
Analyzing Large Microbiome Datasets Using Machine Learning and Big Data

 , , ,
, , ,  ,
,  and
and 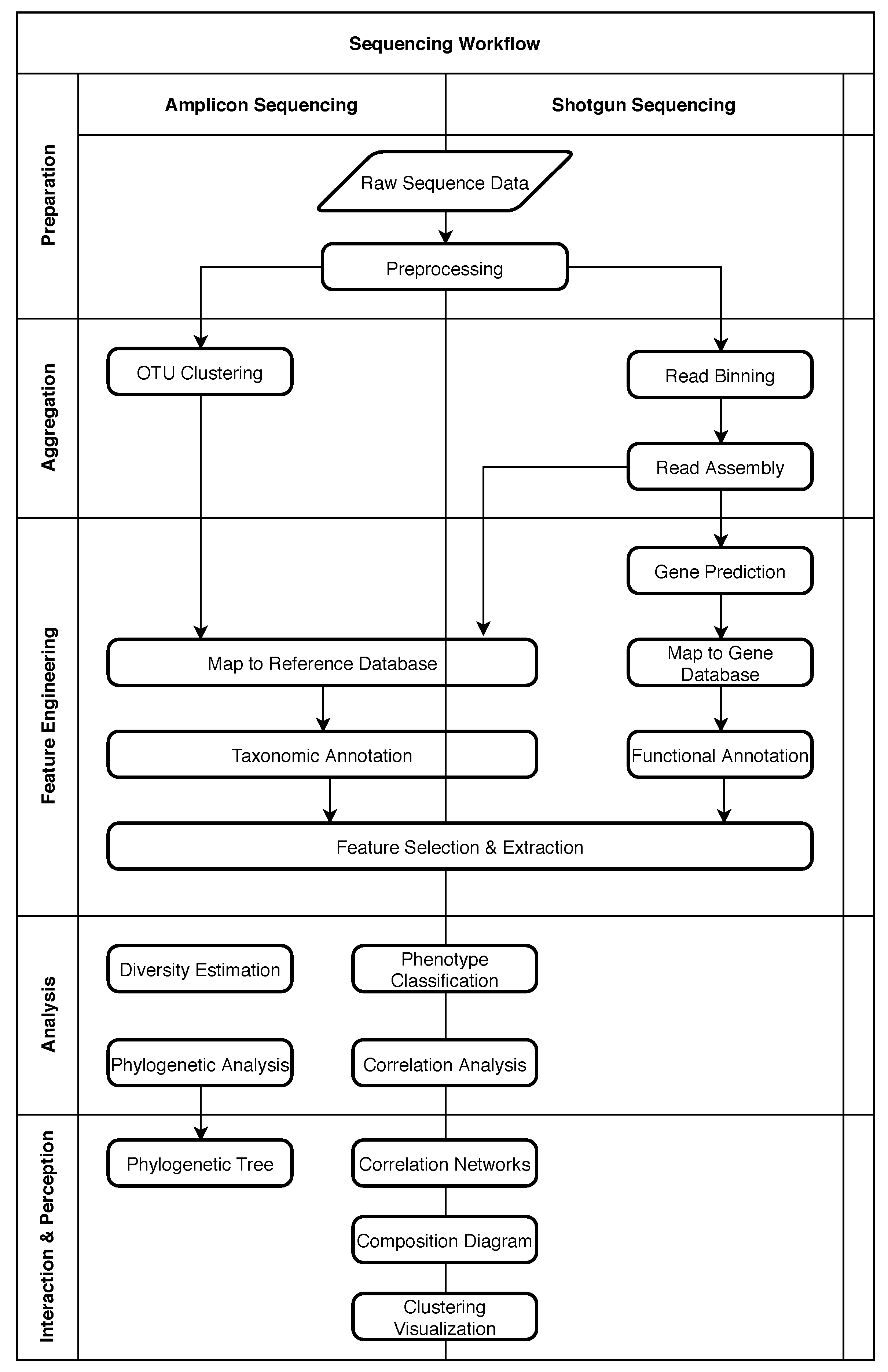{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Machine Learning Trends
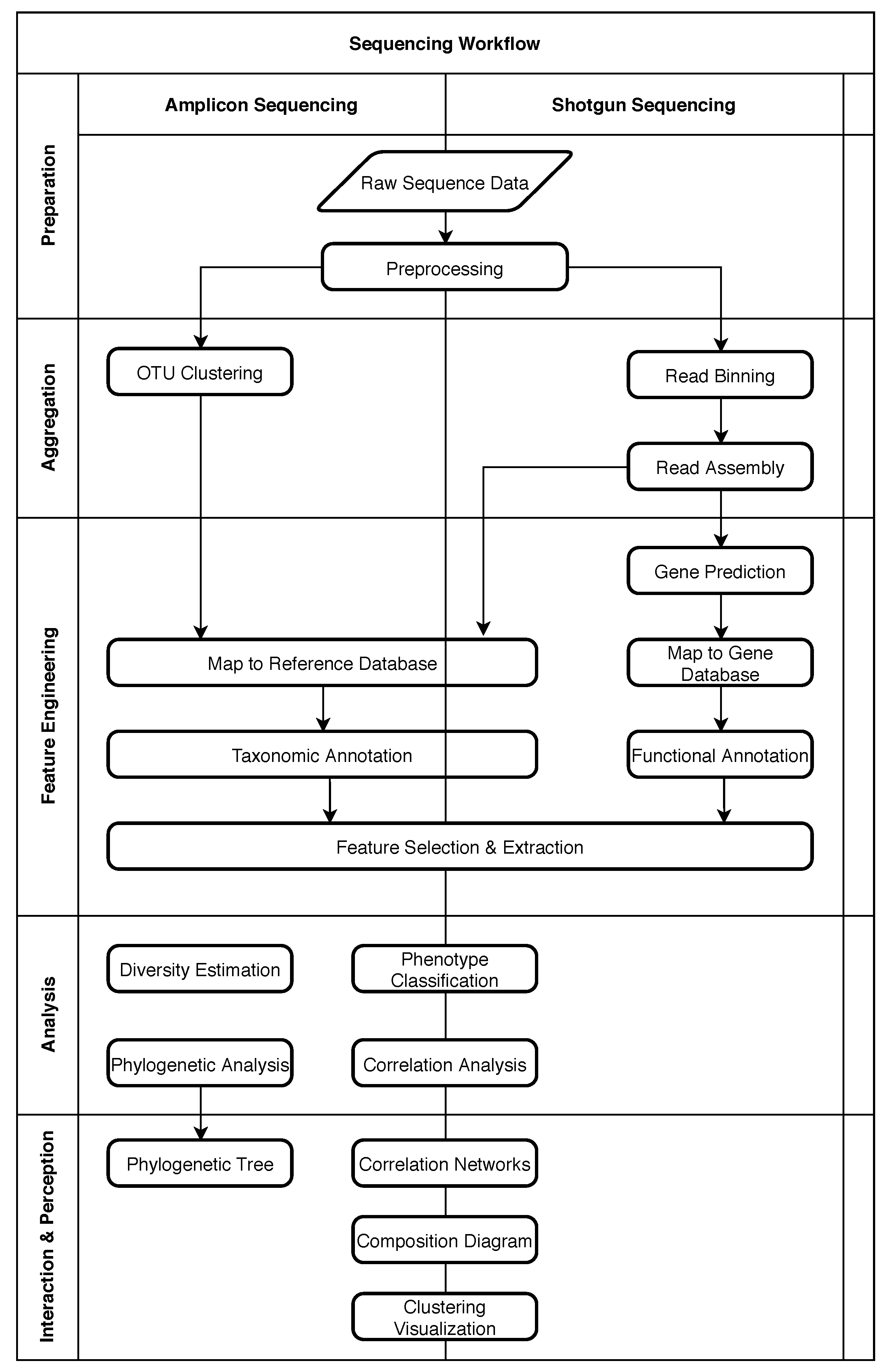
2. Structure of Metagenomic Studies
2.1. Five Phases of Metagenomic Studies
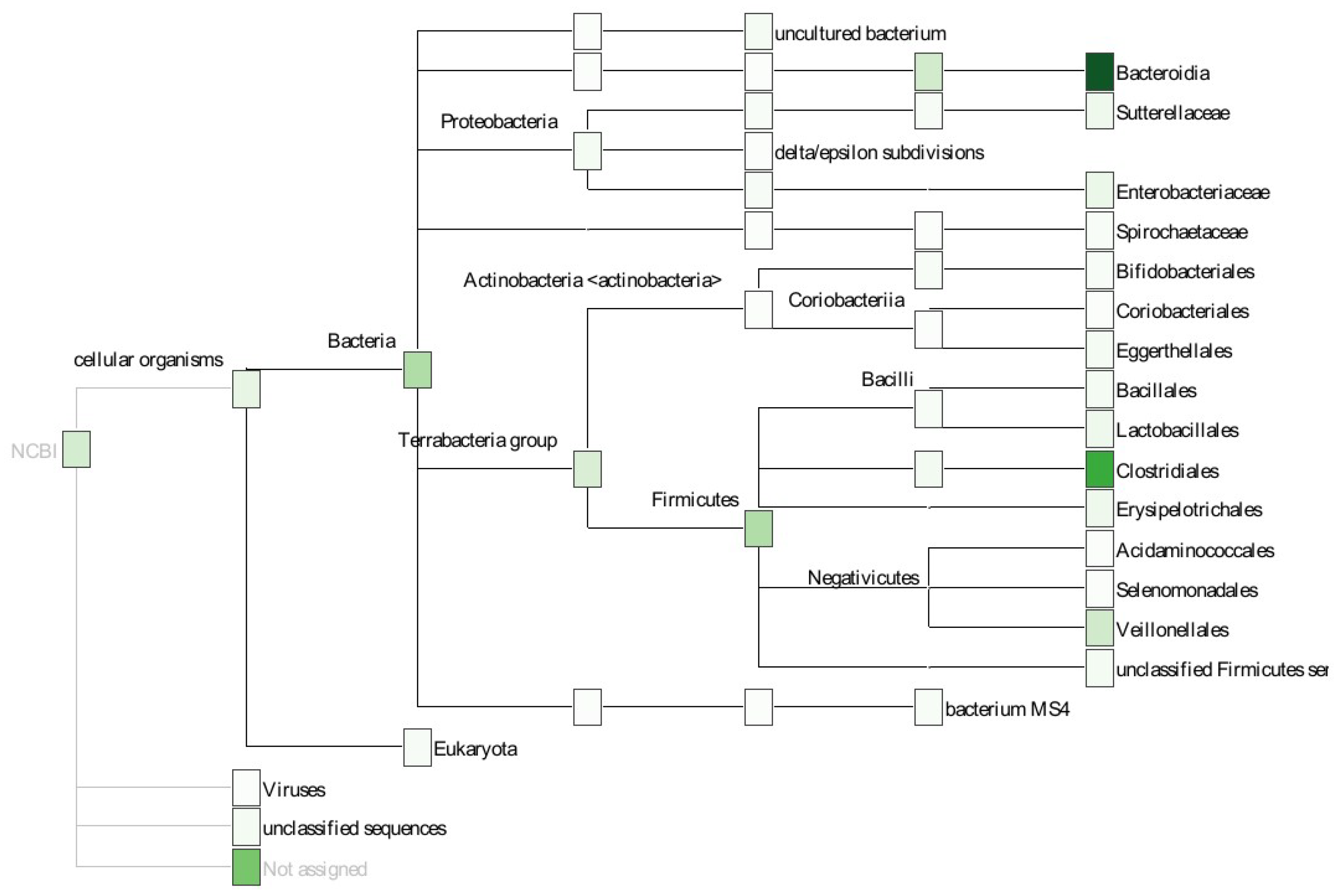
2.2. Example: Microbiome Analysis on Four Human Body Sites
3. Machine Learning
3.1. Vector Space Transformations
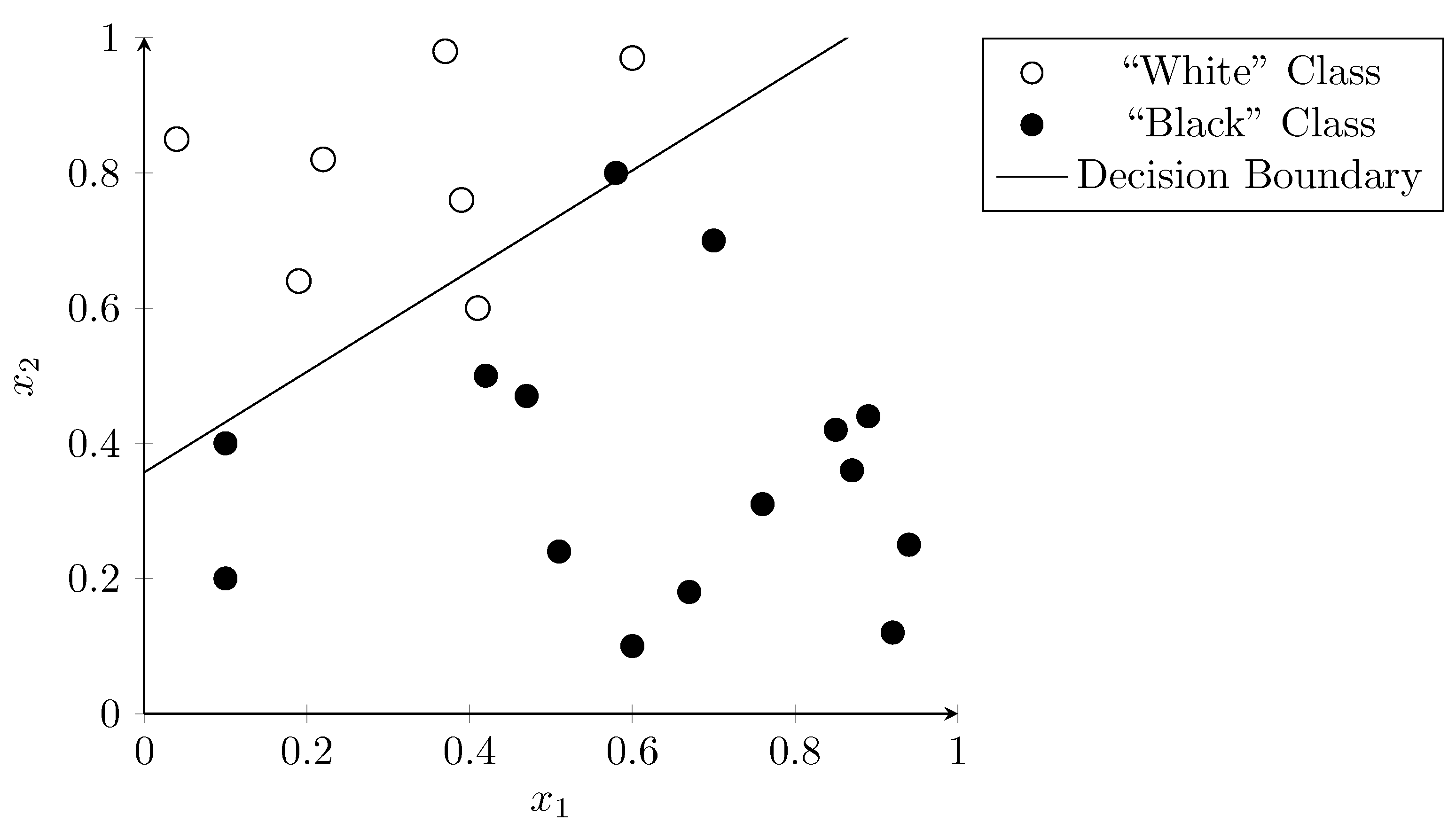
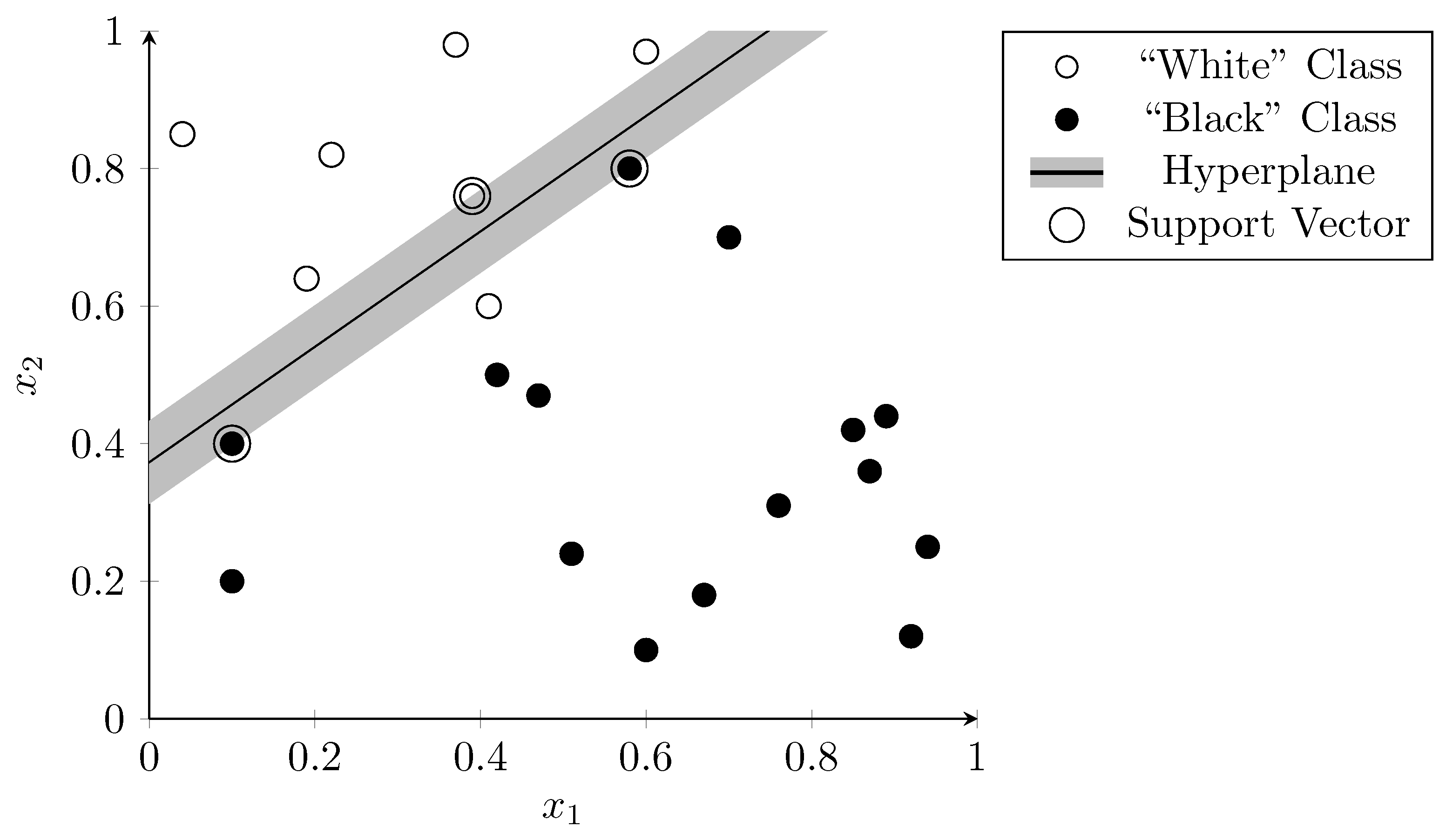
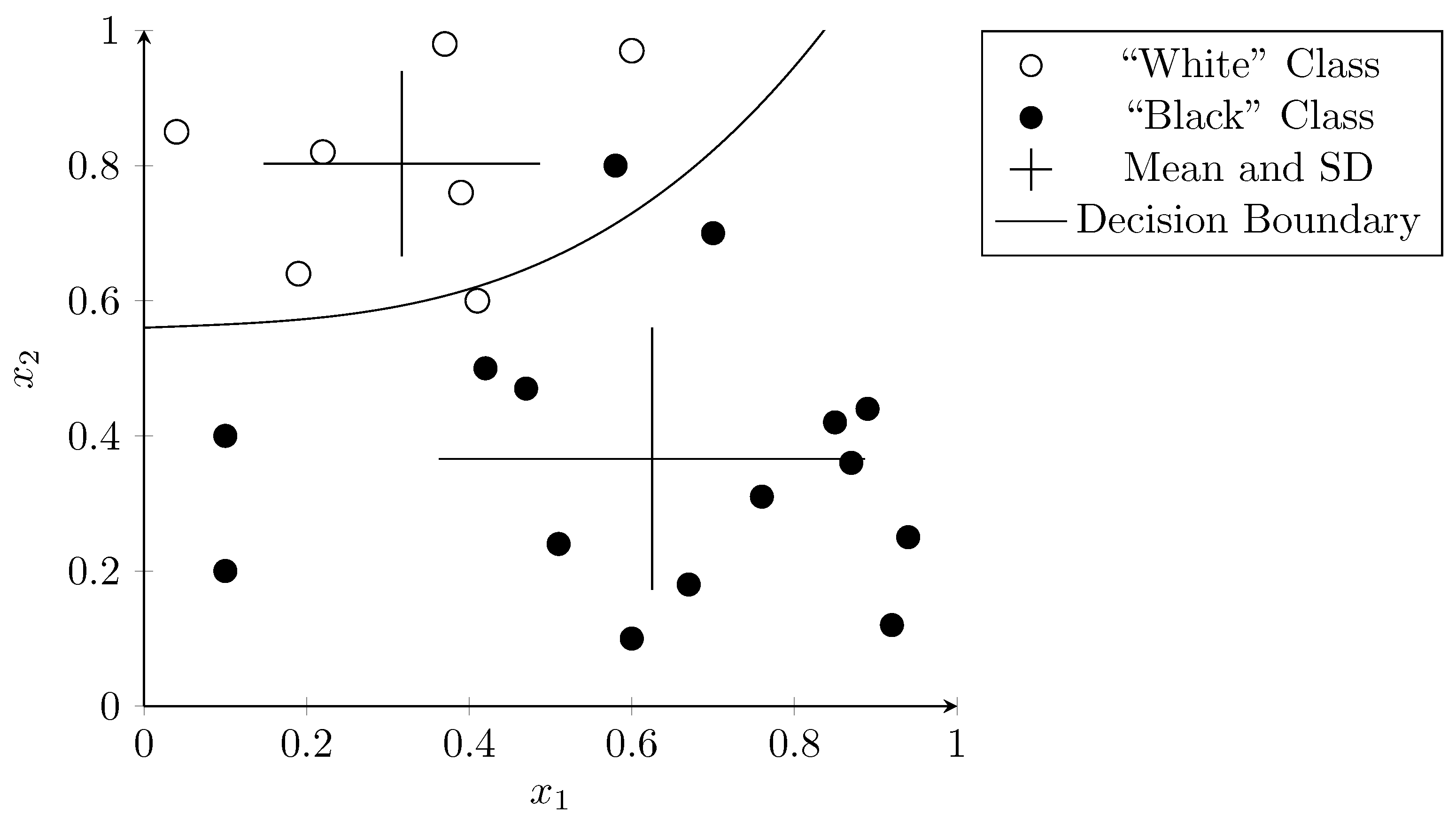
3.2. Support Vector Machines
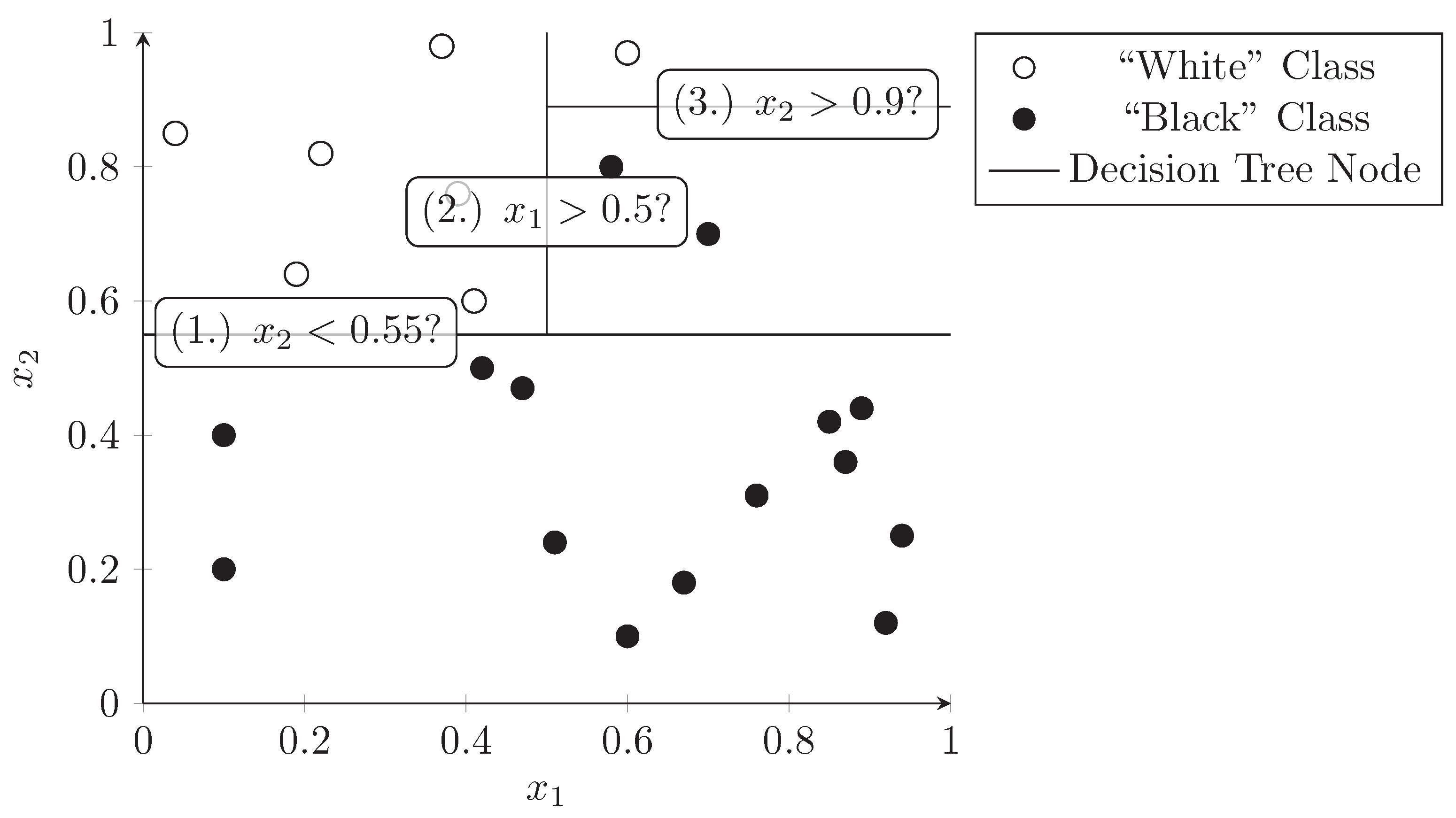
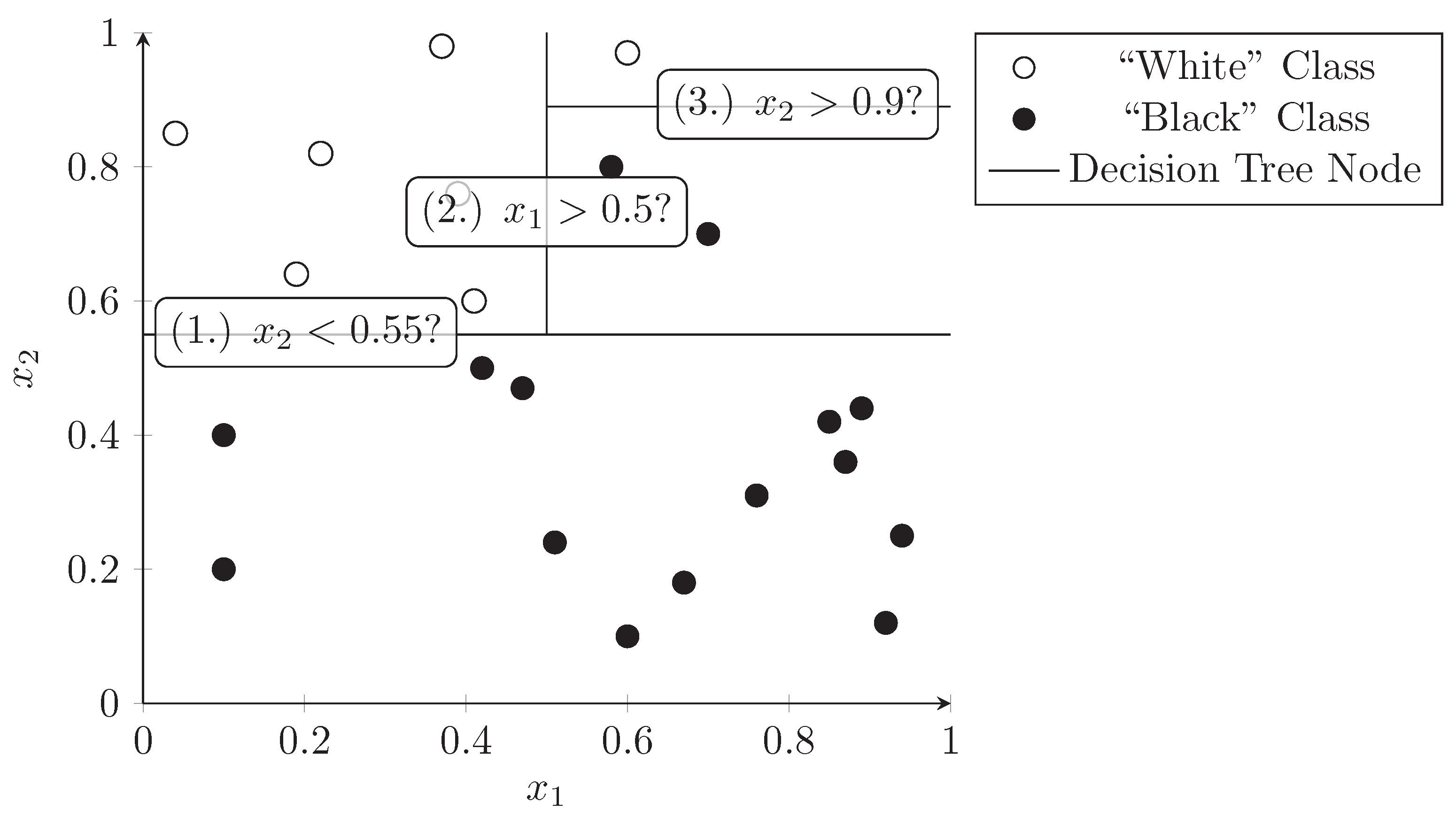
3.3. Decision Trees
3.4. Random Forest
3.5. Naïve Bayes Classifier
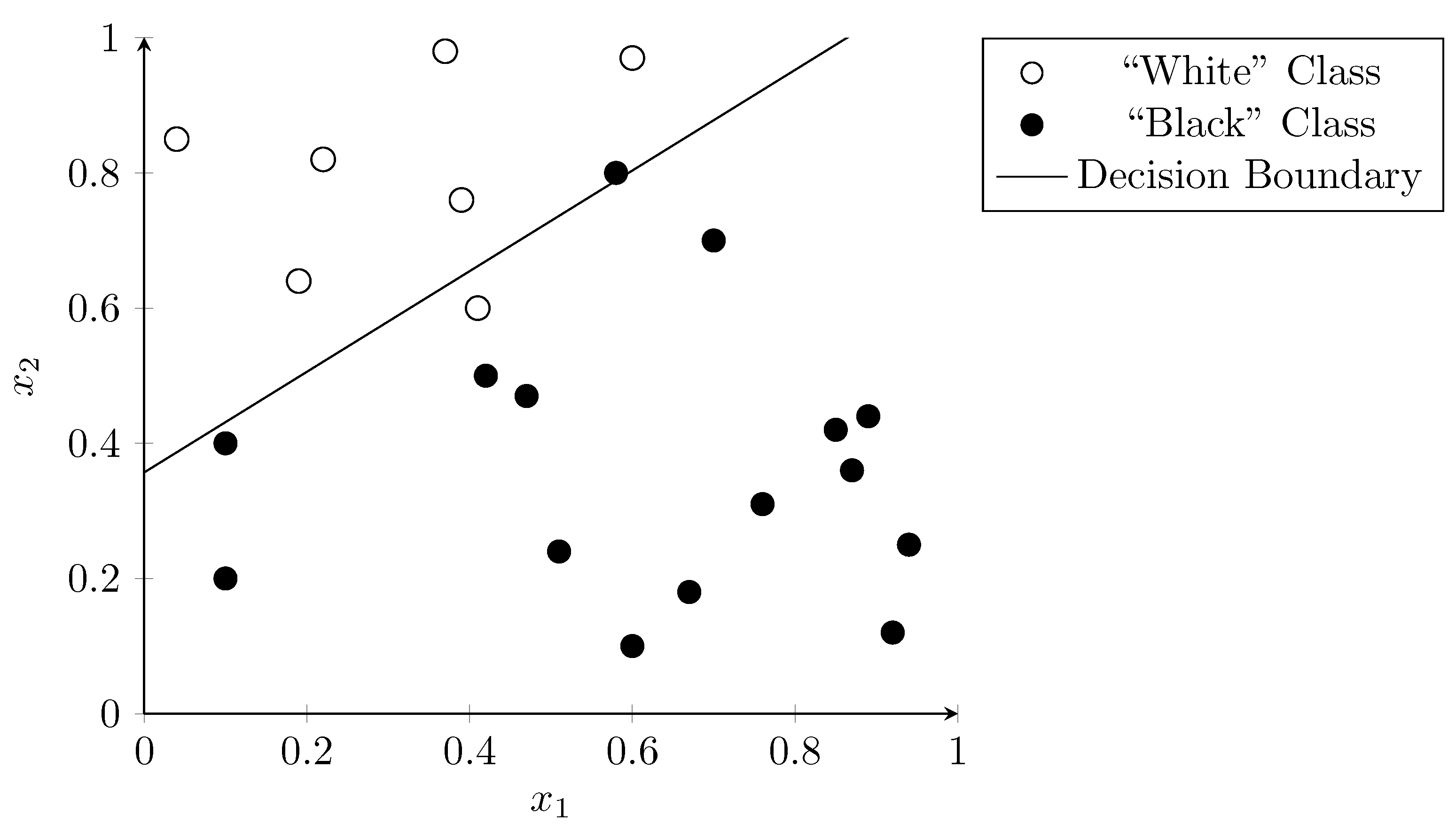
3.6. Logistic Regression
3.7. Clustering Algorithms
3.8. Neural Networks
3.9. Deep Learning
- Increasing the number of neurons leads to a higher number of variables which increases the risk of overfitting [51].
- The processing power for training large networks was not available [15].
- The popular “activation functions” used in neurons to compute the final output value of the neuron based on its inputs were proven to perform poorly in the context of many layers (known as the vanishing and exploding gradient problems in the literature) [52].
- Deep learning networks are often trained with a significantly larger quantity of data. Many breakthroughs come from big tech companies like Google that have access to huge datasets (e.g., billions of images) which decreases the risk that the network only memorizes its training input and fails to generalize. Some computational techniques were also developed which keep the network from overfitting (e.g., “Dropout”, see Srivastava et al. [51]).
- The processing power was increased by offloading the training to the Graphics Processing Unit (GPU) and utilizing large computing clusters. The advent of cloud computing enabled this possibility not only for big companies but also for smaller research teams.
- The problem of vanishing or exploding gradients was mitigated by using vastly simpler activation functions, which do not exhibit this problem.
4. Role of Machine Learning in Metagenomics
4.1. Obtaining Raw Sequence Data
4.2. Preprocessing
4.3. OTU Clustering
4.4. Read Binning
4.5. Read Assembly
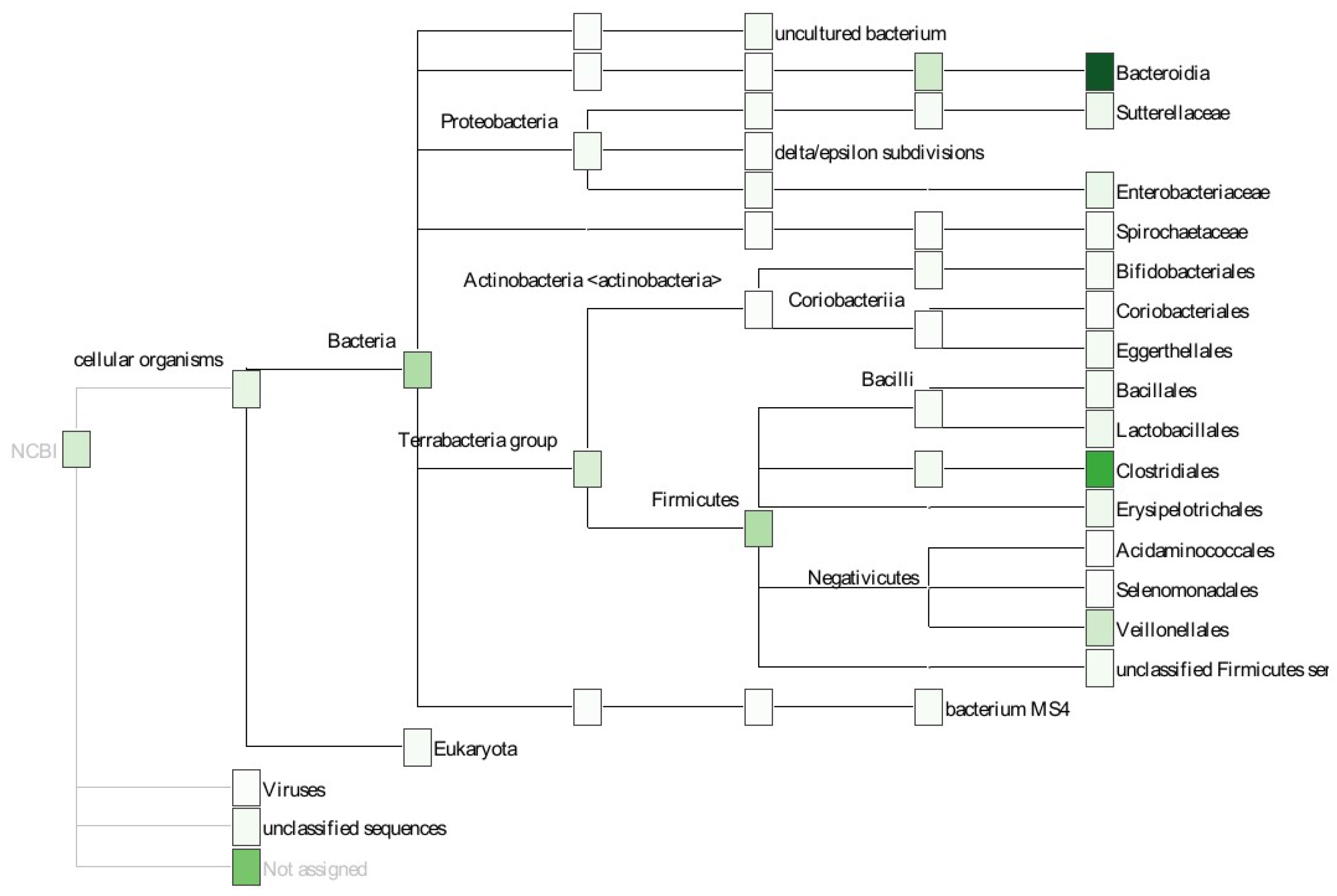
4.6. Taxonomic Annotation
4.7. Functional Annotation
4.8. Gene Prediction
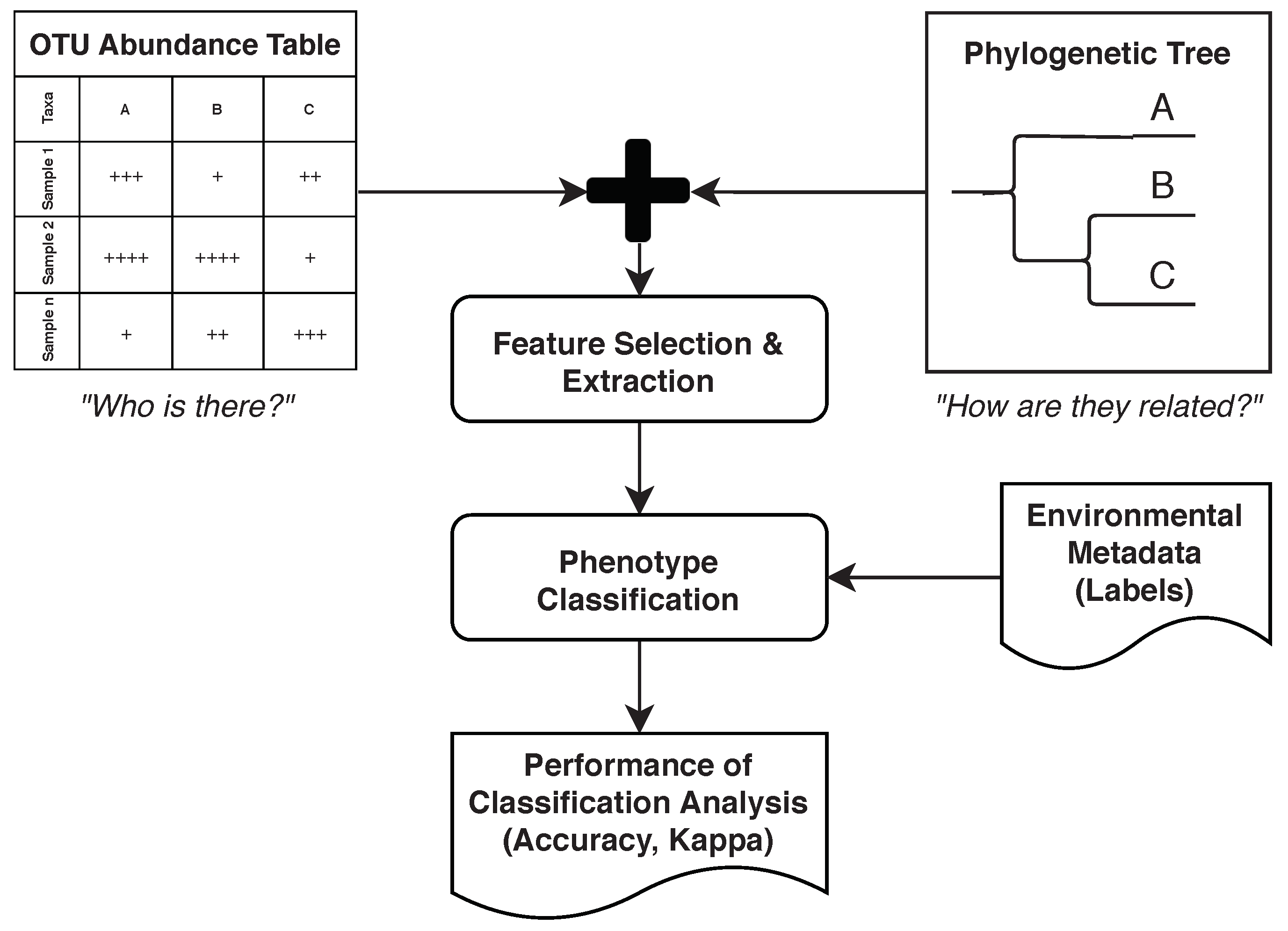
4.9. Feature Selection and Extraction
4.10. Phenotype Classification
4.11. Other Common Analysis Tasks
4.12. Interaction and Perception
5. Challenges for Machine Learning in Metagenomics
5.1. Model Selection
5.2. Deep Learning and Feature Engineering
5.3. Accessibility
5.4. Explainability
5.5. Reproducibility
5.6. Biological Diversity
5.7. High Dimensionality and Low Number of Samples
5.8. Big Data
6. Metagenomic Processing Pipelines
6.1. Galaxy
6.2. MG-RAST and MGnify
6.3. QIIME 2
6.4. MetaPlat and Successors
- Sample gut collection, from cattle, for sequencing
- Collection of publicly available databases to create a new classification of previously unclassified sequences, using ML algorithms
- Development of accurate classification algorithms
- Real-time or time-efficient comparison analyses
- Production of statistical and visual representations, conveying more useful information
- Platform integration
- Insights into probiotic supplement usage, methane production and feed conversion efficiency in cattle
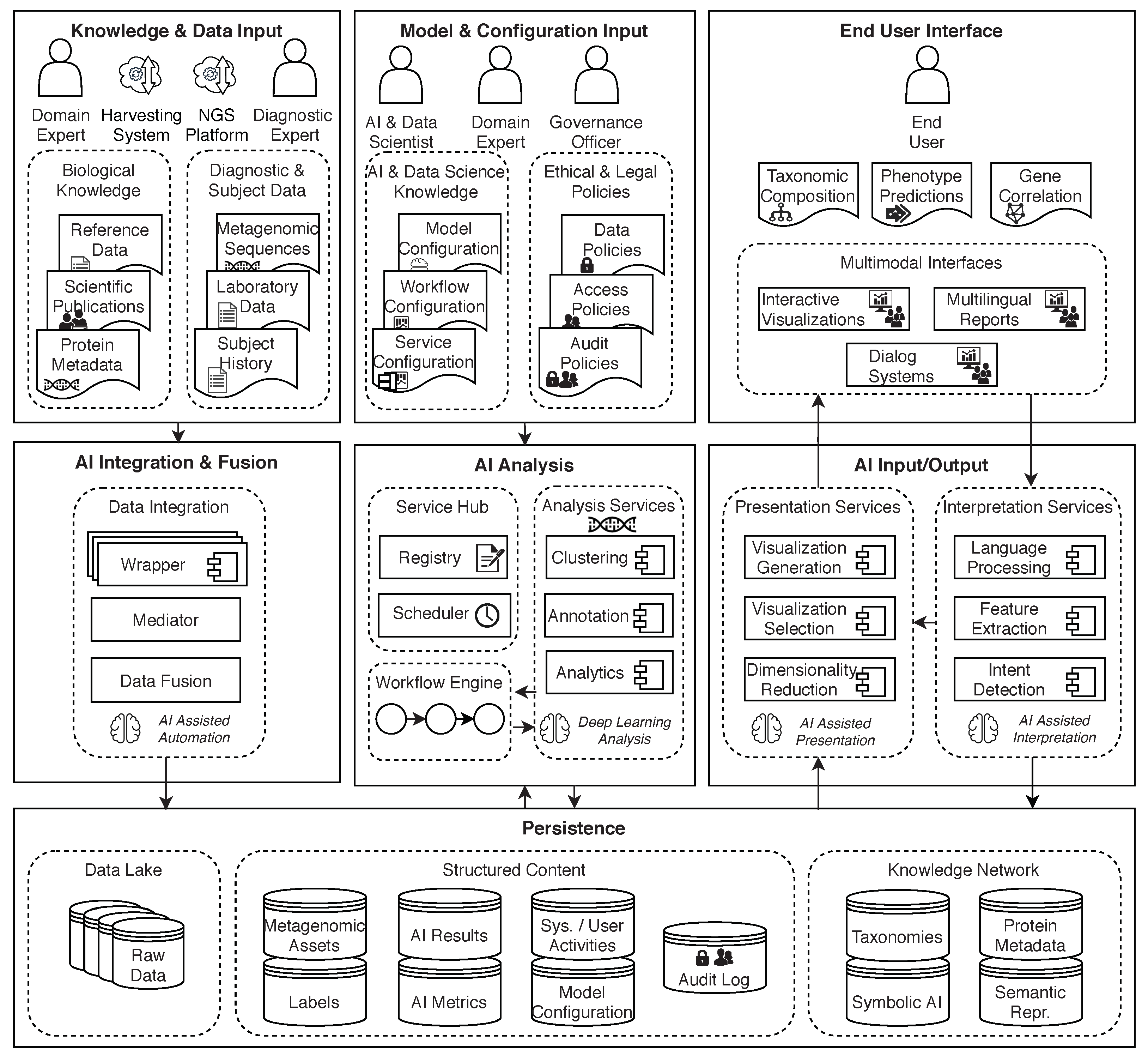
7. AI2VIS4BigData Conceptual Architecture for Metagenomics Supporting Human Medicine
7.1. Description of the Conceptual Architecture
7.2. Use in Clinical Settings
8. Remaining Challenges, Conclusions, and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Charbonneau, M.R.; Blanton, L.V.; DiGiulio, D.B.; Relman, D.A.; Lebrilla, C.B.; Mills, D.A.; Gordon, J.I. A microbial perspective of human developmental biology. Nature 2016, 535, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Bäumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef]
- Nazir, A. Review on Metagenomics and its Applications. Imp. J. Interdiscip. Res. 2016, 2, 277–286. [Google Scholar]
- Nagarajan, M. (Ed.) Metagenomics: Perspectives, Methods, and Applications; Academic Press: London, UK, 2018. [Google Scholar]
- Mardanov, A.V.; Kadnikov, V.V.; Ravin, N.V. Metagenomics: A Paradigm Shift in Microbiology. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 1–13. [Google Scholar] [CrossRef]
- Wetterstrand, K.A. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). 2019. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 4 November 2021).
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef]
- Vu, B.; Wu, Y.; Afli, H.; Mc Kevitt, P.; Walsh, P.; Engel, F.C.; Fuchs, M.; Hemmje, M.L. A Metagenomic Content and Knowledge Management Ecosystem Platform. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2019, San Diego, CA, USA, 18–21 November 2019; Yoo, I., Bi, J., Hu, X., Eds.; IEEE: Piscataway, NJ, USA, 2019. [Google Scholar]
- Soueidan, H.; Nikolski, M. Machine Learning for Metagenomics: Methods and Tools. arXiv 2015, arXiv:1510.06621. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA; London, UK, 2016. [Google Scholar]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Al-Ajlan, A.; El Allali, A. CNN-MGP: Convolutional Neural Networks for Metagenomics Gene Prediction. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [Green Version]
- Méndez-García, C.; Bargiela, R.; Martínez-Martínez, M.; Ferrer, M. Metagenomic Protocols and Strategies. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 15–54. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Buermans, H.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [Green Version]
- Ramazzotti, M.; Bacci, G. Chapter 5—16S rRNA-Based Taxonomy Profiling in the Metagenomics Era. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 103–119. [Google Scholar] [CrossRef]
- Krause, T.; Andrade, B.G.N.; Afli, H.; Wang, H.; Zheng, H.; Hemmje, M.L. Understanding the Role of (Advanced) Machine Learning in Metagenomic Workflows; Advanced Visual Interfaces; Lecture Notes in Computer Science; Reis, T., Bornschlegl, M.X., Angelini, M., Hemmje, M.L., Eds.; Springer: Ischia, Italy, 2021; pp. 56–82. [Google Scholar]
- Zhbannikov, I.Y.; Foster, J.A. Chapter 6—Analyzing High-Throughput Microbial Amplicon Sequence Data Using Multiple Markers. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 121–138. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J. Strategies for Taxonomic and Functional Annotation of Metagenomes. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 55–79. [Google Scholar] [CrossRef]
- Guyon, I.; Elisseeff, A. An Introduction to Variable and Feature Selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Reis, T.; Bornschlegl, M.X.; Hemmje, M.L. Towards a Reference Model for Artificial Intelligence Supporting Big Data Analysis. In Proceedings of the 2020 International Conference on Data Science (ICDATA’20), Las Vegas, NV, USA, 27–30 July 2020. [Google Scholar]
- Wassan, J.T.; Wang, H.; Browne, F.; Zheng, H. Phy-PMRFI: Phylogeny-Aware Prediction of Metagenomic Functions Using Random Forest Feature Importance. IEEE Trans. Nanobioscience 2019, 18, 273–282. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Aaronson, S.; Hutner, S.H. Biochemical markers and microbial phylogeny. Q. Rev. Biol. 1966, 41, 13–46. [Google Scholar] [CrossRef]
- Knights, D.; Costello, E.K.; Knight, R. Supervised classification of human microbiota. FEMS Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef] [Green Version]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef]
- Calle, M.L. Statistical Analysis of Metagenomics Data. Genom. Informatics 2019, 17, e6. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Corrected at 8th Printing 2009 ed.; Information Science and Statistics; Springer: New York, NY, USA, 2006. [Google Scholar]
- Naumov, M. On the Dimensionality of Embeddings for Sparse Features and Data. arXiv 2019, arXiv:1901.02103. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Weston, J.; Mukherjee, S.; Chapelle, O.; Pontil, M.; Poggio, T.; Vapnik, V. Feature Selection for SVMs. In Proceedings of the 13th International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 6–12 December 2000; Leen, T., Dietterich, T., Tresp, V., Eds.; 2000; pp. 668–674. [Google Scholar]
- Bramer, M.A. Principles of Data Mining; Undergraduate Topics in Computer Science; Springer: London, UK, 2007. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Berkson, J. Application to the Logistic Function to Bio-Assay. J. Am. Stat. Assoc. 1944, 39, 357. [Google Scholar] [CrossRef]
- Arthur, D.; Vassilvitskii, S. How slow is the k-means method? In Proceedings of the 22nd Annual Symposium on Computational Geometry, Sedona, AZ, USA, 5–7 June 2006; Amenta, N., Ed.; ACM: New York, NY, USA, 2006; pp. 144–153. [Google Scholar] [CrossRef]
- Kröse, B.; van der Smagt, P. An Introduction to Neural Networks; University of Amsterdam: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Rojas, R. Theorie der neuronalen Netze: Eine Systematische Einführung; Springer: Berlin, Germany, 1996. [Google Scholar]
- Rey, G.D.; Wender, K.F. Neuronale Netze: Eine Einführung in die Grundlagen, Anwendungen und Datenauswertung; Springer: Berlin, Germany, 2011. [Google Scholar]
- Collobert, R.; Bengio, S. Links between perceptrons, MLPs and SVMs. In Proceedings of the Twenty-First International Conference on Machine Learning–ICML ’04, Banff, AB, Canada, 4–8 July 2004; Brodley, C., Ed.; ACM Press: New York, NY, USA, 2004; p. 23. [Google Scholar] [CrossRef] [Green Version]
- Habibi, M.; Weber, L.; Neves, M.; Wiegandt, D.L.; Leser, U. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics 2017, 33, i37–i48. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Hochreiter, S.; Bengio, Y.; Frasconi, P.; Schmidhuber, J. Gradient Flow in Recurrent Nets: The Difficulty of Learning LongTerm Dependencies. In A Field Guide to Dynamical Recurrent Networks; Kolen, J.F., Kremer, S.C., Eds.; IEEE Press: New York, NY, USA; IEEE Xplore: Piscataway, NJ, USA, 2001; pp. 237–243. [Google Scholar] [CrossRef] [Green Version]
- Cacho, A.; Smirnova, E.; Huzurbazar, S.; Cui, X. A Comparison of Base-calling Algorithms for Illumina Sequencing Technology. Briefings Bioinform. 2016, 17, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Cao, M.D.; Hall, M.B.; Duarte, T.; Wang, S.; Coin, L.J.M. Chiron: Translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience 2018, 7. [Google Scholar] [CrossRef]
- Boža, V.; Brejová, B.; Vinař, T. DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Abe, T.; Kanaya, S.; Kinouchi, M.; Ichiba, Y.; Kozuki, T.; Ikemura, T. Informatics for unveiling hidden genome signatures. Genome Res. 2003, 13, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Padovani de Souza, K.; Setubal, J.C.; Ponce de Leon F de Carvalho, A.C.; Oliveira, G.; Chateau, A.; Alves, R. Machine learning meets genome assembly. Briefings Bioinform. 2019, 20, 2116–2129. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vervier, K.; Mahé, P.; Tournoud, M.; Veyrieras, J.B.; Vert, J.P. Large-scale machine learning for metagenomics sequence classification. Bioinformatics 2016, 32, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Tech, M.; Lingner, T.; Daniel, R.; Morgenstern, B.; Meinicke, P. Gene prediction in metagenomic fragments: A large scale machine learning approach. BMC Bioinform. 2008, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.W.; Jin, X.Y.; Zhang, T. Gene Prediction in Metagenomic Fragments with Deep Learning. BioMed Res. Int. 2017, 2017, 4740354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassan, J.T.; Wang, H.; Browne, F.; Zheng, H. A Comprehensive Study on Predicting Functional Role of Metagenomes Using Machine Learning Methods. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In KDD2016; Krishnapuram, B., Shah, M., Smola, A., Aggarwal, C., Shen, D., Rastogi, R., Eds.; Association for Computing Machinery Inc. (ACM): New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Walsh, P.; Andrade, B.G.N.; Palu, C.; Wu, J.; Lawlor, B.; Kelly, B.; Hemmje, M.L.; Kramer, M. ImmunoAdept—bringing blood microbiome profiling to the clinical practice. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine, Madrid, Spain, 3–6 December 2018; Zheng, H., Ed.; IEEE: Piscataway, NJ, USA, 2018; pp. 1577–1581. [Google Scholar] [CrossRef]
- Wang, H.; Pujos-Guillot, E.; Comte, B.; de Miranda, J.L.; Spiwok, V.; Chorbev, I.; Castiglione, F.; Tieri, P.; Watterson, S.; McAllister, R.; et al. Deep learning in Systems Medicine. Briefings Bioinform. 2020, 22, 1543–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, B.; He, T.; Li, G.; Jiang, X. Robust biomarker discovery for microbiome-wide association studies. Methods 2020, 173, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Feng, J. Deep Forest: Towards An Alternative to Deep Neural Networks. In Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence, Melbourne, Australia, 19–25 August 2017; Sierra, C., Ed.; 2017; pp. 3553–3559. [Google Scholar]
- Sardaraz, M.; Tahir, M.; Ikram, A.A.; Bajwa, H. Applications and Algorithms for Inference of Huge Phylogenetic Trees: A Review. Am. J. Bioinform. Res. 2012, 2, 21–26. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Louis, S.; Tappu, R.M.; Damms-Machado, A.; Huson, D.H.; Bischoff, S.C. Characterization of the Gut Microbial Community of Obese Patients Following a Weight-Loss Intervention Using Whole Metagenome Shotgun Sequencing. PLoS ONE 2016, 11, e0149564. [Google Scholar] [CrossRef]
- Laczny, C.C.; Sternal, T.; Plugaru, V.; Gawron, P.; Atashpendar, A.; Margossian, H.H.; Coronado, S.; van der Maaten, L.; Vlassis, N.; Wilmes, P. VizBin—An application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 2015, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zela, A.; Klein, A.; Falkner, S.; Hutter, F. Towards Automated Deep Learning: Efficient Joint Neural Architecture and Hyperparameter Search. In Proceedings of the ICML 2018 AutoML Workshop, Stockholm, Sweden, 10–15 July 2018. [Google Scholar]
- Hamon, R.; Junklewitz, H.; Sanchez, I. Robustness and Explainability of Artificial Intelligence: From Technical to Policy Solutions; Publications Office of the European Union: Luxembourg, 2020; Volume 30040. [Google Scholar]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef] [Green Version]
- London, A.J. Artificial Intelligence and Black-Box Medical Decisions: Accuracy versus Explainability. Hastings Cent. Rep. 2019, 49, 15–21. [Google Scholar] [CrossRef]
- Eck, S.H. Challenges in data storage and data management in a clinical diagnostic setting. LaboratoriumsMedizin 2018, 42, 219–224. [Google Scholar] [CrossRef]
- Kothari, R.K.; Nathani, N.M.; Mootapally, C.; Rank, J.K.; Gosai, H.B.; Dave, B.P.; Joshi, C.G. Comprehensive Exploration of the Rumen Microbial Ecosystem with Advancements in Metagenomics. In Metagenomics; Nagarajan, M., Ed.; Academic Press: London, UK, 2018; pp. 215–229. [Google Scholar] [CrossRef]
- Zhu, X.; Vondrick, C.; Fowlkes, C.; Ramanan, D. Do We Need More Training Data? Int. J. Comput. Vis. 2016, 119, 76–92. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.W.; Lin, X. Big Data Deep Learning: Challenges and Perspectives. IEEE Access 2014, 2, 514–525. [Google Scholar] [CrossRef]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I. Language Models are Unsupervised Multitask Learners. 2019. Available online: https://openai.com/blog/better-language-models/ (accessed on 4 November 2021).
- Shoeybi, M.; Patwary, M.; Puri, R.; LeGresley, P.; Casper, J.; Catanzaro, B. Megatron-LM: Training Multi-Billion Parameter Language Models Using Model Parallelism. arXiv 2019, arXiv:1909.08053. [Google Scholar]
- Pan, S.J.; Yang, Q. A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. [Google Scholar] [CrossRef]
- Abawajy, J. Comprehensive analysis of big data variety landscape. Int. J. Parallel Emergent Distrib. Syst. 2015, 30, 5–14. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.; Wadhawan, S.; Chiaromonte, F.; Ananda, G.; Chung, W.Y.; Taylor, J.; Nekrutenko, A. Windshield splatter analysis with the Galaxy metagenomic pipeline. Genome Res. 2009, 19, 2144–2153. [Google Scholar] [CrossRef] [Green Version]
- Batut, B.; Gravouil, K.; Defois, C.; Hiltemann, S.; Brugère, J.F.; Peyretaillade, E.; Peyret, P. ASaiM: A Galaxy-based framework to analyze raw shotgun data from microbiota. bioRxiv 2017, 183970. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://github.com/fchollet/keras (accessed on 1 November 2021).
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2020, 48, D570–D578. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Dillon, M.R.; Bolyen, E.; Kaehler, B.D.; Huttley, G.A.; Caporaso, J.G. q2-sample-classifier: Machine-learning tools for microbiome classification and regression. bioRxiv 2018, 306167. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidou, N.; Walsh, P.; Lu, X.; Bekaert, M.; Lawlor, B.; Kelly, B.; Zheng, H.; Browne, F.; Dewhurst, R.J.; Roehe, R.; et al. MetaPlat: A Cloud based Platform for Analysis and Visualisation of Metagenomics Data. In Proceedings of the Collaborative European Research Conference (CERC 2016), Cork, Ireland, 23–24 September 2016; Bleimann, U., Humm, B., Loew, R., Stengel, I., Walsh, P., Eds.; 2016. [Google Scholar]
- Wassan, J.T.; Zheng, H.; Browne, F.; Bowen, J.; Walsh, P.; Roehe, R.; Dewhurst, R.J.; Palu, C.; Kelly, B.; Wang, H. An Integrative Framework for Functional Analysis of Cattle Rumen Microbiomes. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 1854–1860. [Google Scholar] [CrossRef]
- Walsh, P.; Carroll, J.; Sleator, R.D. Accelerating in silico research with workflows: A lesson in Simplicity. Comput. Biol. Med. 2013, 43, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Reis, T.; Krause, T.; Bornschlegl, M.X.; Hemmje, M.L. A Conceptual Architecture for AI-based Big Data Analysis and Visualization Supporting Metagenomics Research. In Proceedings of the Collaborative European Research Conference (CERC 2020), Belfast, Northern-Ireland, UK, 10–11 September 2020. [Google Scholar]
- Dijkstra, E.W. On the Role of Scientific Thought. In Selected Writings on Computing: A personal Perspective; Dijkstra, E.W., Ed.; Texts and Monographs in Computer Science; Springer: New York, NY, USA, 1982; pp. 60–66. [Google Scholar] [CrossRef]
- Fowler, M.; Rice, D. Patterns of Enterprise Application Architecture; The Addison-Wesley Signature Series; Addison-Wesley: Boston, MA, USA, 2002. [Google Scholar]
- Schmatz, K.D. Konzeption, Implementierung und Evaluierung einer Datenbasierten Schnittstelle für Heterogene Quellsysteme Basierend auf der Mediator-Wrapper-Architektur Innerhalb Eines Hadoop-Ökosystems. Ph.D. Thesis, Fernuniversität Hagen, Hagen, Germany, 2018. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krause, T.; Wassan, J.T.; Mc Kevitt, P.; Wang, H.; Zheng, H.; Hemmje, M. Analyzing Large Microbiome Datasets Using Machine Learning and Big Data. BioMedInformatics 2021, 1, 138-165. https://doi.org/10.3390/biomedinformatics1030010
Krause T, Wassan JT, Mc Kevitt P, Wang H, Zheng H, Hemmje M. Analyzing Large Microbiome Datasets Using Machine Learning and Big Data. BioMedInformatics. 2021; 1(3):138-165. https://doi.org/10.3390/biomedinformatics1030010
Chicago/Turabian StyleKrause, Thomas, Jyotsna Talreja Wassan, Paul Mc Kevitt, Haiying Wang, Huiru Zheng, and Matthias Hemmje. 2021. "Analyzing Large Microbiome Datasets Using Machine Learning and Big Data" BioMedInformatics 1, no. 3: 138-165. https://doi.org/10.3390/biomedinformatics1030010
APA StyleKrause, T., Wassan, J. T., Mc Kevitt, P., Wang, H., Zheng, H., & Hemmje, M. (2021). Analyzing Large Microbiome Datasets Using Machine Learning and Big Data. BioMedInformatics, 1(3), 138-165. https://doi.org/10.3390/biomedinformatics1030010