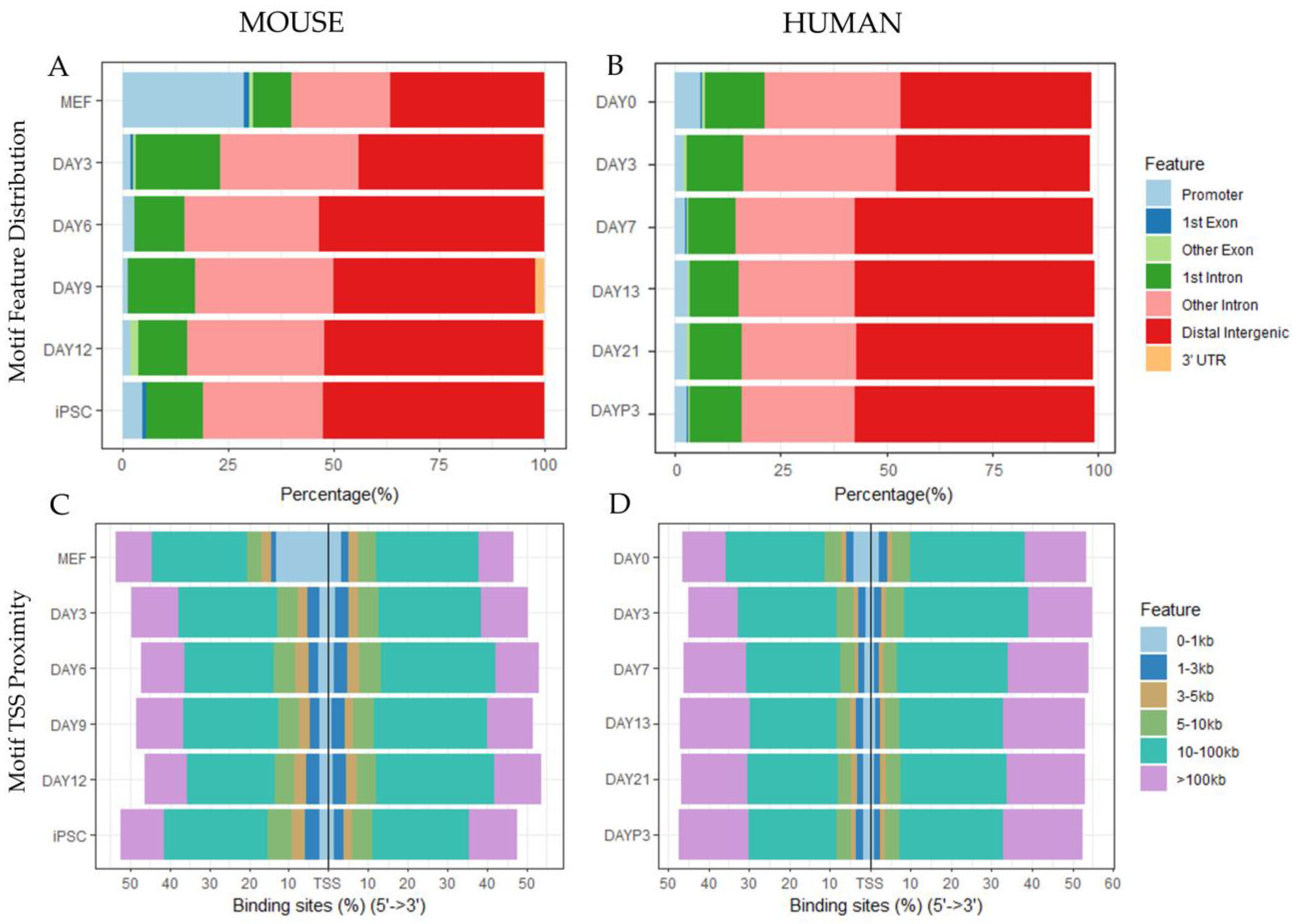
3.1.1. Peak Locations Show Diverse Changes in Chromatin Accessibility across Species and Datasets
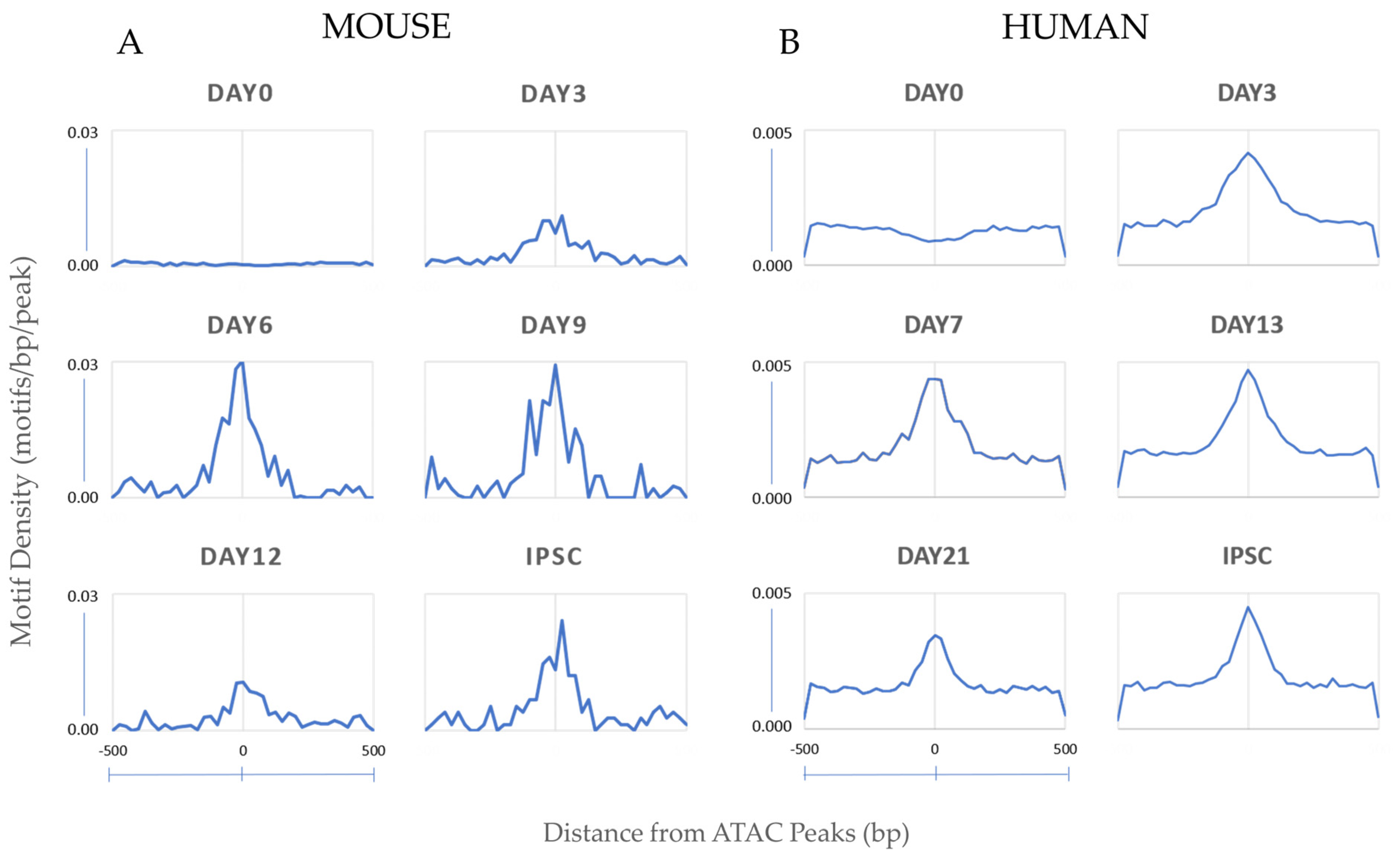
3.1.2. Motif Enrichment Analysis Reveals Common and Species-Specific Motifs across Timepoints
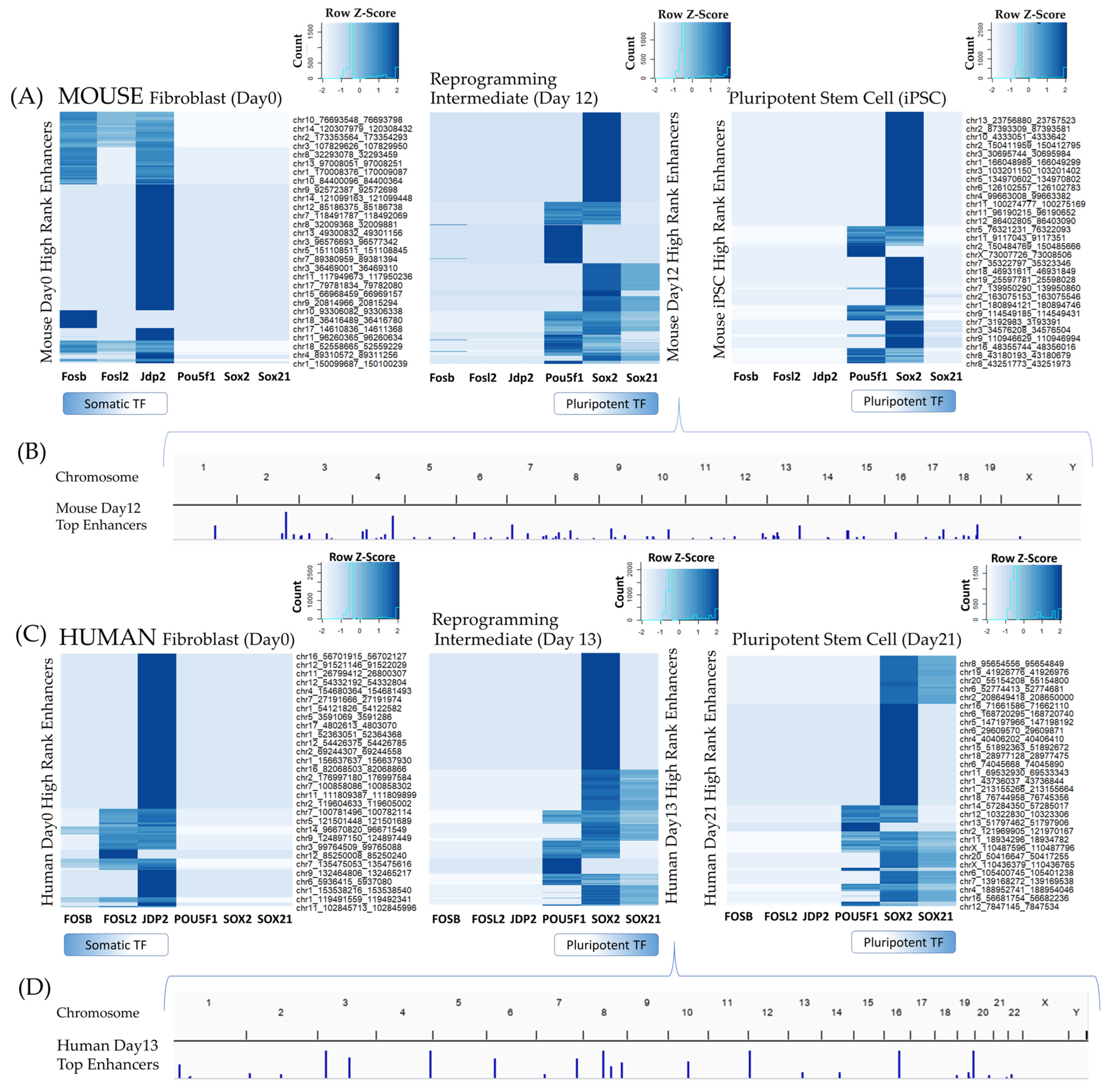
3.1.3. Oct4-Sox2-Tcf-Nanog (OSTN) Orchestrates Changes from Distal Enhancers
3.2. Cis-Regulatory Network Analysis
3.2.1. Enhancers Mediating TF–TG Interactions during Reprogramming
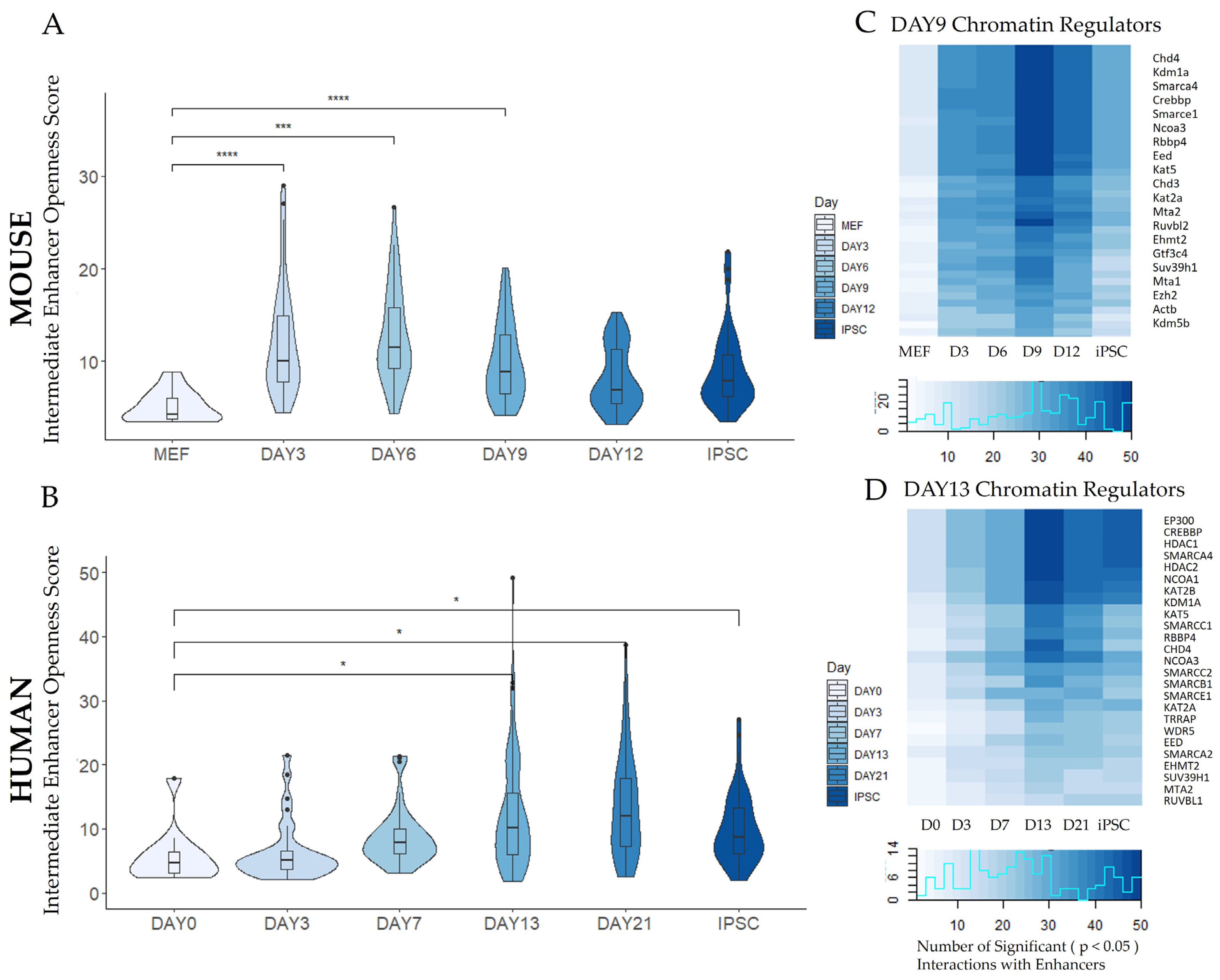
3.2.2. Enhancer Dynamics during Reprogramming
3.3. Trans-Regulatory Network Analysis during Reprogramming
3.3.1. Construction of Trans-Regulatory Networks
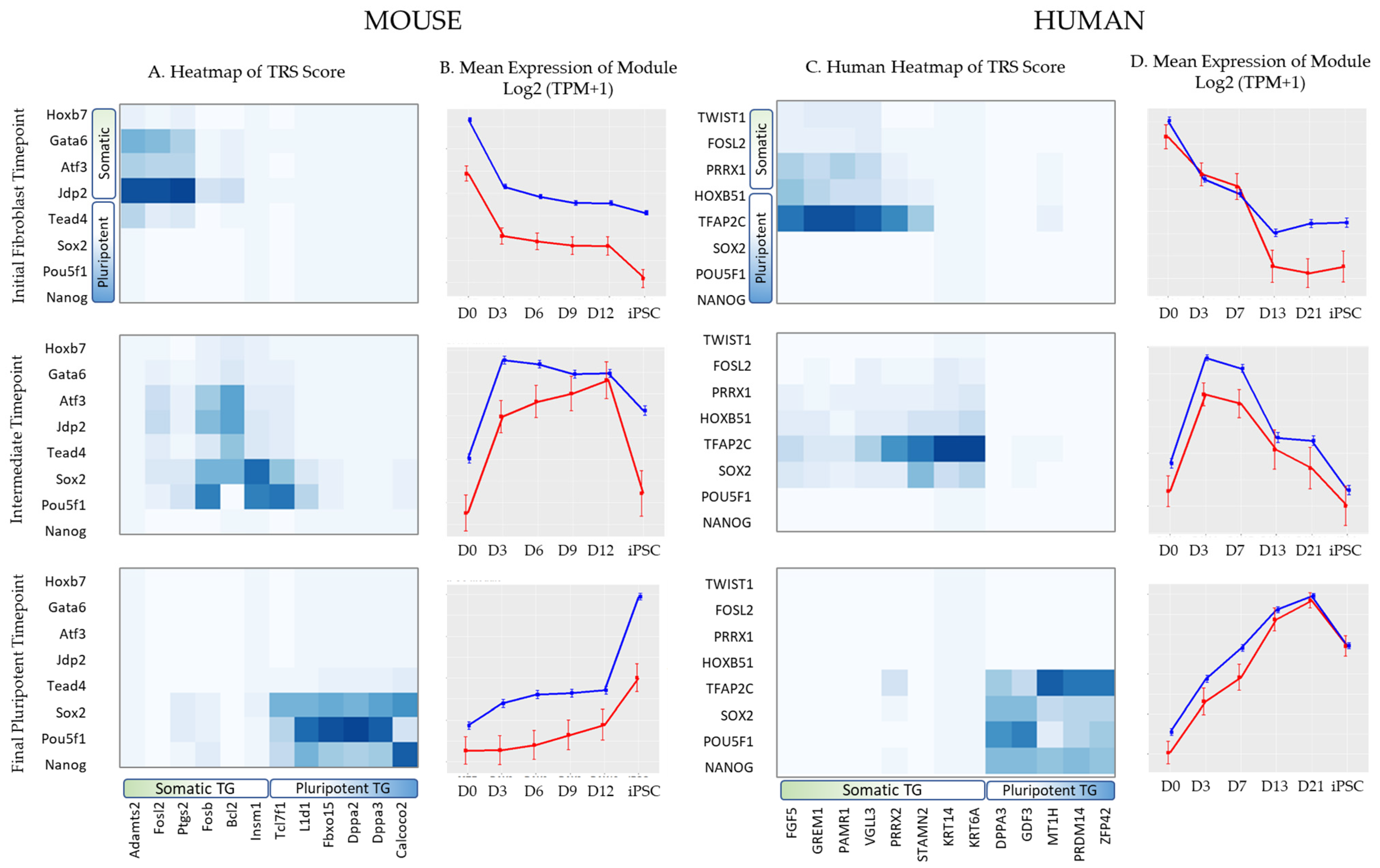
3.3.2. Paired Expression and Chromatin Accessibility Analysis Reveals Dynamic TF–TG Networks
3.3.3. Trans-Regulatory Networks Involved in Reprogramming Efficiency
3.3.4. Inter-Species Conservation of Trans-Regulatory Networks
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seah, Y.F.S.; EL Farran, C.A.; Warrier, T.; Xu, J.; Loh, Y.-H. Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges. Int. J. Mol. Sci. 2015, 16, 28614–28634. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Cheow, L.F.; Courtois, E.T.; Tan, Y.; Viswanathan, R.; Xing, Q.; Tan, R.Z.; Tan, D.S.W.; Robson, P.; Loh, Y.-H.; Quake, S.R.; et al. Single-Cell Multimodal Profiling Reveals Cellular Epigenetic Heterogeneity. Nat. Methods 2016, 13, 833–836. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Hochedlinger, K. Induced Pluripotency: History, Mechanisms, and Applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into IPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-Resolution Analysis with Novel Cell-Surface Markers Identifies Routes to IPS Cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef]
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.e20. [Google Scholar] [CrossRef]
- Knaupp, A.S.; Buckberry, S.; Pflueger, J.; Lim, S.M.; Ford, E.; Larcombe, M.R.; Rossello, F.J.; de Mendoza, A.; Alaei, S.; Firas, J.; et al. Transient and Permanent Reconfiguration of Chromatin and Transcription Factor Occupancy Drive Reprogramming. Cell Stem Cell 2017, 21, 834–845.e6. [Google Scholar] [CrossRef]
- Schiebinger, G.; Shu, J.; Tabaka, M.; Cleary, B.; Subramanian, V.; Solomon, A.; Gould, J.; Liu, S.; Lin, S.; Berube, P.; et al. Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 2019, 176, 1517. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, C.; Hou, Z.; Yang, Y.; Bi, Y.; Wang, H.; Zhang, Y.; Gao, S. Unique Molecular Events during Reprogramming of Human Somatic Cells to Induced Pluripotent Stem Cells (IPSCs) at Naïve State. eLife 2018, 7, e29518. [Google Scholar] [CrossRef]
- Theunissen, T.W.; Friedli, M.; He, Y.; Planet, E.; O’Neil, R.C.; Markoulaki, S.; Pontis, J.; Wang, H.; Iouranova, A.; Imbeault, M.; et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 2016, 19, 502–515. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining Molecular Cornerstones during Fibroblast to IPS Cell Reprogramming in Mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef]
- Xing, Q.R.; El Farran, C.A.; Gautam, P.; Chuah, Y.S.; Warrier, T.; Toh, C.-X.D.; Kang, N.-Y.; Sugii, S.; Chang, Y.-T.; Xu, J.; et al. Diversification of Reprogramming Trajectories Revealed by Parallel Single-Cell Transcriptome and Chromatin Accessibility Sequencing. Sci. Adv. 2020, 6, eaba1190. [Google Scholar] [CrossRef] [PubMed]
- Toh, C.-X.D.; Chan, J.-W.; Chong, Z.-S.; Wang, H.F.; Guo, H.C.; Satapathy, S.; Ma, D.; Goh, G.Y.L.; Khattar, E.; Yang, L.; et al. RNAi Reveals Phase-Specific Global Regulators of Human Somatic Cell Reprogramming. Cell Rep. 2016, 15, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Chang, K.-Y.; Rana, T.M. Genome-Wide Functional Analysis Reveals Factors Needed at the Transition Steps of Induced Reprogramming. Cell Rep. 2014, 8, 327–337. [Google Scholar] [CrossRef]
- Fang, H.-T.; El Farran, C.A.; Xing, Q.R.; Zhang, L.-F.; Li, H.; Lim, B.; Loh, Y.-H. Global H3.3 Dynamic Deposition Defines Its Bimodal Role in Cell Fate Transition. Nat. Commun. 2018, 9, 1537. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G.; Boyes, J.; Chung, J.; Clark, D.; Studitsky, V. Chromatin Structure and Gene Expression. Proc. Natl. Acad. Sci. USA 1996, 93, 9384–9388. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The Accessible Chromatin Landscape of the Human Genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef]
- McVean, G.A.; Altshuler (Co-Chair), D.M.; Durbin (Co-Chair), R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. An Integrated Map of Genetic Variation from 1,092 Human Genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An Expansive Human Regulatory Lexicon Encoded in Transcription Factor Footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gusmao, E.G.; Allhoff, M.; Zenke, M.; Costa, I.G. Analysis of Computational Footprinting Methods for DNase Sequencing Experiments. Nat. Methods 2016, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pique-Regi, R.; Degner, J.F.; Pai, A.A.; Gaffney, D.J.; Gilad, Y.; Pritchard, J.K. Accurate Inference of Transcription Factor Binding from DNA Sequence and Chromatin Accessibility Data. Genome Res. 2011, 21, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, W.W.Y.; D’Antonio-Chronowska, A.; Benaglio, P.; Matsui, H.; Smith, E.N.; D’Antonio, M.; Frazer, K.A. Chromatin Co-Accessibility Is Highly Structured, Spans Entire Chromosomes, and Mediates Long Range Regulatory Genetic Effects. bioRxiv 2019, 604371. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-Seq and RNA-Seq Identifies Human Alpha Cell and Beta Cell Signature Genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From Reads to Insight: A Hitchhiker’s Guide to ATAC-Seq Data Analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Duren, Z.; Chen, X.; Jiang, R.; Wang, Y.; Wong, W.H. Modeling Gene Regulation from Paired Expression and Chromatin Accessibility Data. Proc. Natl. Acad. Sci. USA 2017, 114, E4914–E4923. [Google Scholar] [CrossRef] [PubMed]
- Gate, R.E.; Cheng, C.S.; Aiden, A.P.; Siba, A.; Tabaka, M.; Lituiev, D.; Machol, I.; Gordon, M.G.; Subramaniam, M.; Shamim, M.; et al. Genetic Determinants of Co-Accessible Chromatin Regions in Activated T Cells across Humans. Nat. Genet. 2018, 50, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Kumasaka, N.; Knights, A.J.; Gaffney, D.J. High-Resolution Genetic Mapping of Putative Causal Interactions between Regions of Open Chromatin. Nat. Genet. 2019, 51, 128–137. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, J.F.; Rossello, F.J.; Tan, J.P.; Davidson, K.C.; Valdes, D.S.; Schröder, J.; Sun, Y.B.Y.; Chen, J.; Knaupp, A.S.; et al. Reprogramming Roadmap Reveals Route to Human Induced Trophoblast Stem Cells. Nature 2020, 586, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, J.; Yang, X.; Zhou, C.; Guo, J.; Wu, C.; Qin, Y.; Guo, L.; He, J.; Yu, S.; et al. Chromatin Accessibility Dynamics during IPSC Reprogramming. Cell Stem Cell 2017, 21, 819–833.e6. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinform. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinform. Oxf. Engl. 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Duren, Z.; Chen, X.; Xin, J.; Wang, Y.; Wong, W.H. Time Course Regulatory Analysis Based on Paired Expression and Chromatin Accessibility Data. Genome Res. 2020, 30, 622–634. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinform. Oxf. Engl. 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome wide annotation for Human. Bioconductor. Available online: http://bioconductor.org/packages/org.Hs.eg.db/ (accessed on 16 August 2023).
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Zmrzlikar, J.; Žganec, M.; Ausec, L.; Štajdohar, M. RNAnorm: RNA-seq data normalization in Python. GitHub. Available online: https://github.com/genialis/RNAnorm/blob/main/CITATION.cff (accessed on 16 August 2023).
- SRA Toolkit Develoment Team; SBGrid Consortium. SRA Toolkit. GitHub. Available online: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software (accessed on 16 August 2023).
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinform. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinform. Oxf. Engl. 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinform. Oxf. Engl. 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Kumar, V.; Muratani, M.; Rayan, N.A.; Kraus, P.; Lufkin, T.; Ng, H.H.; Prabhakar, S. Uniform, Optimal Signal Processing of Mapped Deep-Sequencing Data. Nat. Biotechnol. 2013, 31, 615–622. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinform. Oxf. Engl. 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Wu, T.; Zhan, L.; Li, L.; Chen, M.; Xie, W.; Xie, Z.; Hu, E.; Xu, S.; et al. Exploring Epigenomic Datasets by ChIPseeker. Curr. Protoc. 2022, 2, e585. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S. Gplots: Various R Programming Tools for Plotting Data. R Package Version 2009, 2, 1. [Google Scholar]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. Available online: https://rpkgs.datanovia.com/ggpubr/authors.html#citation (accessed on 16 August 2023).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. CytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S.P.; Barabási, A.-L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Pržulj, N.; Wigle, D.A.; Jurisica, I. Functional Topology in a Network of Protein Interactions. Bioinformatics 2004, 20, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Sabidussi, G. The Centrality Index of a Graph. Psychometrika 1966, 31, 581–603. [Google Scholar] [CrossRef]
- Chin, C.-S.; Samanta, M.P. Global Snapshot of a Protein Interaction Network—a Percolation Based Approach. Bioinformatics 2003, 19, 2413–2419. [Google Scholar] [CrossRef]
- Shimbel, A. Structural Parameters of Communication Networks. Bull. Math. Biophys. 1953, 15, 501–507. [Google Scholar] [CrossRef]
- Wickham, H.; Sievert, C. Ggplot2: Elegant Graphics for Data Analysis, 1st ed.; Vols. 1–1; Springer: New York, NY, USA, 2016; Volume 1. [Google Scholar]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.F.; Johnstone, S.E.; Newman, J.J.; Kagey, M.H.; Young, R.A. Tcf3 Is an Integral Component of the Core Regulatory Circuitry of Embryonic Stem Cells. Genes Dev. 2008, 22, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Martello, G.; Sugimoto, T.; Diamanti, E.; Joshi, A.; Hannah, R.; Ohtsuka, S.; Göttgens, B.; Niwa, H.; Smith, A. Esrrb Is a Pivotal Target of the Gsk3/Tcf3 Axis Regulating Embryonic Stem Cell Self-Renewal. Cell Stem Cell 2012, 11, 491–504. [Google Scholar] [CrossRef]
- Tam, W.-L.; Lim, C.Y.; Han, J.; Zhang, J.; Ang, Y.-S.; Ng, H.-H.; Yang, H.; Lim, B. T-Cell Factor 3 Regulates Embryonic Stem Cell Pluripotency and Self-Renewal by the Transcriptional Control of Multiple Lineage Pathways. Stem Cells 2008, 26, 2019–2031. [Google Scholar] [CrossRef]
- Yi, F.; Pereira, L.; Merrill, B.J. Tcf3 Functions as a Steady-State Limiter of Transcriptional Programs of Mouse Embryonic Stem Cell Self-Renewal. Stem Cells 2008, 26, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Papp, B.; Hoffman, J.A.; Merrill, B.J.; Plath, K. Stage-Specific Regulation of Reprogramming to Induced Pluripotent Stem Cells by Wnt Signaling and T Cell Factor Proteins. Cell Rep. 2013, 3, 2113–2126. [Google Scholar] [CrossRef]
- Zhao, H.; Han, Z.; Liu, X.; Gu, J.; Tang, F.; Wei, G.; Jin, Y. The Chromatin Remodeler Chd4 Maintains Embryonic Stem Cell Identity by Controlling Pluripotency- and Differentiation-Associated Genes. J. Biol. Chem. 2017, 292, 8507–8519. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.A.; Dhele, N.; Cheemadan, S.; Ketkar, A.; Jayandharan, G.R.; Palakodeti, D.; Rampalli, S. Ezh2 Mediated H3K27me3 Activity Facilitates Somatic Transition during Human Pluripotent Reprogramming. Sci. Rep. 2015, 5, 8229. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Maia, A.; Alajem, A.; Polesso, F.; Sridharan, R.; Mason, M.J.; Heidersbach, A.; Ramalho-Santos, J.; McManus, M.T.; Plath, K.; Meshorer, E.; et al. Chd1 Regulates Open Chromatin and Pluripotency of Embryonic Stem Cells. Nature 2009, 460, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Ang, Y.-S.; Tsai, S.-Y.; Lee, D.-F.; Monk, J.; Su, J.; Ratnakumar, K.; Ding, J.; Ge, Y.; Darr, H.; Chang, B.; et al. Wdr5 Mediates Self-Renewal and Reprogramming via the Embryonic Stem Cell Core Transcriptional Network. Cell 2011, 145, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gafni, O.; Weinberger, L.; Zviran, A.; Ayyash, M.; Rais, Y.; Krupalnik, V.; Zerbib, M.; Amann-Zalcenstein, D.; Maza, I.; et al. The H3K27 Demethylase Utx Regulates Somatic and Germ Cell Epigenetic Reprogramming. Nature 2012, 488, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-C.; Yen, J.-M.; Gong, K.-B.; Hsu, T.-T.; Chen, L.-R. IGF-1/IGFBP-1 Increases Blastocyst Formation and Total Blastocyst Cell Number in Mouse Embryo Culture and Facilitates the Establishment of a Stem-Cell Line. BMC Cell Biol. 2003, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Leitch, H.G.; McEwen, K.R.; Turp, A.; Encheva, V.; Carroll, T.; Grabole, N.; Mansfield, W.; Nashun, B.; Knezovich, J.G.; Smith, A.; et al. Naive Pluripotency Is Associated with Global DNA Hypomethylation. Nat. Struct. Mol. Biol. 2013, 20, 311–316. [Google Scholar] [CrossRef]
- Eiselleova, L.; Matulka, K.; Kriz, V.; Kunova, M.; Schmidtova, Z.; Neradil, J.; Tichy, B.; Dvorakova, D.; Pospisilova, S.; Hampl, A.; et al. A Complex Role for FGF-2 in Self-Renewal, Survival, and Adhesion of Human Embryonic Stem Cells. Stem Cells Dayt. Ohio 2009, 27, 1847–1857. [Google Scholar] [CrossRef]
- Chan, E.M.; Ratanasirintrawoot, S.; Park, I.-H.; Manos, P.D.; Loh, Y.-H.; Huo, H.; Miller, J.D.; Hartung, O.; Rho, J.; Ince, T.A.; et al. Live Cell Imaging Distinguishes Bona Fide Human IPS Cells from Partially Reprogrammed Cells. Nat. Biotechnol. 2009, 27, 1033–1037. [Google Scholar] [CrossRef]
- Lv, Y.; Dai, H.; Yan, G.; Meng, G.; Zhang, X.; Guo, Q. Downregulation of Tumor Suppressing STF CDNA 3 Promotes Epithelial-Mesenchymal Transition and Tumor Metastasis of Osteosarcoma by the Wnt/GSK-3β/β-Catenin/Snail Signaling Pathway. Cancer Lett. 2016, 373, 164–173. [Google Scholar] [CrossRef]
- Jiao, J.; Dang, Y.; Yang, Y.; Gao, R.; Zhang, Y.; Kou, Z.; Sun, X.-F.; Gao, S. Promoting Reprogramming by FGF2 Reveals That the Extracellular Matrix Is a Barrier for Reprogramming Fibroblasts to Pluripotency. Stem Cells 2013, 31, 729–740. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, D.; Wei, B.; Jiang, J.; Kang, J.; Ling, K.; Gu, Y.; Li, J.; Xiao, L.; Pei, G. E-Cadherin-Mediated Cell–Cell Contact Is Critical for Induced Pluripotent Stem Cell Generation. Stem Cells 2010, 28, 1315–1325. [Google Scholar] [CrossRef]
- Lehembre, F.; Yilmaz, M.; Wicki, A.; Schomber, T.; Strittmatter, K.; Ziegler, D.; Kren, A.; Went, P.; Derksen, P.W.B.; Berns, A.; et al. NCAM-Induced Focal Adhesion Assembly: A Functional Switch upon Loss of E-Cadherin. EMBO J. 2008, 27, 2603–2615. [Google Scholar] [CrossRef]
- Mah, N.; Wang, Y.; Liao, M.-C.; Prigione, A.; Jozefczuk, J.; Lichtner, B.; Wolfrum, K.; Haltmeier, M.; Flöttmann, M.; Schaefer, M.; et al. Molecular Insights into Reprogramming-Initiation Events Mediated by the OSKM Gene Regulatory Network. PLoS ONE 2011, 6, e24351. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization Eliminates a Roadblock during Cellular Reprogramming into IPS Cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen Activator Inhibitor-1 Is a Critical Downstream Target of P53 in the Induction of Replicative Senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New Cell Lines from Mouse Epiblast Share Defining Features with Human Embryonic Stem Cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.-L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The Ground State of Embryonic Stem Cell Self-Renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef]
- ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic Stem Cells Require Wnt Proteins to Prevent Differentiation to Epiblast Stem Cells. Nat. Cell Biol. 2011, 13, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.-K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional Genomics Reveals a BMP-Driven Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and Cellular Reprogramming: Facts, Hypotheses, Unresolved Issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and Impediments of the Pluripotency Reprogramming Factors’ Initial Engagement with the Genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef]
- Koche, R.P.; Smith, Z.D.; Adli, M.; Gu, H.; Ku, M.; Gnirke, A.; Bernstein, B.E.; Meissner, A. Reprogramming Factor Expression Initiates Widespread Targeted Chromatin Remodeling. Cell Stem Cell 2011, 8, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liang, Z.; Zhang, J.; Hu, G.; Wang, J.; Li, Y.; Guo, R.; Dong, X.; Babarinde, I.A.; Ping, W.; et al. CTCF Functions as an Insulator for Somatic Genes and a Chromatin Remodeler for Pluripotency Genes during Reprogramming. Cell Rep. 2022, 39, 110626. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOUSE | |||||
| MEF | Day 3 | Day 6 | Day 9 | Day 12 | iPSC |
| Fos | Klf5 | OSTN 1 | OSTN 1 | OSTN 1 | OSTN 1 |
| Atf3 | Klf6 | Oct4 | Oct6 | Klf5 | Sox3 |
| Fra1 | Sox3 | Oct6 | Sox3 | Sox2 | Sox10 |
| BATF | Klf4 | Sox3 | Oct4 | Sox3 | Sp5 |
| Fra2 | OSTN 1 | Brn1 | Sox2 | Oct4 | Sox21 |
| JunB | Klf1 | Klf5 | Oct1 | Sox21 | Sox6 |
| Fosl2 | Sox2 | Sox10 | Brn1 | Sox6 | Sox2 |
| AP-1 | Klf3 | Sox6 | Sox6 | Klf1 | Sox15 |
| Jun-AP1 | Sox10 | Sox2 | Sox10 | Klf4 | Oct4 |
| NFY | EKlf | Sox21 | Sox21 | Sox10 | Sp2 |
| HUMAN | |||||
| HF | Day 3 | Day 7 | Day 13 | Day 21 | iPSC |
| FOS | FOS | CTCF | CTCF | CTCF | CTCF |
| FRA1 | FRA1 | BORIS | BORIS | BORIS | BORIS |
| ATF3 | ATF3 | KLF5 | OSTN 1 | TEAD1 | OSTN 1 |
| BATF | BATF | OSTN 1 | KLF5 | TEAD3 | SOX3 |
| JUNB | FRA-2 | KLF1 | KLF1 | OSTN 1 | OCT4 |
| AP1 | AP-1 | KLF6 | OCT4 | TEAD4 | BRN1 |
| FRA2 | JUNB | SP2 | KLF6 | TEAD | SOX6 |
| FOSL2 | KLF5 | KLF4 | SP5 | TEAD2 | SOX21 |
| JUN-AP1 | FOSL2 | KLF14 | KLF14 | SOX3 | OCT6 |
| CTCF | JUN-AP-1 | EKLF | KLF4 | JUN-AP1 | SOX2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thangavelu, C.S.; Norden-Krichmar, T.M. Integrative Meta-Analysis during Induced Pluripotent Stem Cell Reprogramming Reveals Conserved Networks and Chromatin Accessibility Signatures in Human and Mouse. BioMedInformatics 2023, 3, 1015-1039. https://doi.org/10.3390/biomedinformatics3040061
Thangavelu CS, Norden-Krichmar TM. Integrative Meta-Analysis during Induced Pluripotent Stem Cell Reprogramming Reveals Conserved Networks and Chromatin Accessibility Signatures in Human and Mouse. BioMedInformatics. 2023; 3(4):1015-1039. https://doi.org/10.3390/biomedinformatics3040061
Chicago/Turabian StyleThangavelu, Chloe S., and Trina M. Norden-Krichmar. 2023. "Integrative Meta-Analysis during Induced Pluripotent Stem Cell Reprogramming Reveals Conserved Networks and Chromatin Accessibility Signatures in Human and Mouse" BioMedInformatics 3, no. 4: 1015-1039. https://doi.org/10.3390/biomedinformatics3040061
APA StyleThangavelu, C. S., & Norden-Krichmar, T. M. (2023). Integrative Meta-Analysis during Induced Pluripotent Stem Cell Reprogramming Reveals Conserved Networks and Chromatin Accessibility Signatures in Human and Mouse. BioMedInformatics, 3(4), 1015-1039. https://doi.org/10.3390/biomedinformatics3040061