
The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor





Abstract
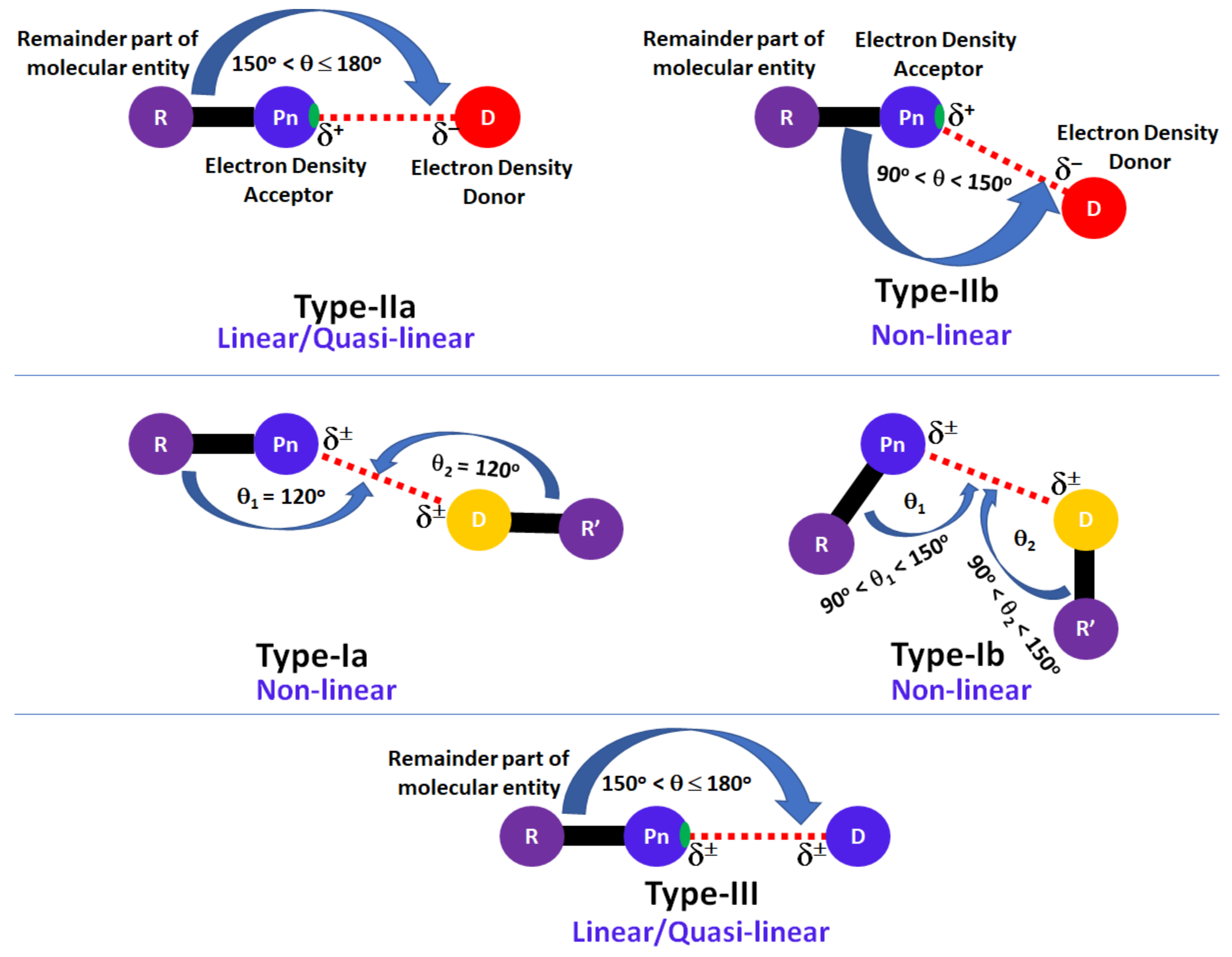
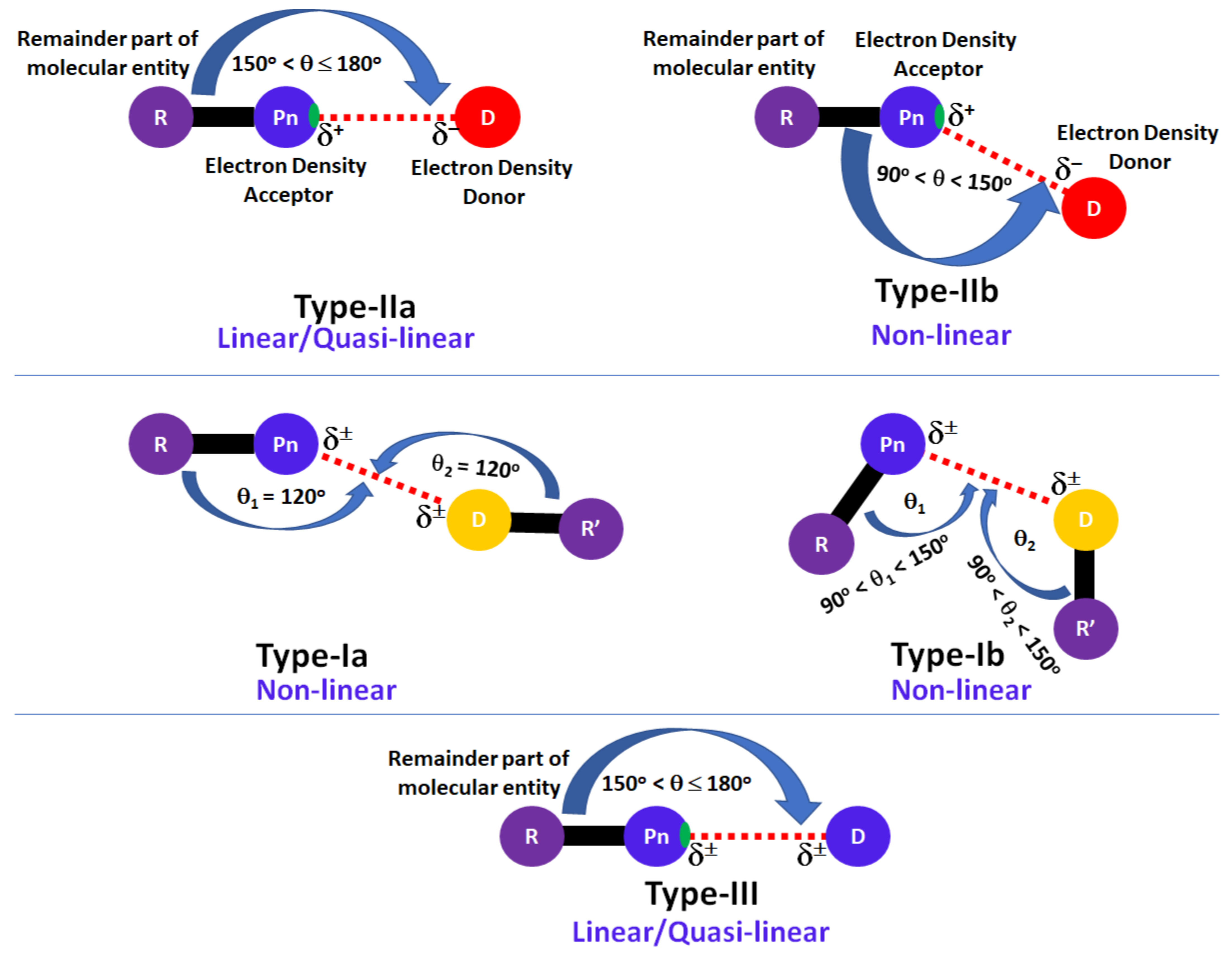
:1. Introduction
2. Computational Details
3. Illustrative Chemical Systems in the Crystalline Phase
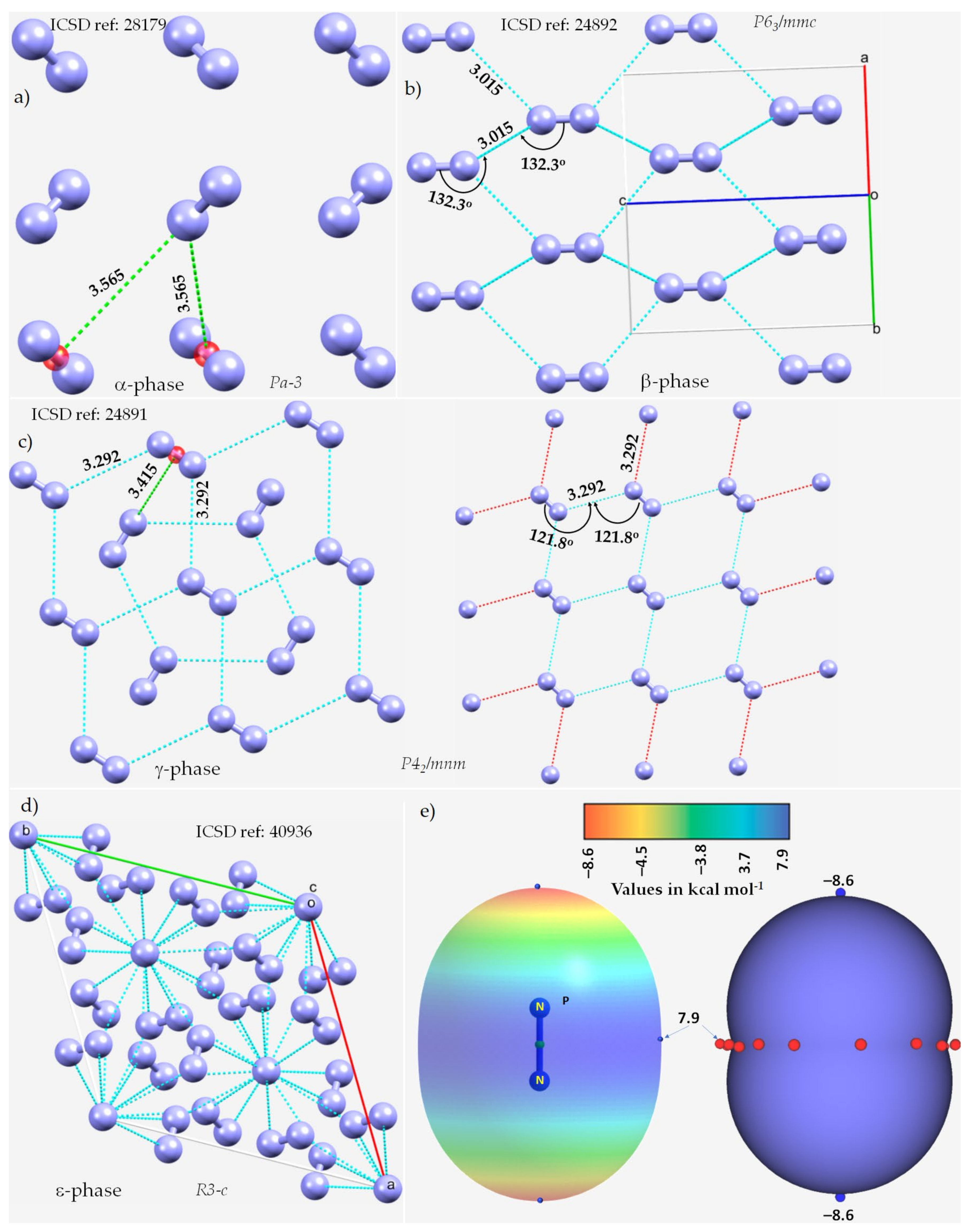
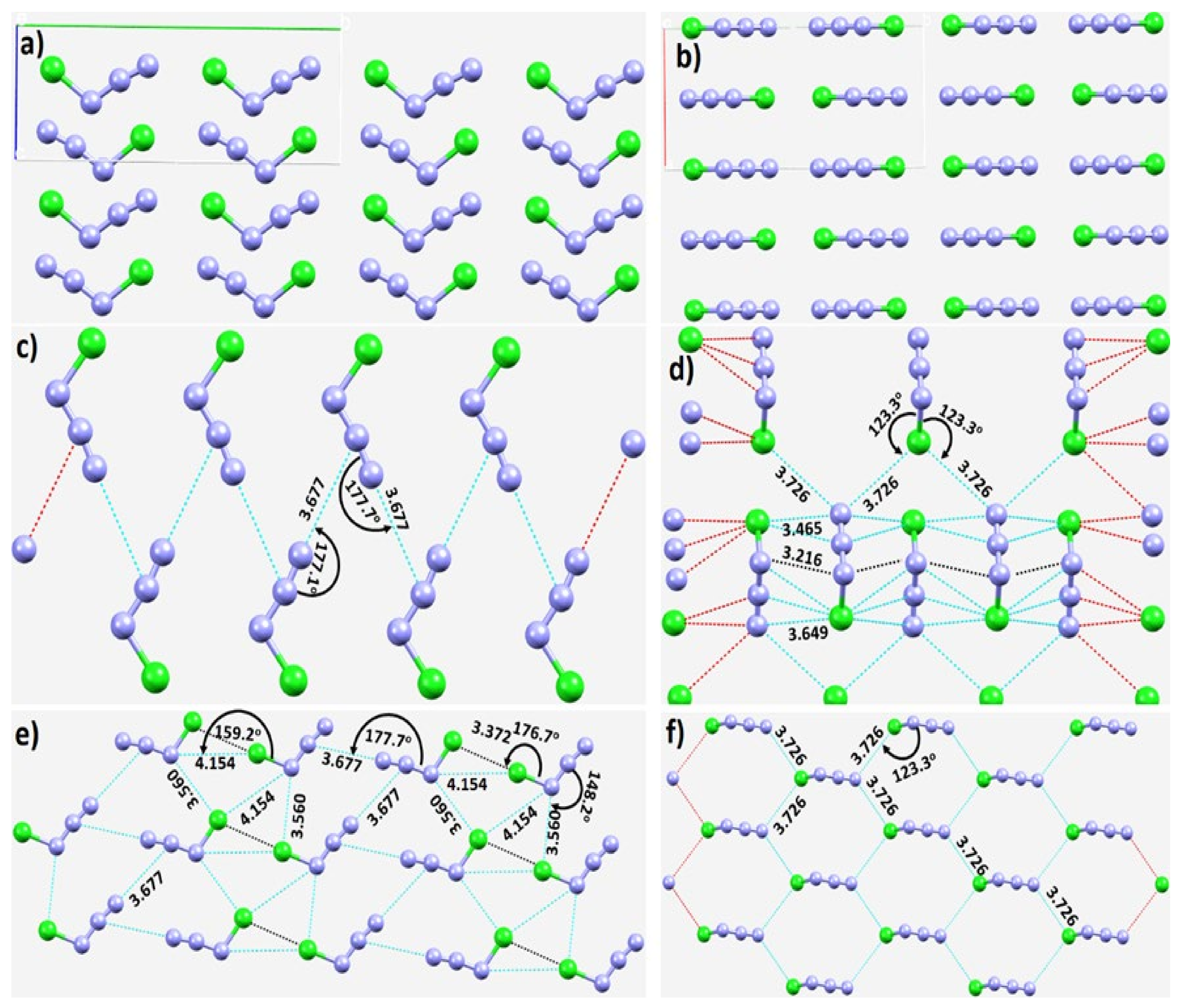
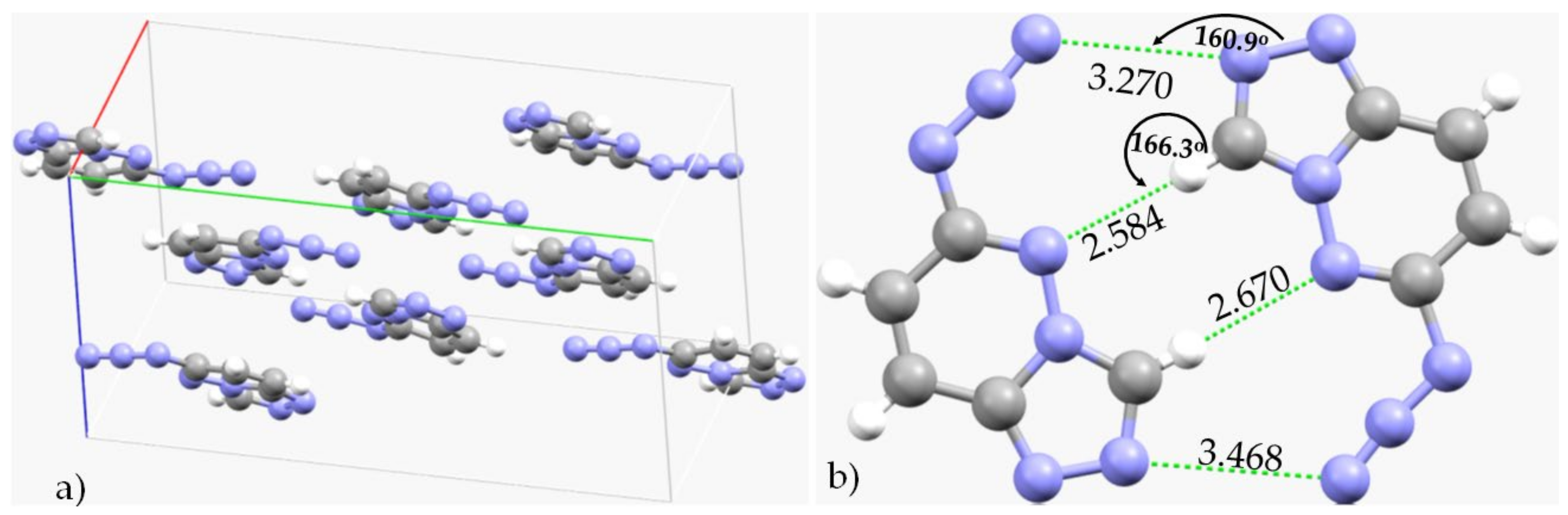
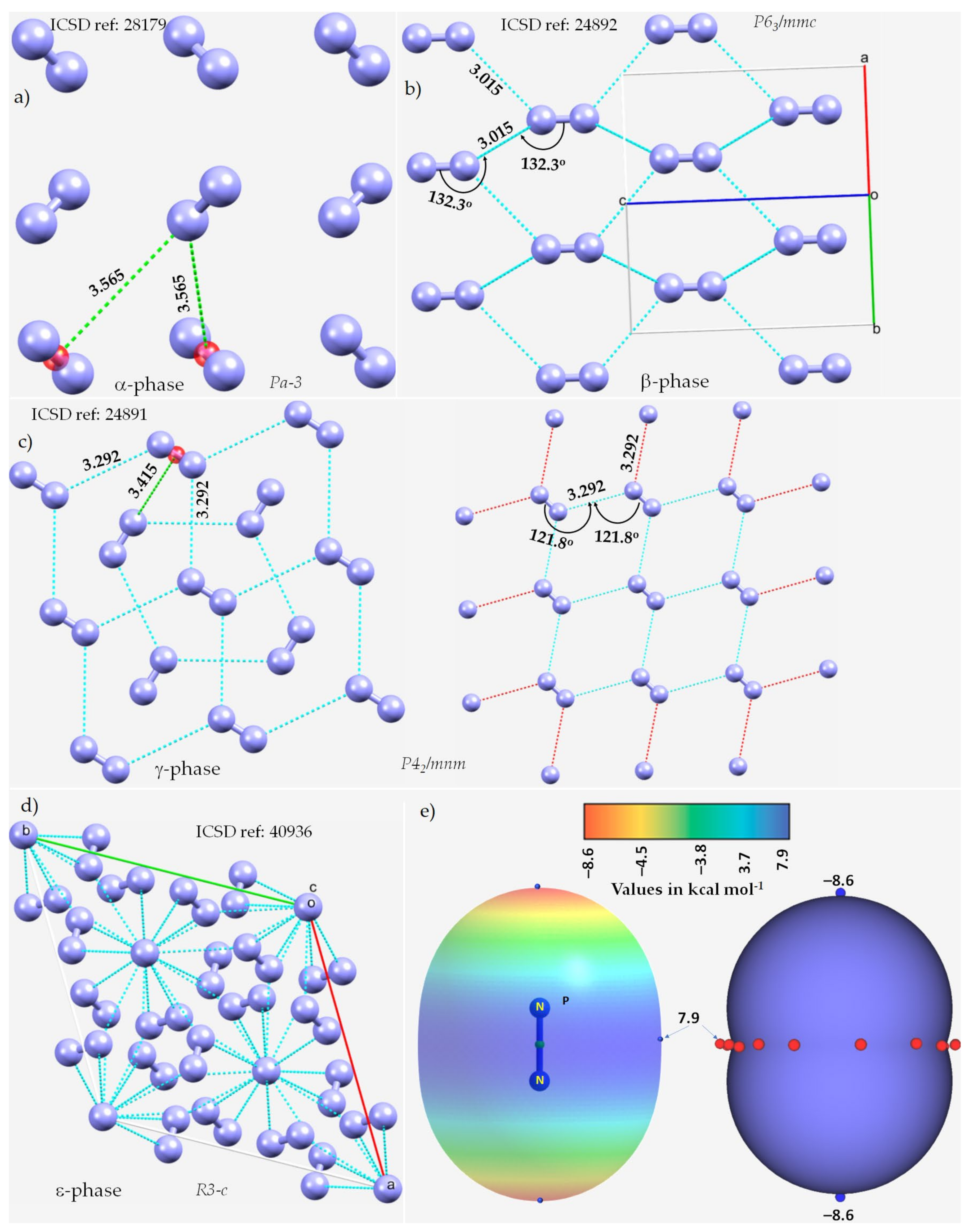
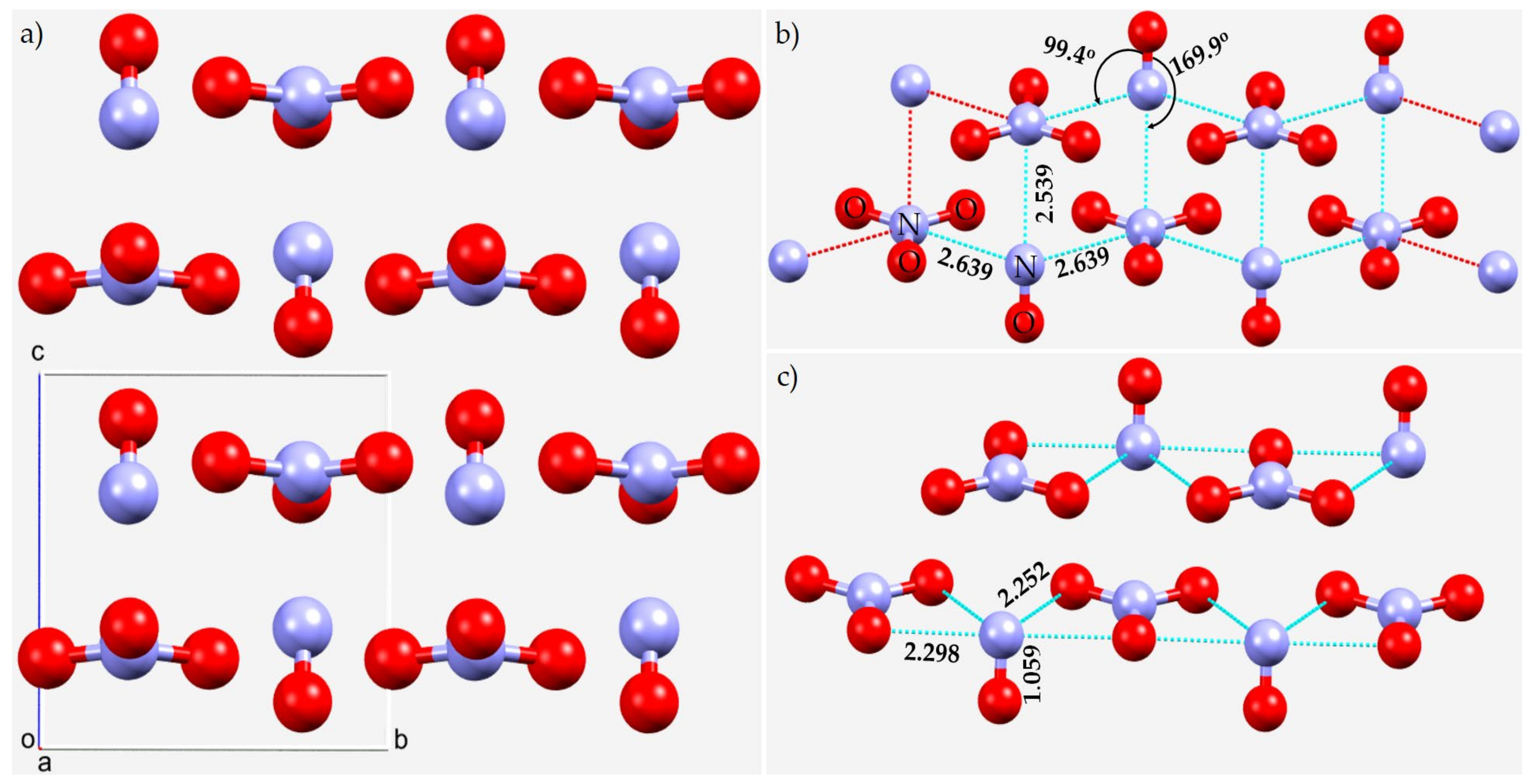
3.1. The Solid-State Structure of Dinitrogen, N2
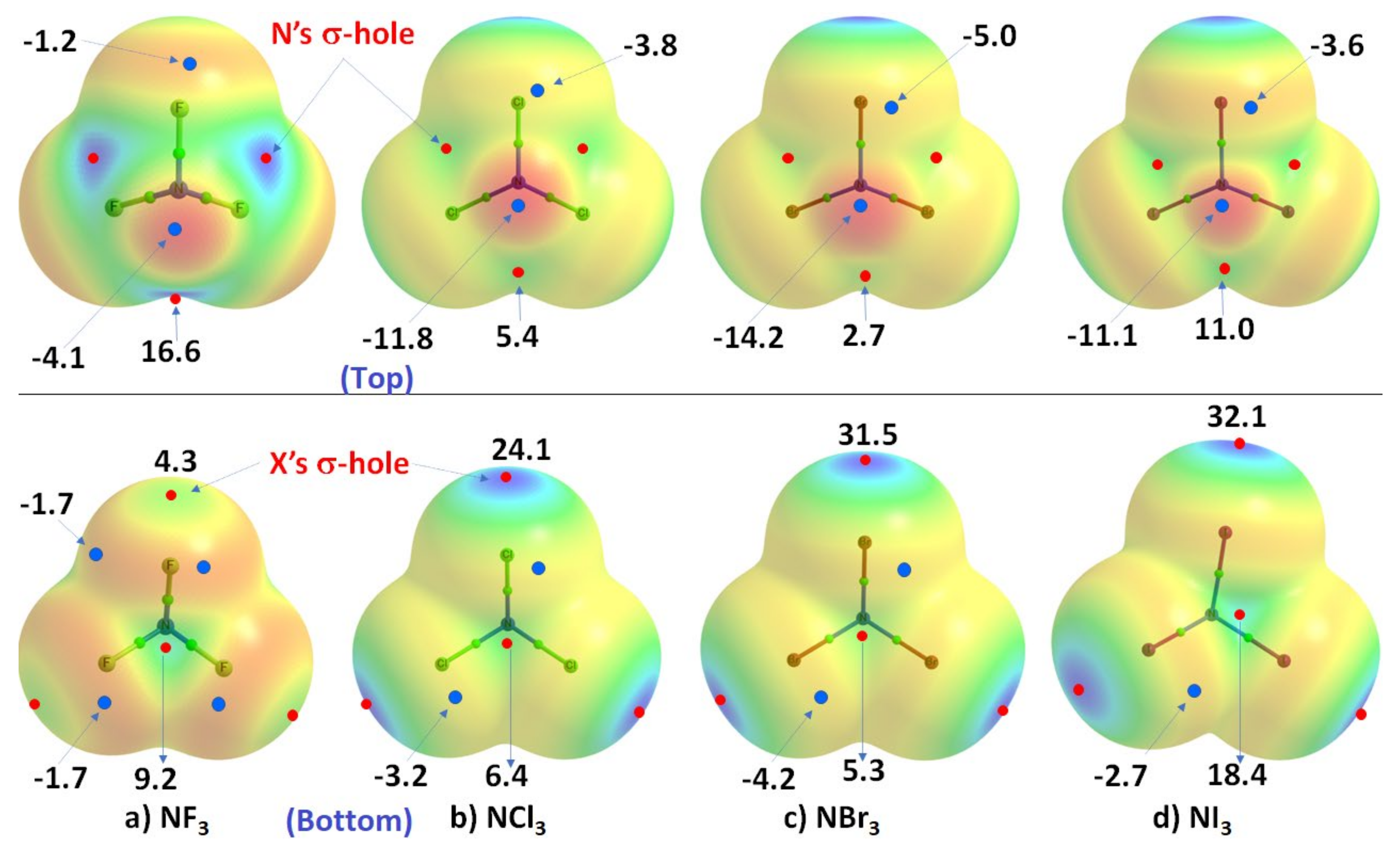
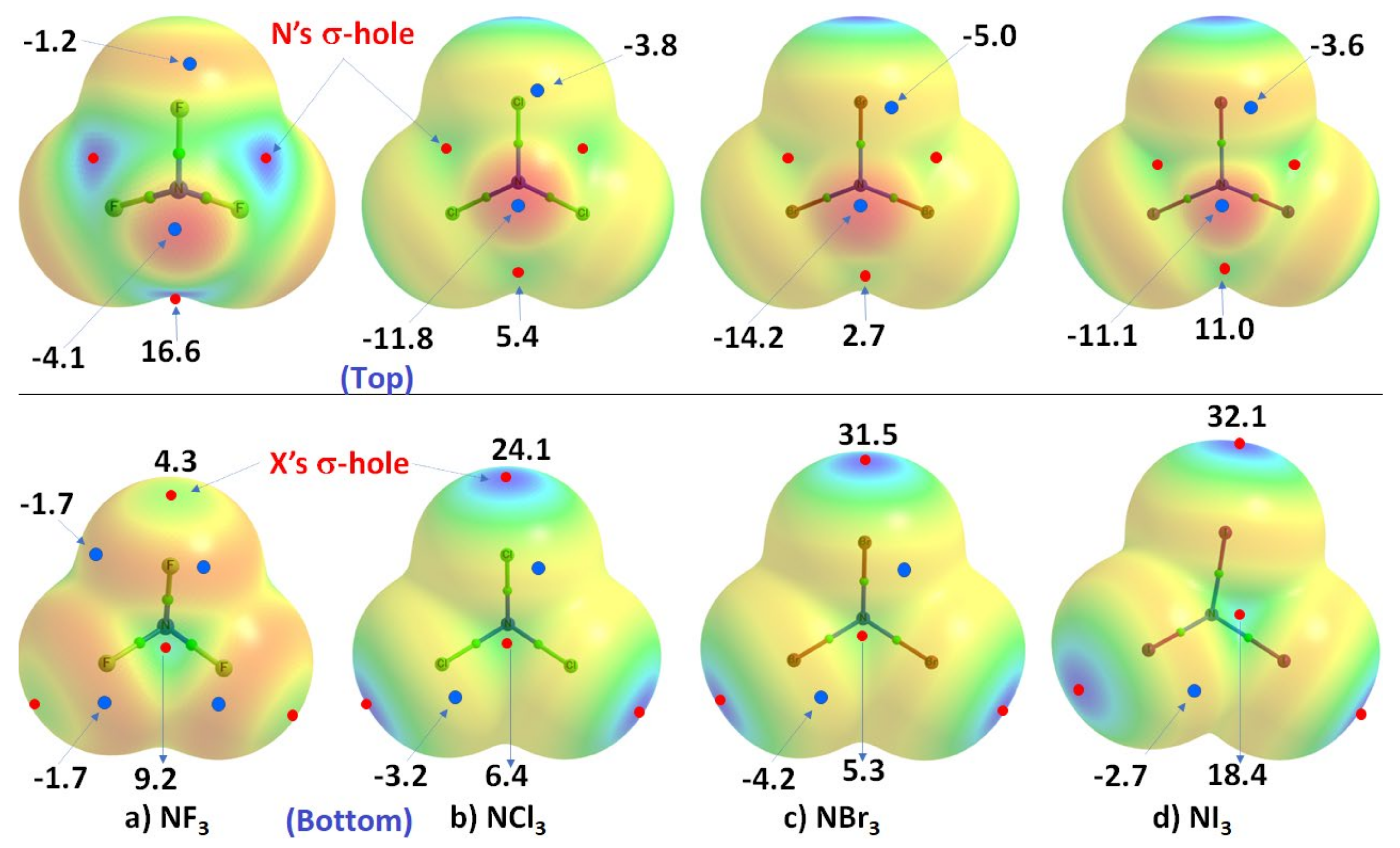
3.2. The Nitrogen Trihalides, NX3 and Their Crystal Structures
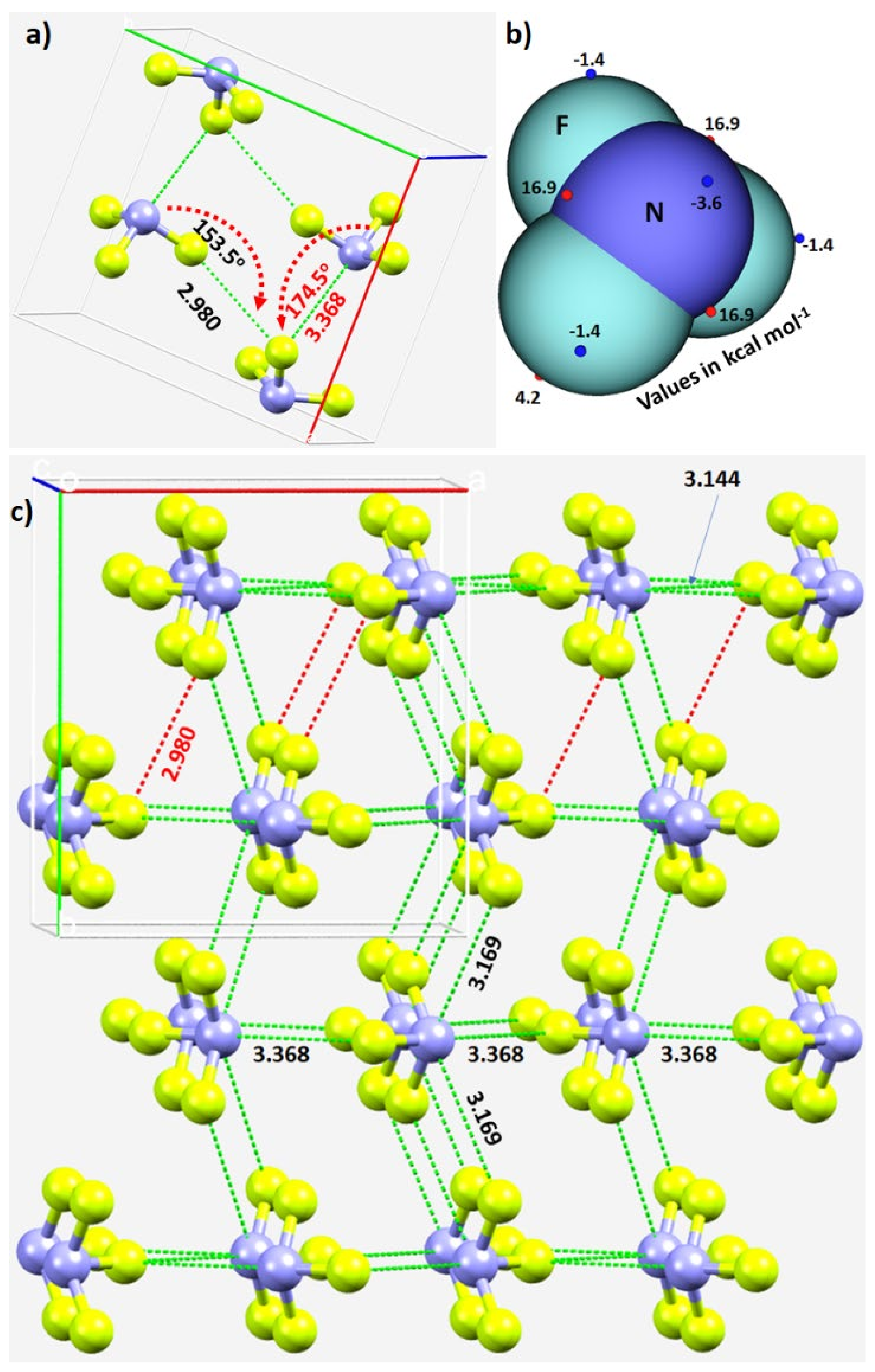
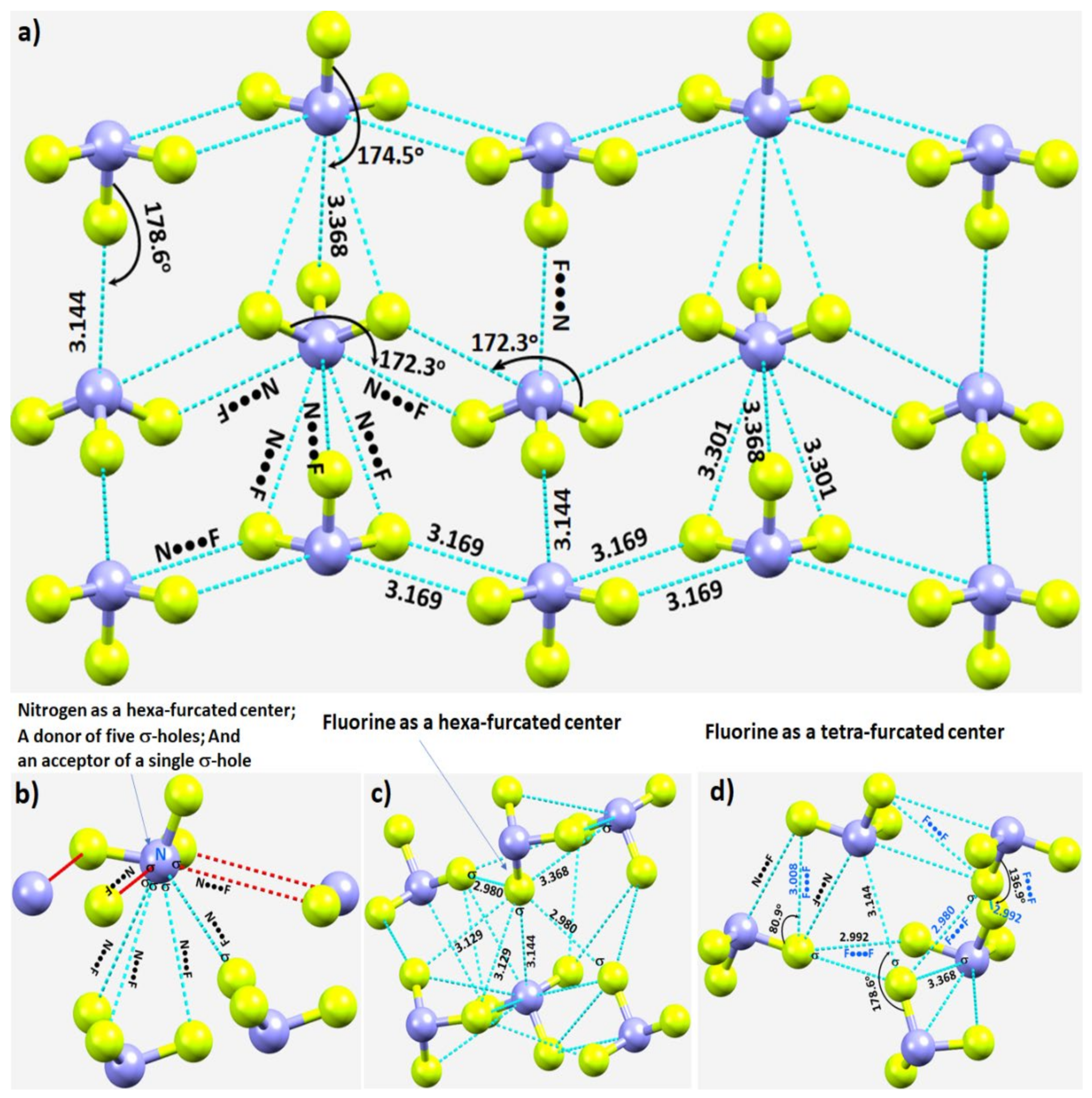
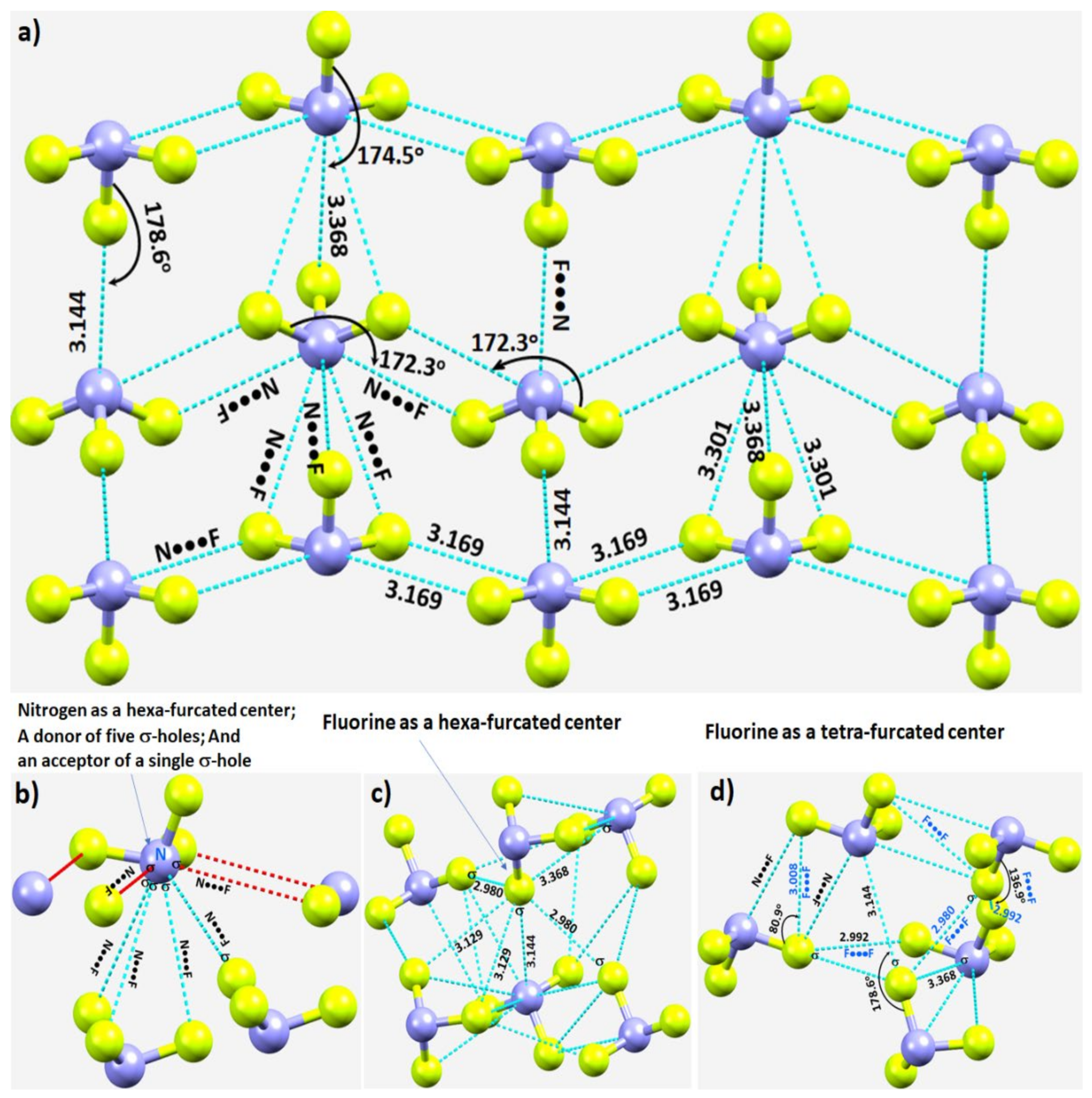
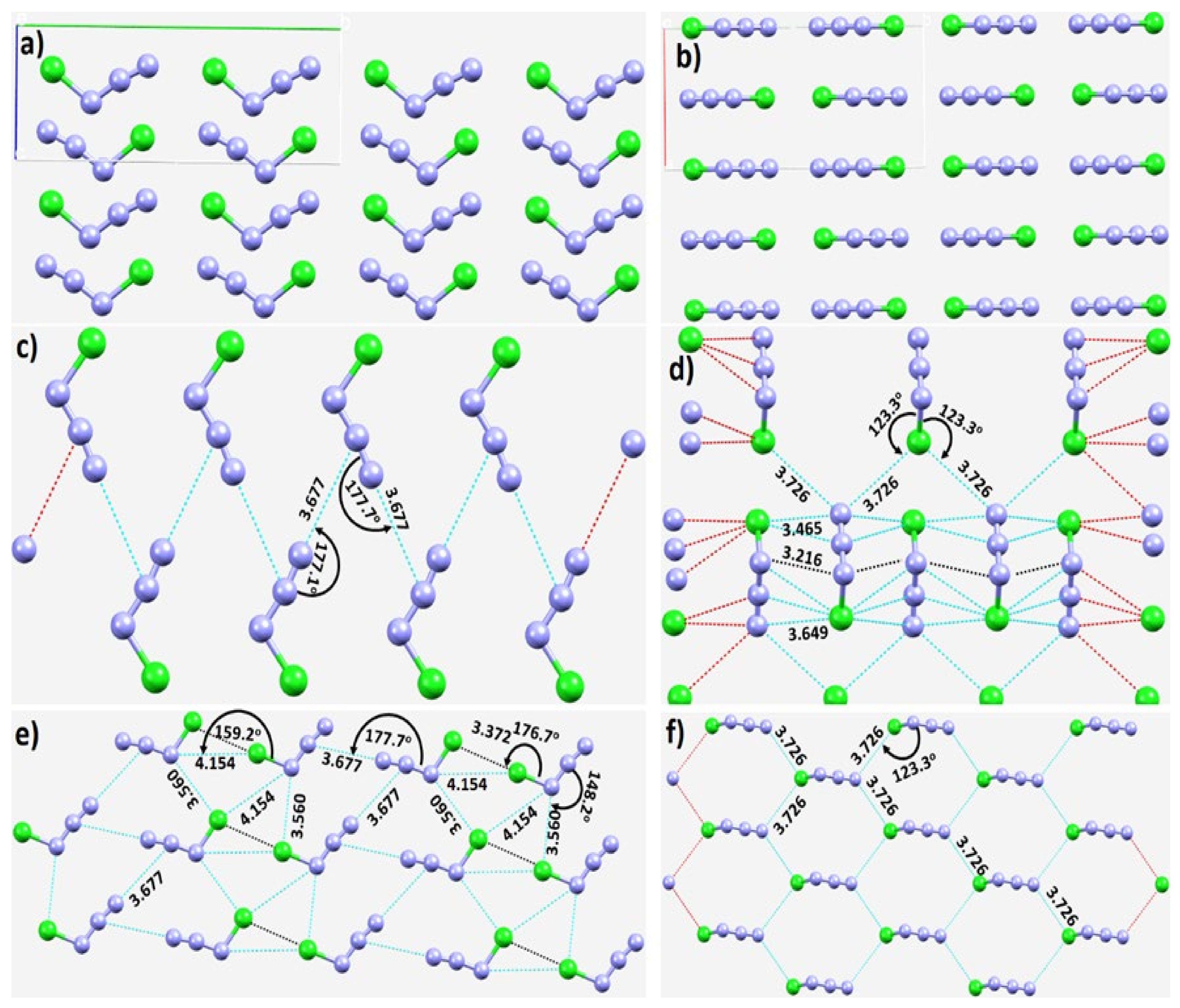
3.2.1. Nitrogen Trifluoride, NF3
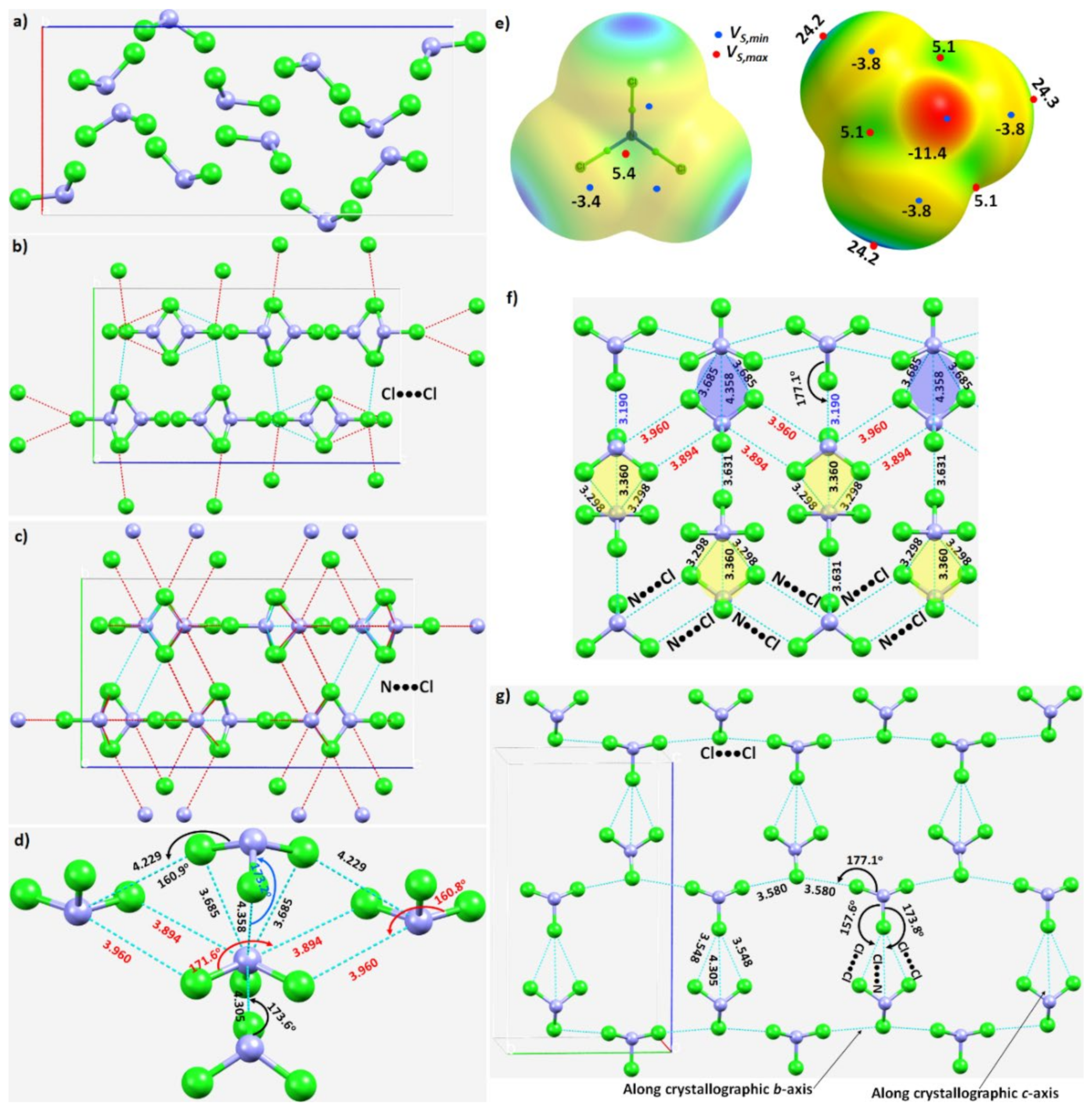
3.2.2. Nitrogen Trichloride, NCl3
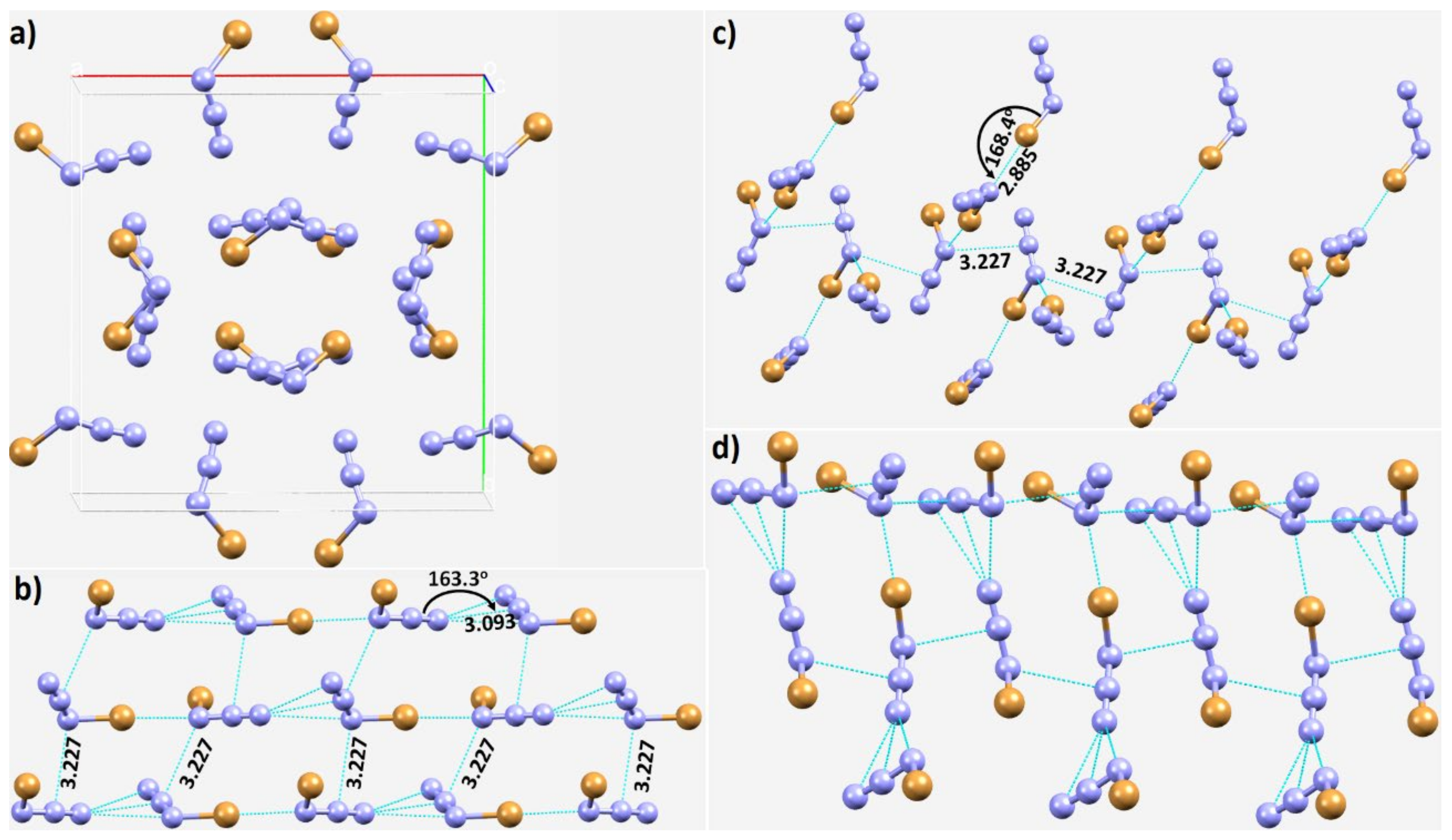
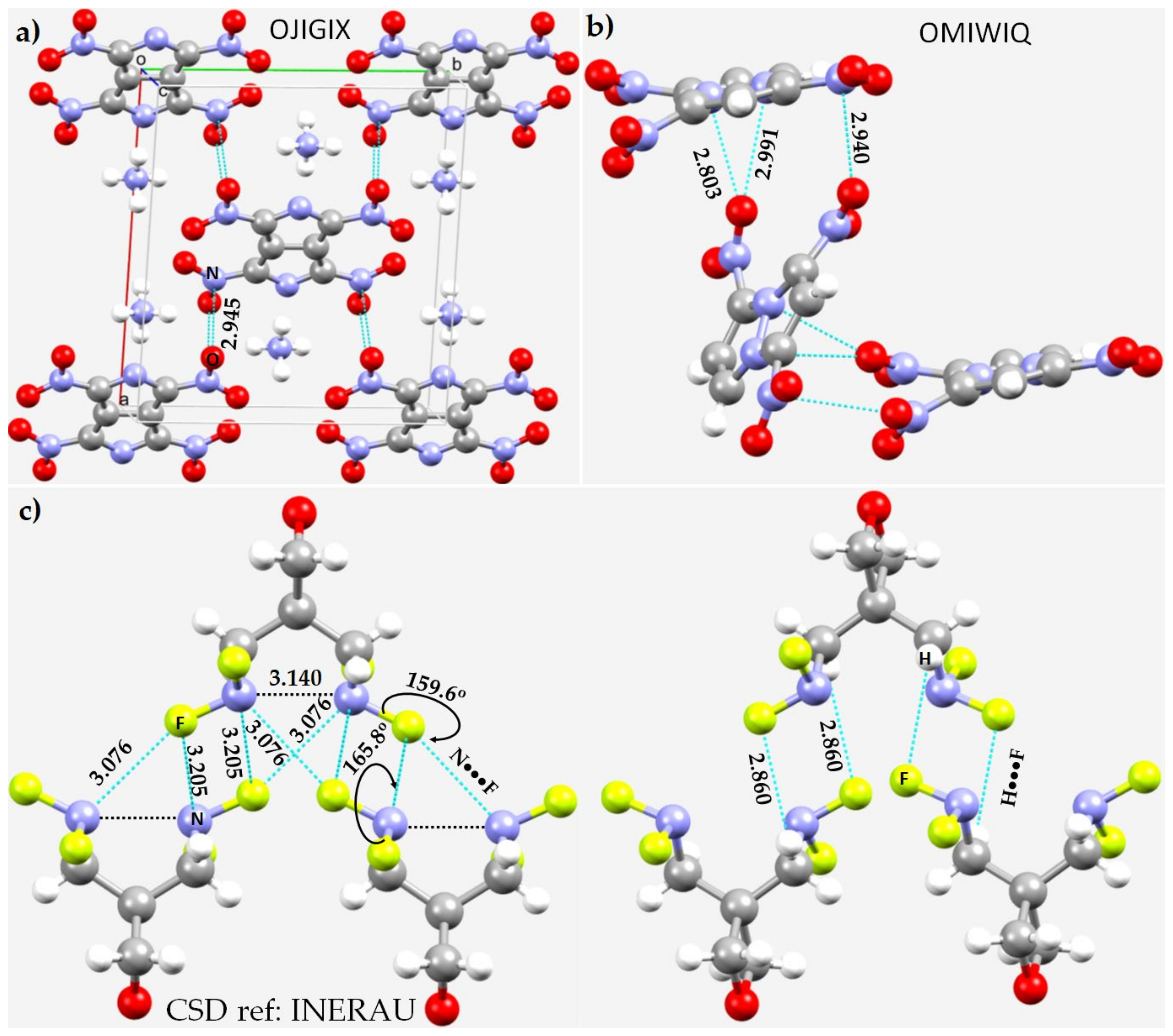
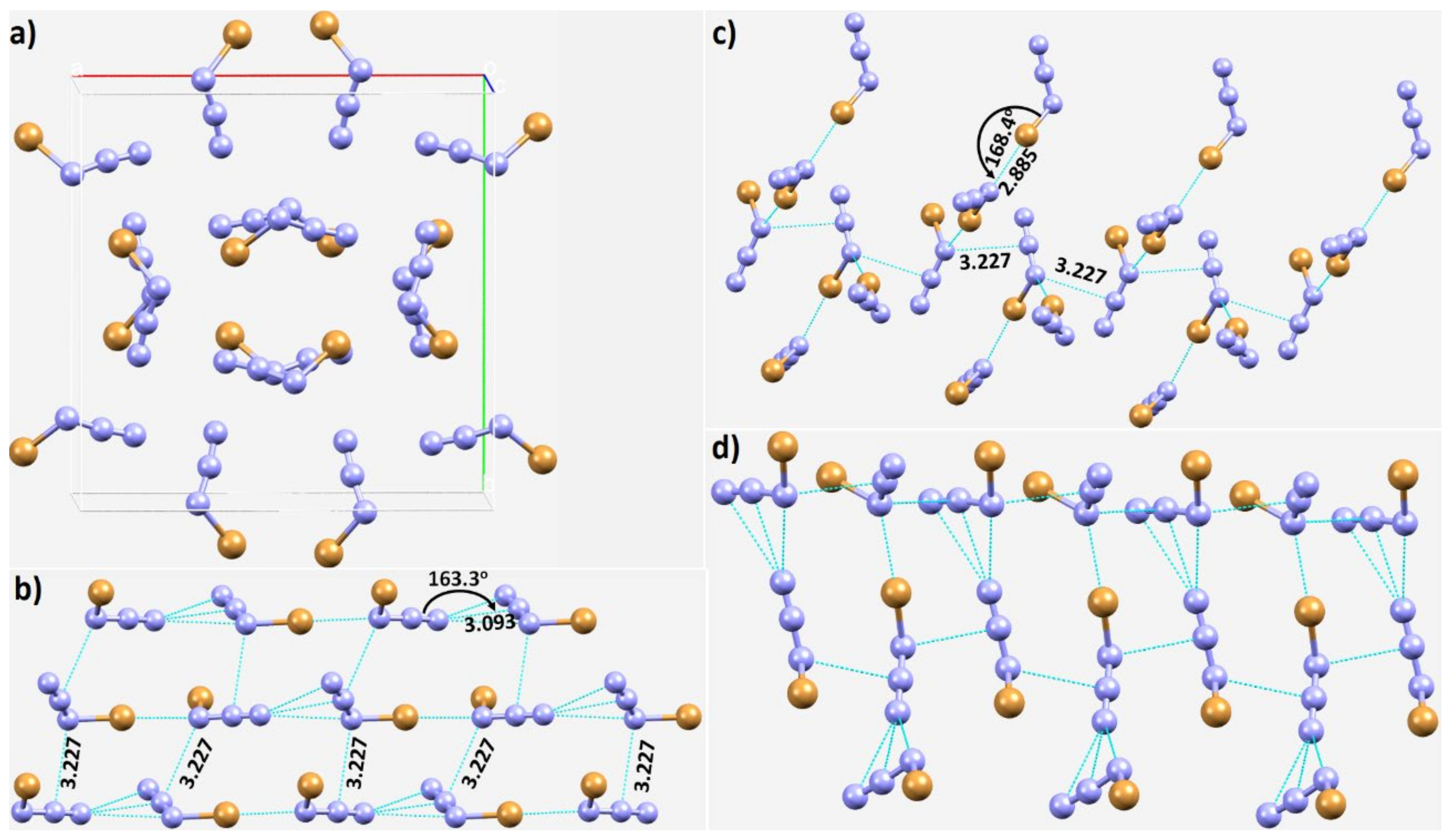
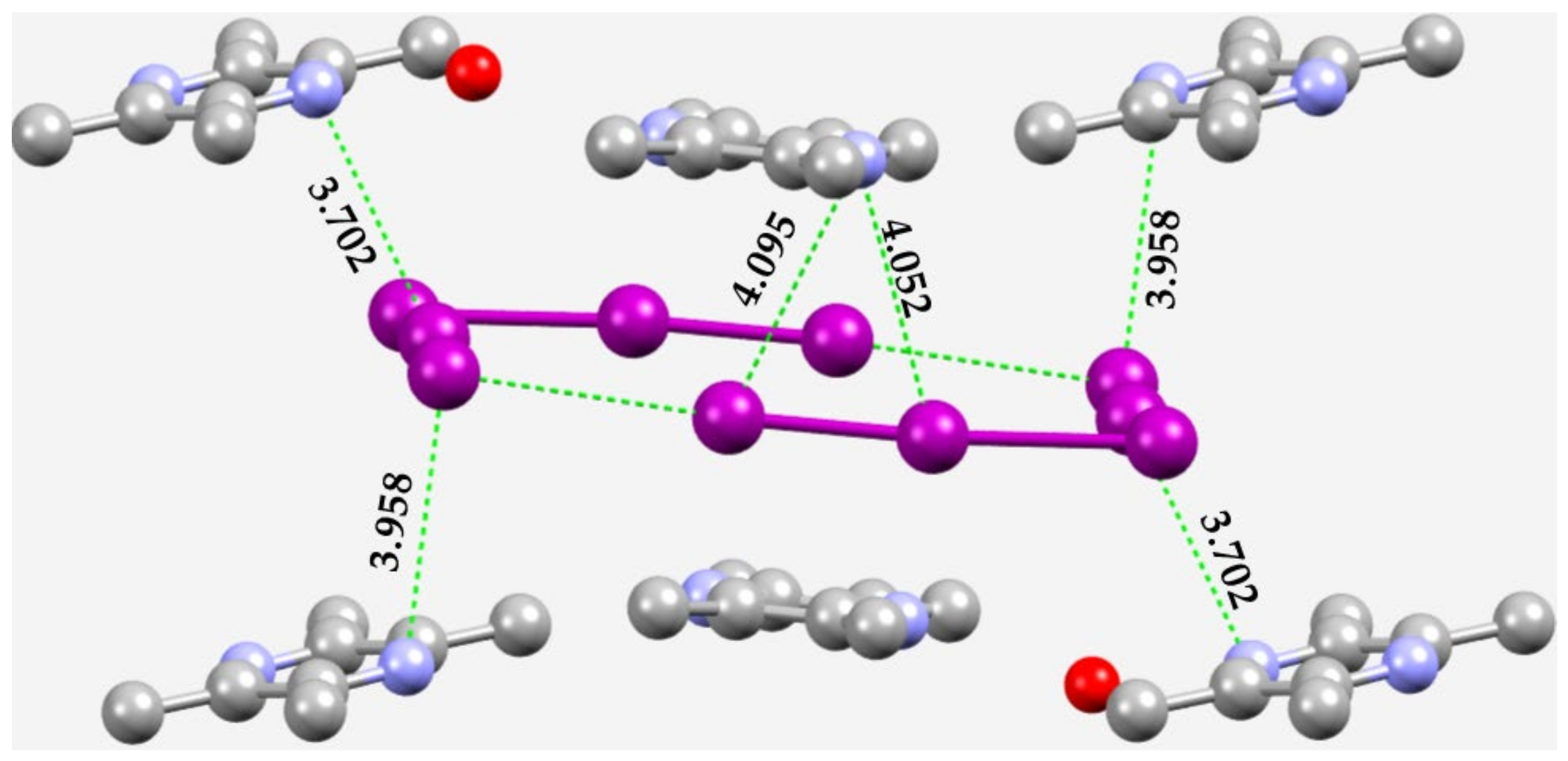
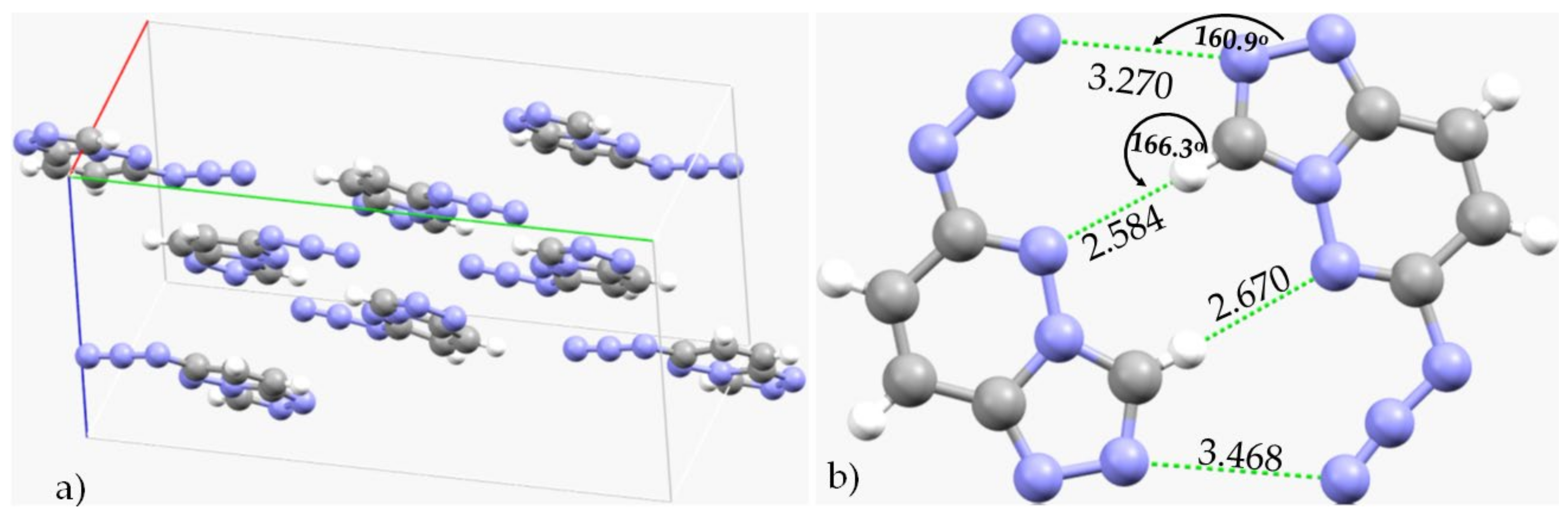
3.3. Halogen Azides, XN3
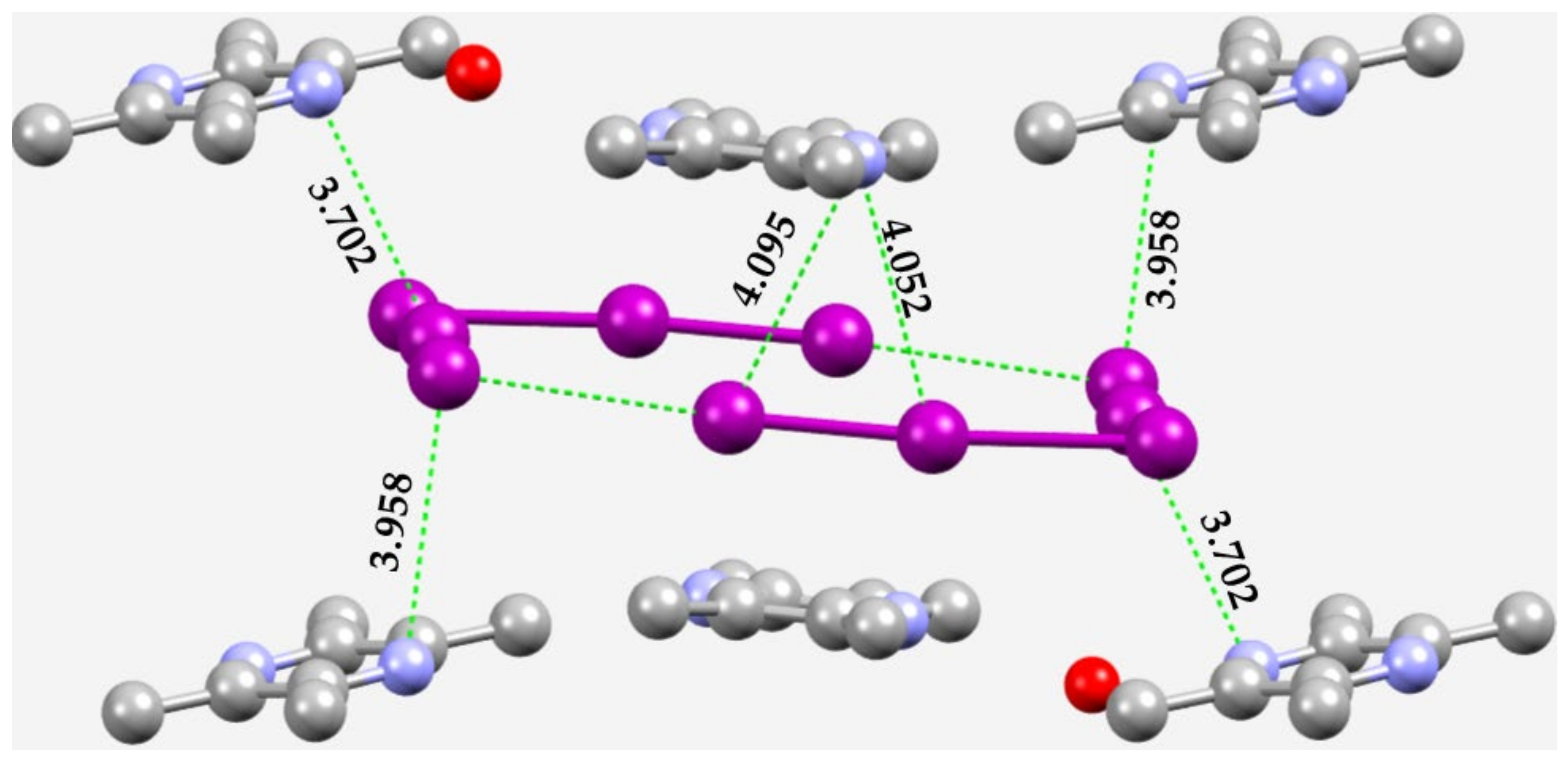
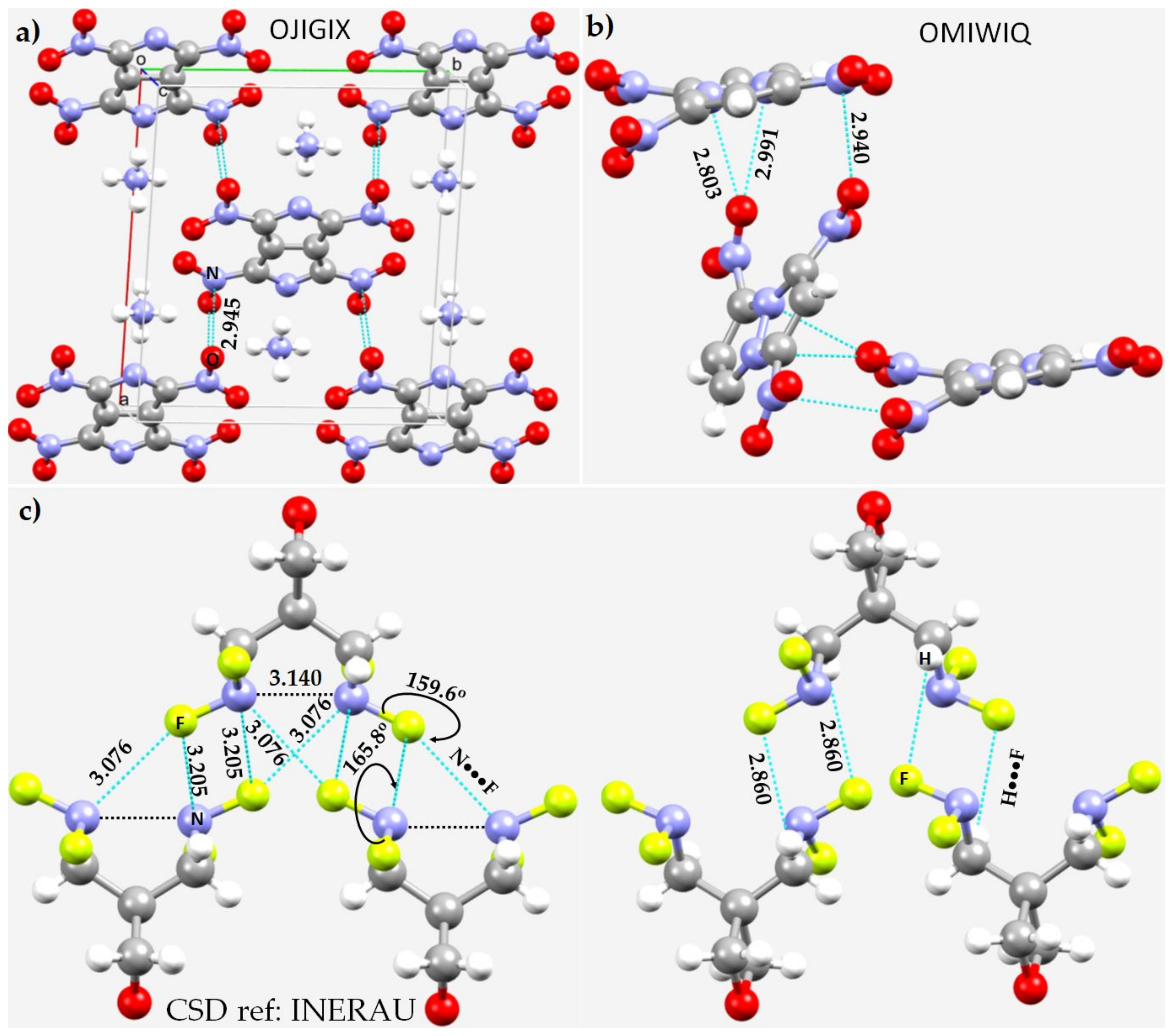
3.4. Other Azides
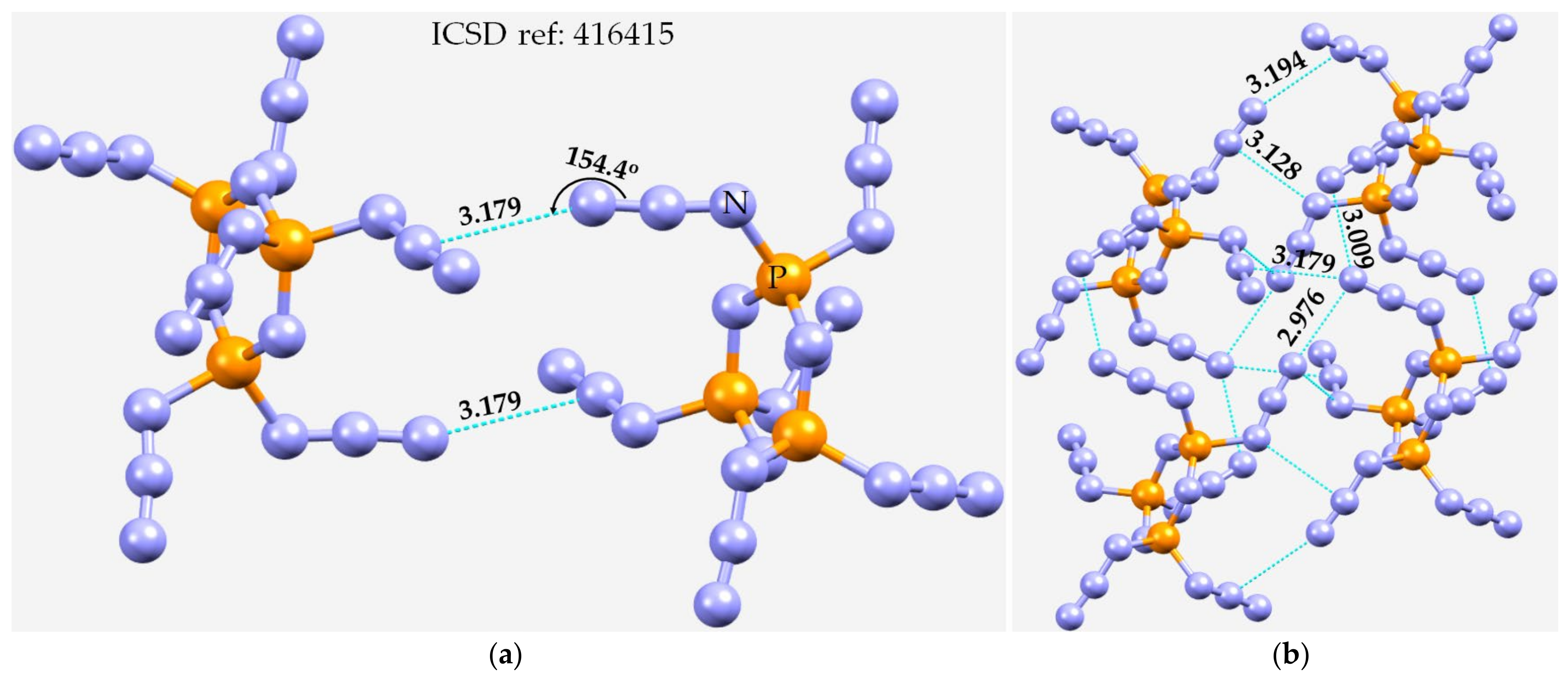
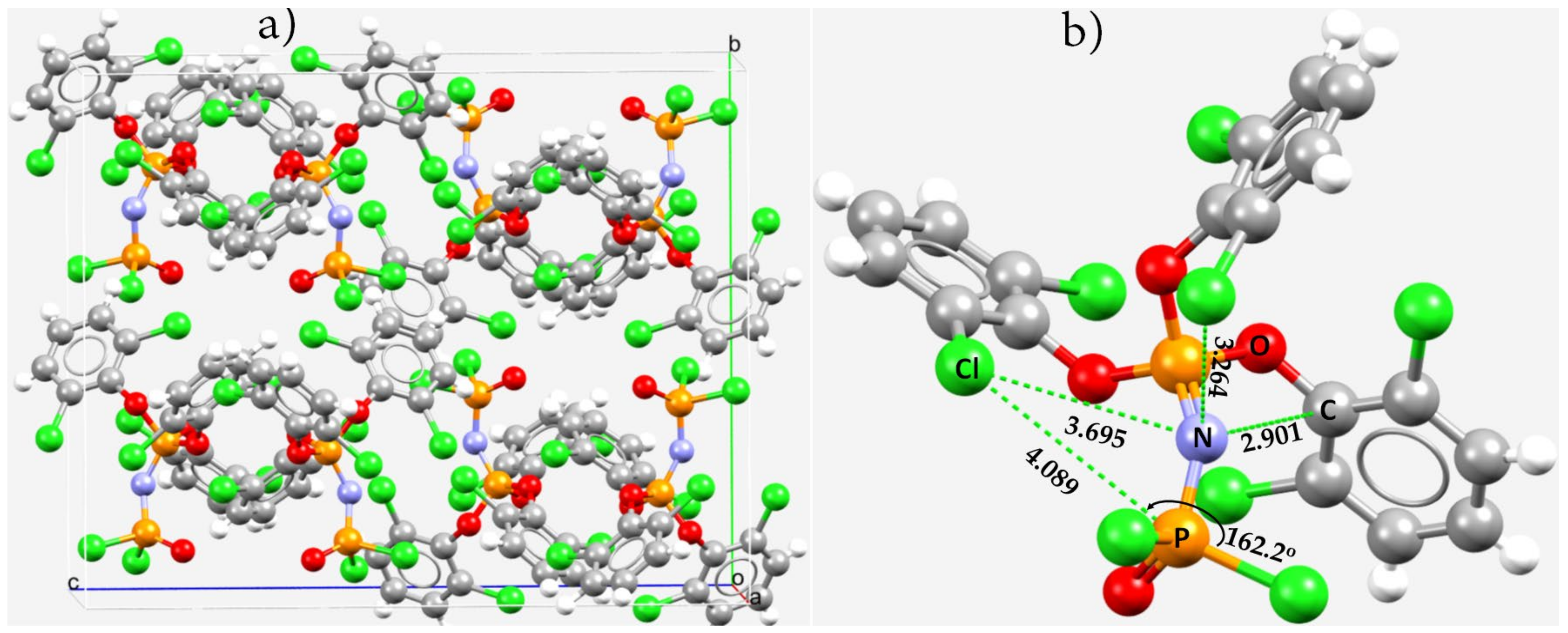
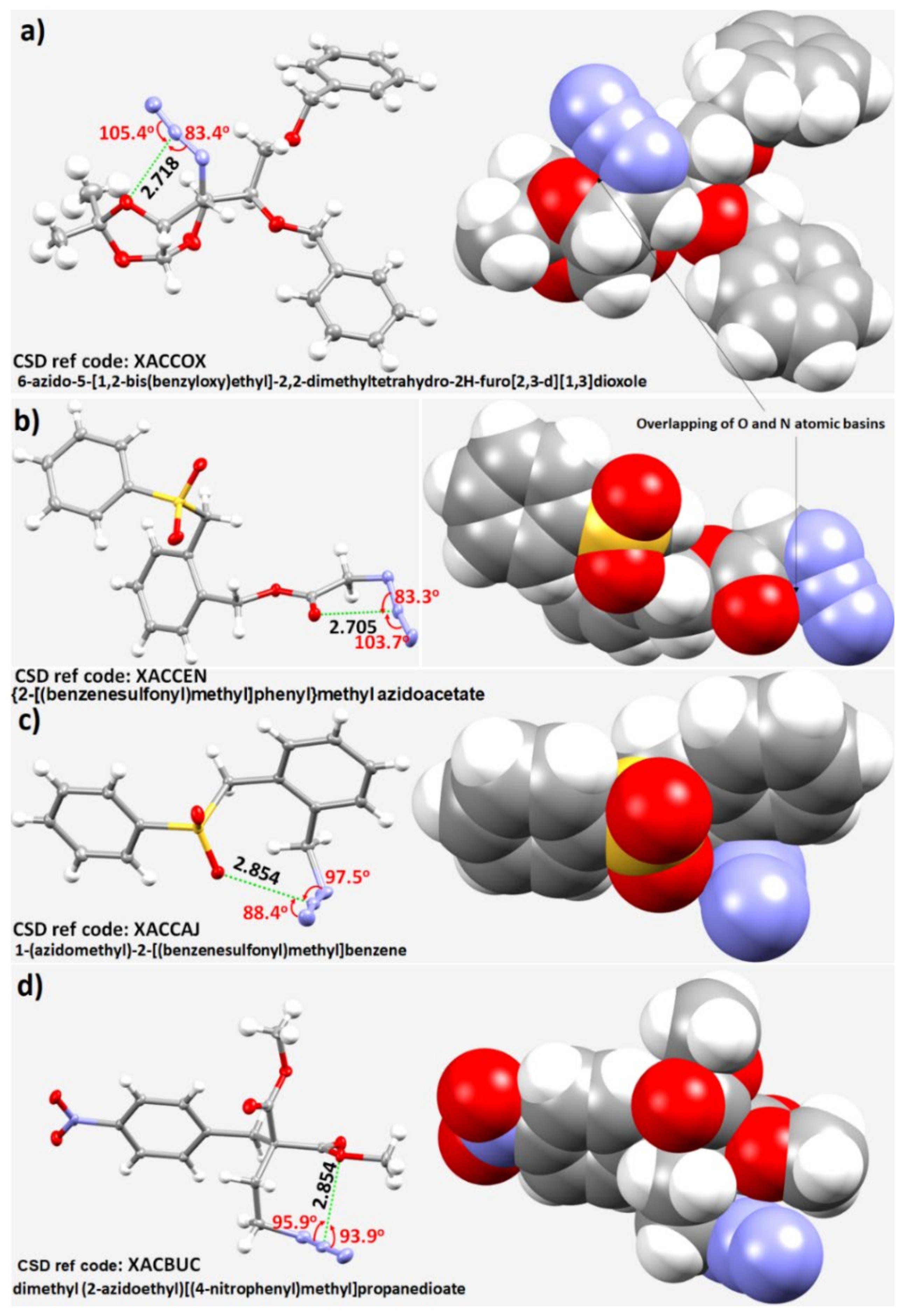
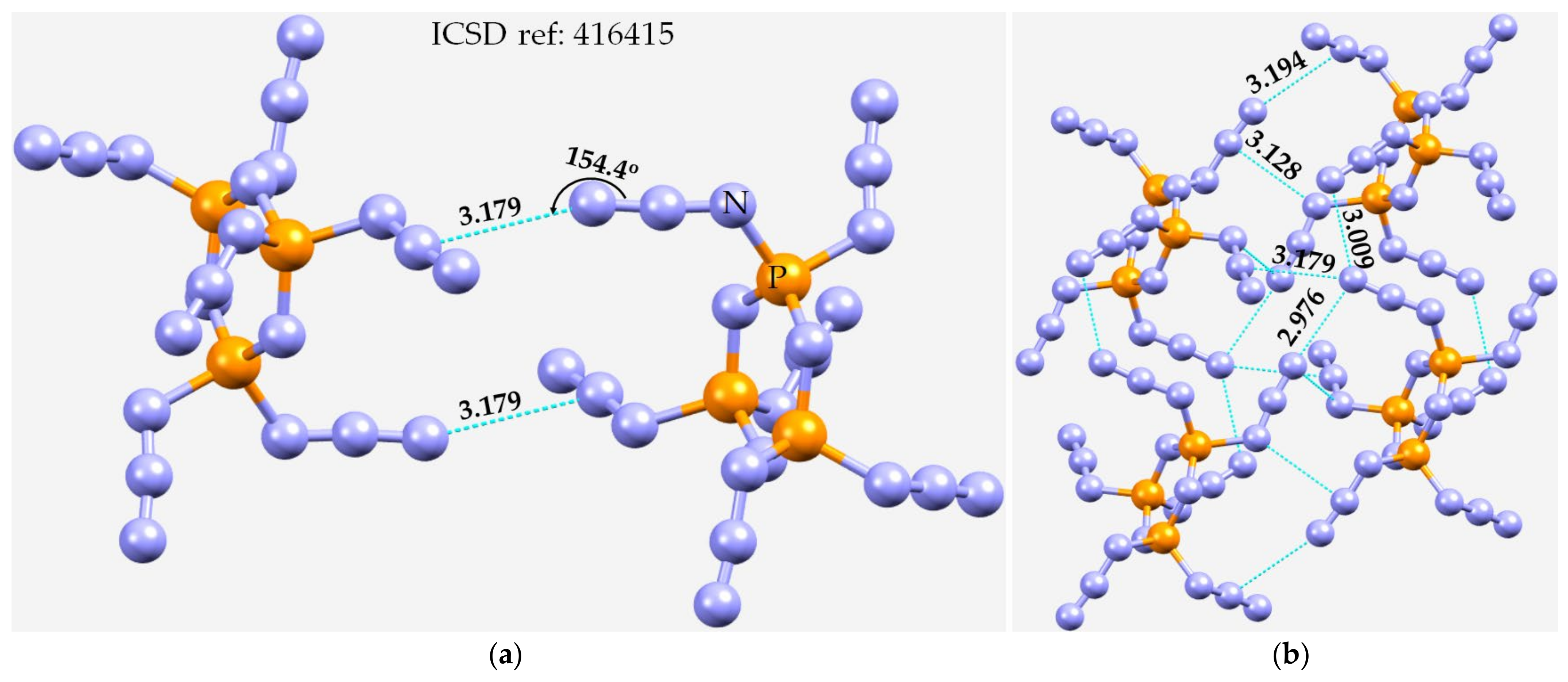
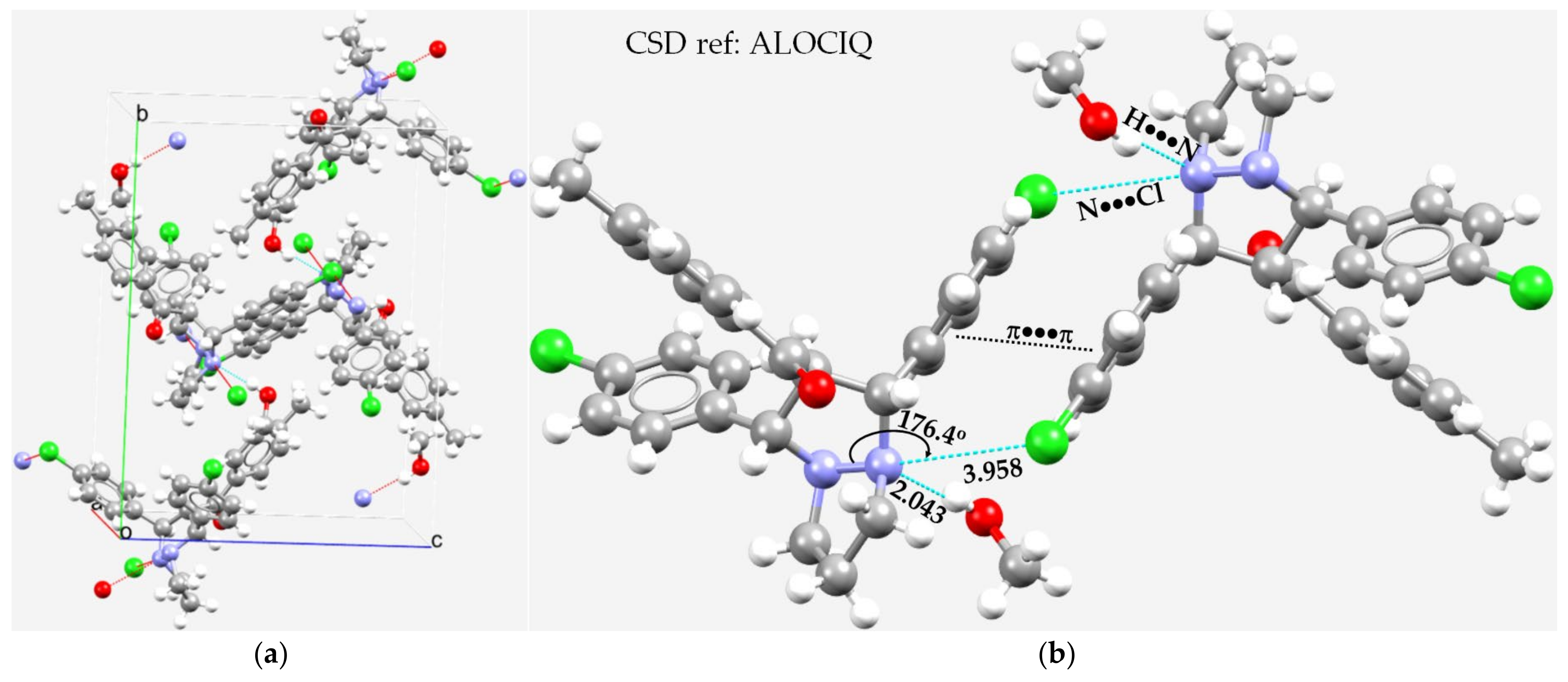
3.5. Miscellaneous Examples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Shing Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, R.G.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- IUPAC Project: Categorizing Chalcogen, Pnictigen, and Tetrel Bonds, and Other Interactions Involving Group 14–16 Elements. Available online: https://iupac.org/projects/project-details/?project_nr=2016-001-2-300 (accessed on 31 January 2022).
- Bakker, D.J.; Peters, A.; Yatsyna, V.; Zhaunerchyk, V.; Rijs, A.M. Far-Infrared Signatures of Hydrogen Bonding in Phenol Derivatives. J. Phys. Chem. Lett. 2016, 7, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Vasylyeva, V.; Catalano, L.; Nervi, C.; Gobetto, R.; Metrangolo, P.; Resnati, G. Characteristic redshift and intensity enhancement as far-IR fingerprints of the halogen bond involving aromatic donors. CrystEngComm 2016, 18, 2247–2250. [Google Scholar] [CrossRef]
- Hardin, A.E.S.; Ellington, T.L.; Nguyen, S.T.; Rheingold, A.L.; Tschumper, G.S.; Watkins, D.L.; Hammer, N.I. A Raman Spectroscopic and Computational Study of New Aromatic Pyrimidine-Based Halogen Bond Acceptors. Inorganics 2019, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Messina, M.T.; Metrangolo, P.; Navarrini, W.; Radice, S.; Resnati, G.; Zerbi, G. Infrared and Raman analyses of the halogen-bonded non-covalent adducts formed by α,ω-diiodoperfluoroalkanes with DABCO and other electron donors. J. Mol. Struct. 2000, 524, 87–94. [Google Scholar] [CrossRef]
- Shen, Q.J.; Jin, W.J. Strong halogen bonding of 1,2-diiodoperfluoroethane and 1,6-diiodoperfluorohexane with halide anions revealed by UV-Vis, FT-IR, NMR spectroscopes and crystallography. Phys. Chem. Chem. Phys. 2011, 13, 13721–13729. [Google Scholar] [CrossRef] [PubMed]
- Widner, D.L.; Robinson, E.R.; Perez, A.B.; Vang, H.G.; Thorson, R.A.; Driscoll, Z.L.; Giebel, S.M.; Berndt, C.W.; Bosch, E.; Speetzen, E.D.; et al. Comparing Strong and Weak Halogen Bonding in Solution: 13C NMR, UV/Vis, Crystallographic, and Computational Studies of an Intramolecular Model. Eur. J. Org. Chem. 2017, 2017, 5739–5749. [Google Scholar] [CrossRef]
- Hakkert, S.B.; Gräfenstein, J.; Erdelyi, M. The 15N NMR chemical shift in the characterization of weak halogen bonding in solution. Faraday Discuss. 2017, 203, 333–346. [Google Scholar] [CrossRef]
- Gomila, R.M.; Frontera, A. Charge assisted halogen and pnictogen bonds: Insights from the Cambridge Structural Database and DFT calculations. CrystEngComm 2020, 22, 7162–7169. [Google Scholar] [CrossRef]
- De Azevedo Santos, L.; Hamlin, T.A.; Ramalho, T.C.; Bickelhaupt, F.M. The pnictogen bond: A quantitative molecular orbital picture. Phys. Chem. Chem. Phys. 2021, 23, 13842–13852. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Zierkiewicz, W.; Švec, P.; Růžičková, Z.; Řezáč, J.; Michalczyk, M.; Růžička, A.; Michalska, D.; Hobza, P. Pnictogen bonding in pyrazine·PnX5 (Pn = P, As, Sb and X = F, Cl, Br) complexes. J. Mol. Model. 2017, 23, 328. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs. π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W. The σ-hole···σ-hole stacking interaction: An unrecognized type of noncovalent interaction. J. Chem. Phys. 2020, 153, 214302. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Mallada, B.; Gallardo, A.; Lamanec, M.; Torre, B.d.l.; Špirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-holes and π-holes: Similarities and differences. J. Comp. Chem. 2018, 39, 464–471. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Fluorines in tetrafluoromethane as halogen bond donors: Revisiting address the nature of the fluorine’s σ-hole. Int. J. Quantum Chem. 2015, 115, 453–470. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Halogen bonding interaction of chloromethane with several nitrogen donating molecules: Addressing the nature of the chlorine surface σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 19573–19589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.A.A.; Moussa, N.A.M. Unconventional Type III Halogen···Halogen Interactions: A Quantum Mechanical Elucidation of σ-Hole∙··σ-Hole and Di-σ-Hole Interactions. ACS Omega 2020, 5, 21824–21835. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals 2020, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Uyanik, M.; Kato, T.; Sahara, N.; Katade, O.; Ishihara, K. High-Performance Ammonium Hypoiodite/Oxone Catalysis for Enantioselective Oxidative Dearomatization of Arenols. ACS Catal. 2019, 9, 11619–11626. [Google Scholar] [CrossRef]
- Hinokuma, S.; Sato, K. Ammonia Combustion Catalysts. Chem. Lett. 2021, 50, 752–759. [Google Scholar] [CrossRef]
- Lamb, K.E.; Dolan, M.D.; Kennedy, D.F. Ammonia for hydrogen storage; A review of catalytic ammonia decomposition and hydrogen separation and purification. Int. J. Hydrogen Energy 2019, 44, 3580–3593. [Google Scholar] [CrossRef]
- US Geological Survey. Mineral Commodity Summaries 2020; US Geological Survey: Reston, VA, USA, 2020. [CrossRef] [Green Version]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Ogasawara, K.; Nakao, T.; Kishida, K.; Ye, T.-N.; Lu, Y.; Abe, H.; Niwa, Y.; Sasase, M.; Kitano, M.; Hosono, H. Ammonia Decomposition over CaNH-Supported Ni Catalysts via an NH2−-Vacancy-Mediated Mars–van Krevelen Mechanism. ACS Catal. 2021, 11, 11005–11015. [Google Scholar] [CrossRef]
- Mukherjee, S.; Devaguptapu, S.V.; Sviripa, A.; Lund, C.R.F.; Wu, G. Low-temperature ammonia decomposition catalysts for hydrogen generation. Appl. Catal. B Environ. 2018, 226, 162–181. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Engelmann, Y.; van ‘t Veer, K.; Postma, R.S.; Bogaerts, A.; Lefferts, L. Plasma-driven catalysis: Green ammonia synthesis with intermittent electricity. Green Chem. 2020, 22, 6258–6287. [Google Scholar] [CrossRef]
- Gini, A.; Paraja, M.; Galmés, B.; Besnard, C.; Poblador-Bahamonde, A.I.; Sakai, N.; Frontera, A.; Matile, S. Pnictogen-bonding catalysis: Brevetoxin-type polyether cyclizations. Chem. Sci. 2020, 11, 7086–7091. [Google Scholar] [CrossRef] [PubMed]
- Humeniuk, H.V.; Gini, A.; Hao, X.; Coelho, F.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis and Transport Combined: Polyether Transporters Made In Situ. JACS Au 2021, 1, 1588–1593. [Google Scholar] [CrossRef]
- Paraja, M.; Gini, A.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis: An Interactive Tool to Uncover Unorthodox Mechanisms in Polyether Cascade Cyclizations. Chem. A Eur. J. 2020, 26, 15471–15476. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Qiu, J.; Song, B.; Li, X.; Cozzolino, A.F. Solution and gas phase evidence of anion binding through the secondary bonding interactions of a bidentate bis-antimony(iii) anion receptor. Phys. Chem. Chem. Phys. 2018, 20, 46–50. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Lee, L.M.; Tsemperouli, M.; Poblador-Bahamonde, A.I.; Benz, S.; Sakai, N.; Sugihara, K.; Matile, S. Anion Transport with Pnictogen Bonds in Direct Comparison with Chalcogen and Halogen Bonds. J. Am. Chem. Soc. 2019, 141, 810–814. [Google Scholar] [CrossRef]
- Moaven, S.; Andrews, M.C.; Polaske, T.J.; Karl, B.M.; Unruh, D.K.; Bosch, E.; Bowling, N.P.; Cozzolino, A.F. Triple-Pnictogen Bonding as a Tool for Supramolecular Assembly. Inorg. Chem. 2019, 58, 16227–16235. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Huseynov, F.E.; Aliyeva, V.A.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Noncovalent Interactions at Lanthanide Complexes. Chem. Eur. J. 2021, 27, 14370–14389. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shin, J.; Son, S.; Choe, Y.; Farokhzad, N.; Tang, Z.; Xiao, Y.; Kong, N.; Xie, T.; Kim, J.S.; et al. Pnictogens in medicinal chemistry: Evolution from erstwhile drugs to emerging layered photonic nanomedicine. Chem. Soc. Rev. 2021, 50, 2260–2279. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Hnyk, D. Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes. Crystals 2018, 8, 390. [Google Scholar] [CrossRef] [Green Version]
- Sobalev, S.; Matveychuk, Y.; Bartashevich, E. Features of the Pnictogen Bonds Formed by Neighboring Nitro Groups in Crystals. Bull. South Ural State Univ. Chem. 2019, 11, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Frontera, A.; Bauza, A. On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. Int. J. Mol. Sci. 2021, 22, 12550. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Chalcogen and pnictogen bonds: Insights and relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Can an entirely negative fluorine in a molecule, viz. perfluorobenzene, interact attractively with the entirely negative site (s) on another molecule (s)? Like liking like! RSC Adv. 2016, 6, 19098–19110. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Cukrowski, I.; Marques, H.M. DFT-X3LYP Studies on the Coordination Chemistry of Ni2+. Part 1: Six Coordinate [Ni(NH3)n(H2O)6−n]2+ Complexes. J. Phys. Chem. A 2008, 112, 10657–10666. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Marques, H.M. The physical chemistry of coordinated aqua-, ammine-, and mixed-ligand Co2+ complexes: DFT studies on the structure, energetics, and topological properties of the electron density. Phys. Chem. Chem. Phys. 2010, 12, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. Can Counter-Intuitive Halogen Bonding Be Coulombic? ChemPhysChem 2021, 22, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Suryaprasad, B.; Ramanathan, N.; Sundararajan, K. Nitrogen as a pnicogen? Evidence for π-hole driven novel pnicogen bonding interactions in nitromethane–ammonia aggregates using matrix isolation infrared spectroscopy and ab initio computations. Phys. Chem. Chem. Phys. 2021, 23, 6286–6297. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthi, R.; Chivers, T.; Vargas-Baca, I. S,C,S-Pnictogen bonding in pincer complexes of the methanediide [C(Ph2PS)2]2−. Dalton Trans. 2011, 40, 8086–8088. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. 31P–31P spin–spin coupling constants for pnicogen homodimers. Chem. Phys. Lett. 2011, 512, 184–187. [Google Scholar] [CrossRef]
- Girolami, G.S. Origin of the Terms Pnictogen and Pnictide. J. Chem. Ed. 2009, 86, 1200. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Hellenbrandt, M. The Inorganic Crystal Structure Database (ICSD)—Present and Future. Crystallogr. Rev. 2004, 10, 17–22. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Kar, S.; Das, R.N. Chapter 2—Chemical Information and Descriptors. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Roy, K., Kar, S., Das, R.N., Eds.; Academic Press: Boston, MS, USA, 2015; pp. 47–80. [Google Scholar] [CrossRef]
- Dance, I. Distance criteria for crystal packing analysis of supramolecular motifs. New J. Chem. 2003, 27, 22–27. [Google Scholar] [CrossRef]
- Motiejunas, D.; Wade, R.C. 4.09—Structural, Energetic, and Dynamic Aspects of Ligand–Receptor Interactions. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 193–213. [Google Scholar] [CrossRef]
- Tredwell, M.; Gouverneur, V. 1.5 Fluorine in Medicinal Chemistry: Importance of Chirality. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 70–85. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Desiraju, G.R. Van der Waals and Polar Intermolecular Contact Distances: Quantifying Supramolecular Synthons. Chem. Asian J. 2008, 3, 868–880. [Google Scholar] [CrossRef]
- Desiraju, G.R. C–H…O Hydrogen Bonding and the Deliberate Design of Organic Crystal Structures. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A Mol. Cryst. Liq. Cryst. 1992, 211, 63–74. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Gafner, G.; Herbstein, F.H.; Lee, C.M. Derivation of van der Waals radii from known crystal structures. Acta Crystallogr. A 1972, 28, 422–426. [Google Scholar] [CrossRef]
- Hu, S.-Z.; Zhou, Z.-H.; Xie, Z.-X.; Robertson, B.E. A comparative study of crystallographic van der Waals radii. Z. Kristall.—Cryst. Mater. 2014, 229, 517–523. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The use and misuse of van der Waals radii. Struct. Chem. 2021, 32, 623–629. [Google Scholar] [CrossRef]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiemenz, G.P. The sum of van der Waals radii—A pitfall in the search for bonding. Z. Naturforsch. B 2007, 62, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S. Molecular Electrostatic Potentials and Chemical Reactivity. In Reviews in Computational Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1991; Volume 2, pp. 273–312. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Molecular Electrostatic Potentials: Significance and Applications. In Chemical Reactivity in Confined Systems; Chattaraj, P.K., Chakraborty, D., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2021; pp. 113–134. [Google Scholar] [CrossRef]
- Mishra, P.C.; Kumar, A. Molecular electrostatic potentials and fields: Hydrogen bonding, recognition, reactivity and modelling. In Theoretical and Computational Chemistry; Murray, J.S., Sen, K., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 3, pp. 257–296. [Google Scholar]
- Liu, L.; Miao, L.; Li, L.; Li, F.; Lu, Y.; Shang, Z.; Chen, J. Molecular Electrostatic Potential: A New Tool to Predict the Lithiation Process of Organic Battery Materials. J. Phys. Chem. Lett. 2018, 9, 3573–3579. [Google Scholar] [CrossRef]
- Alipour, M.; Mohajeri, A. Molecular Electrostatic Potential as a tool for Evaluating the Etherification Rate Constant. J. Phys. Chem. A 2010, 114, 7417–7422. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Phosphorous Bond, or the Phosphorous-Centered Pnictogen Bond: The Covalently Bound Phosphorous Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 1487. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Molecular Surfaces, van der Waals Radii and Electrostatic Potentials in Relation to Noncovalent Interactions. Croat. Chem. Acta 2009, 82, 267–275. [Google Scholar]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R. Does Oxygen Feature Chalcogen Bonding? Molecules 2019, 24, 3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Very strong chalcogen bonding: Is oxygen in molecule capable of forming it? A First Princiles Perspective. Authorea, 2020; preprints. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Chalcogen Bonding in the Molecular Dimers of WCh2 (Ch = S, Se, Te): On the Basic Understanding of the Local Interfacial and Interlayer Bonding Environment in 2D Layered Tungsten Dichalcogenides. Int. J. Mol. Sci. 2022, 23, 1263. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Marques, H.M.; Varadwaj, A.; Yamashita, K. Chalcogen···Chalcogen Bonding in Molybdenum Disulfide, Molybdenum Diselenide and Molybdenum Ditelluride Dimers as Prototypes for a Basic Understanding of the Local Interfacial Chemical Bonding Environment in 2D Layered Transition Metal Dichalcogenides. Inorganics 2022, 10, 11. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Politzer, P. Hydrogen Bonding: A Coulombic σ-Hole Interaction. J. Indian Inst. Sci. 2020, 100, 21–30. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Engelage, E.; Weiss, R.; Huber, S.M. “Anti-Electrostatic” Halogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 11150–11157. [Google Scholar] [CrossRef] [Green Version]
- Loy, C.; Holthoff, J.M.; Weiss, R.; Huber, S.M.; Rosokha, S.V. “Anti-electrostatic” halogen bonding in solution. Chem. Sci. 2021, 12, 8246–8251. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? Like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The s-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Clark, T.; Politzer, P.; Murray, J.S. Correct electrostatic treatment of noncovalent interactions: The importance of polarization. WIREs Comput. Mol. Sci. 2015, 5, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Murray, J.S.; Politzer, P. Role of Polarization in Halogen Bonds. Aust. J. Chem. 2014, 67, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Adonin, S.A.; Gorokh, I.D.; Novikov, A.S.; Samsonenko, D.G.; Plyusnin, P.E.; Sokolov, M.N.; Fedin, V.P. Bromine-rich complexes of bismuth: Experimental and theoretical studies. Dalton Trans. 2018, 47, 2683–2689. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon π-Hole···[MII] (d8 M = Pt, Pd) Interactions, where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. 2019, 131, 4208–4212. [Google Scholar] [CrossRef]
- Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Matveychuk, Y.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Bifurcated Halogen Bonding C–Br···η2(Cl–Pt). Cryst. Growth Des. 2019, 19, 1364–1376. [Google Scholar] [CrossRef]
- Kashina, M.V.; Kinzhalov, M.A.; Smirnov, A.S.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Dihalomethanes as Bent Bifunctional XB/XB-Donating Building Blocks for Construction of Metal-involving Halogen Bonded Hexagons. Chem. Asian J. 2019, 14, 3915–3920. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)···[dz2-MII] Interactions of (Isocyanide)[MII] Complexes, in which Positively Charged Metal Centers (d8-M = Pt, Pd) Act as Nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision C.01; Gaussian, Inc.: Wallinford, CT, USA, 2016.
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, version 5.0.9; Semichem, Inc.: Shawnee Mission, KS, USA, 2009.
- Keith, T.A. AIMAll, version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. Available online: https://aim.tkgristmill.com(accessed on 24 February 2022).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Turnbull, R.; Hanfland, M.; Binns, J.; Martinez-Canales, M.; Frost, M.; Marqués, M.; Howie, R.T.; Gregoryanz, E. Unusually complex phase of dense nitrogen at extreme conditions. Nat. Commun. 2018, 9, 4717. [Google Scholar] [CrossRef] [PubMed]
- Hanfland, M.; Lorenzen, M.; Wassilew-Reul, C.; Zontone, F. Structures of Molecular Nitrogen at High Pressures. Rev. High Press. Sci. Tecnol. 1998, 7, 787–789. [Google Scholar] [CrossRef]
- Vos, W.L.; Finger, L.W.; Hemley, R.J.; Hu, J.Z.; Mao, H.K.; Schouten, J.A. A high-pressure van der Waals compound in solid nitrogen-helium mixtures. Nature 1992, 358, 46–48. [Google Scholar] [CrossRef]
- Venables, J.A.; English, C.A. Electron diffraction and the structure of [alpha]-N2. Acta Cryst. B 1974, 30, 929–935. [Google Scholar] [CrossRef]
- Schuch, A.F.; Mills, R.L. Crystal Structures of the Three Modifications of Nitrogen 14 and Nitrogen 15 at High Pressure. J. Chem. Phys. 1970, 52, 6000–6008. [Google Scholar] [CrossRef]
- Mills, R.L.; Olinger, B.; Cromer, D.T. Structures and phase diagrams of N2 and CO to 13 GPa by X-ray diffraction. J. Chem. Phys. 1986, 84, 2837–2845. [Google Scholar] [CrossRef] [Green Version]
- Stinton, G.W.; Loa, I.; Lundegaard, L.F.; McMahon, M.I. The crystal structures of δ and δ∗ nitrogen. J. Chem. Phys. 2009, 131, 104511. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Ivlev, S.I.; Conrad, M.; Hoelzel, M.; Karttunen, A.J.; Kraus, F. Crystal Structures of α- and β-Nitrogen Trifluoride. Inorg. Chem. 2019, 58, 6422–6430. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing Factors Influencing the Fluorine-Centered Non-Covalent Interactions in Some Fluorine-substituted Molecular Complexes: Insights from First-Principles Studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Sadeghi, A.; Xu, F.; Peng, L.; Orita, A.; Otera, J.; Goedecker, S.; Meyer, E. Extended halogen bonding between fully fluorinated aromatic molecules. ACS Nano 2015, 9, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Comment on “Extended Halogen Bonding between Fully Fluorinated Aromatic Molecules: Kawai et al., ACS Nano, 2015, 9, 2574–2583”. CondMat Public Archive. Available online: https://arxiv.org/ftp/arxiv/papers/1802/1802.09995.pdf (accessed on 12 October 2021).
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Unusual bonding modes of perfluorobenzene in its polymeric (dimeric, trimeric and tetrameric) forms: Entirely negative fluorine interacting cooperatively with entirely negative fluorine. Phys. Chem. Chem. Phys. 2015, 17, 31624–31645. [Google Scholar] [CrossRef] [PubMed]
- Hartl, H.; Schöner, J.; Jander, J.; Schulz, H. Die Struktur des festen Stickstofftrichlorids (−125 °C). Z. Anorg. Allg. Chem. 1975, 413, 61–71. [Google Scholar] [CrossRef]
- Dehnicke, K. The Chemistry of Iodine Azide. Angew. Chem. Int. Ed. Engl. 1979, 18, 507–514. [Google Scholar] [CrossRef]
- Dehnicke, K. The Chemistry of the Halogen Azides. Adv. Inorg. Chem. 1983, 26, 169–200. [Google Scholar]
- Tornieporth-Oetting, I.C.; Klapötke, T.M. Covalent Inorganic Nonmetal Azides. In Combustion Efficiency and Air Quality; Hargitta, I., Vidóczy, T., Eds.; Springer: Boston, MA, USA, 1995. [Google Scholar]
- Tornieporth-Oetting, I.C.; Klapötke, T.M. Recent developments in the chemistry of binary nitrogen-halogen species. Comments Inorg. Chem. 1994, 15, 137–169. [Google Scholar] [CrossRef]
- Buzek, P.; Klapötke, T.M.; von Ragué Schleyer, P.; Tornieporth-Oetting, I.C.; White, P.S. Iodine Azide. Angew. Chem. Int. Ed. Engl. 1993, 32, 275–277. [Google Scholar] [CrossRef]
- Lyhs, B.; Bläser, D.; Wölper, C.; Schulz, S.; Jansen, G. A Comparison of the Solid-State Structures of Halogen Azides XN3 (X = Cl, Br, I). Angew. Chem. Int. Ed. 2012, 51, 12859–12863. [Google Scholar] [CrossRef]
- Lyhs, B.; Bläser, D.; Wölper, C.; Schulz, S.; Jansen, G. Solid-State Structure of Bromine Azide. Angew. Chem. Int. Ed. 2012, 51, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.; Ivlev, S.; Schulz, S.; Wölper, C. Automated Crystal Structure Determination Has its Pitfalls: Correction to the Crystal Structures of Iodine Azide. Angew. Chem. Int. Ed. 2021, 60, 17452–17454. [Google Scholar] [CrossRef] [PubMed]
- Bursch, M.; Kunze, L.; Vibhute, A.M.; Hansen, A.; Sureshan, K.M.; Jones, P.G.; Grimme, S.; Werz, D.B. Quantification of Noncovalent Interactions in Azide–Pnictogen, –Chalcogen, and –Halogen Contacts. Chem. Eur. J. 2021, 27, 4627–4639. [Google Scholar] [CrossRef] [PubMed]
- Göbel, M.; Karaghiosoff, K.; Klapötke, T.M. The First Structural Characterization of a Binary P–N Molecule: The Highly Energetic Compound P3N21. Angew. Chem. Int. Ed. 2006, 45, 6037–6040. [Google Scholar] [CrossRef] [PubMed]
- Laniel, D.; Winkler, B.; Koemets, E.; Fedotenko, T.; Chariton, S.; Milman, V.; Glazyrin, K.; Prakapenka, V.; Dubrovinsky, L.; Dubrovinskaia, N. Nitrosonium nitrate (NO+NO3−) structure solution using in situ single-crystal X-ray diffraction in a diamond anvil cell. IUCrJ 2021, 8, 208–214. [Google Scholar] [CrossRef]
- Addison, C.C.; Thompson, R. Ionic Reactions in Liquid Dinitrogen Tetroxide. Nature 1948, 162, 369–370. [Google Scholar] [CrossRef]
- Parts, L.; Miller, J.T., Jr. Nitrosonium Nitrate. Isolation at 79°–205°K and Infrared Spectra of the Polymorphic Compound. J. Chem. Phys. 1965, 43, 136–139. [Google Scholar] [CrossRef]
- Agnew, S.F.; Swanson, B.I.; Jones, L.H.; Mills, R.L.; Schiferl, D. Chemistry of nitrogen oxide (N2O4) at high pressure: Observation of a reversible transformation between molecular and ionic crystalline forms. J. Phys. Chem. 1983, 87, 5065–5068. [Google Scholar] [CrossRef]
- Agnew, S.F.; Swanson, B.I.; Jones, L.H.; Mills, R.L. Disproportionation of nitric oxide at high pressure. J. Phys. Chem. 1985, 89, 1678–1682. [Google Scholar] [CrossRef]
- Sihachakr, D.; Loubeyre, P. High-pressure transformation of N2/O2 mixtures into ionic compounds. Phys. Rev. B 2006, 74, 064113. [Google Scholar] [CrossRef]
- Somayazulu, M.; Madduri, A.; Goncharov, A.F.; Tschauner, O.; McMillan, P.F.; Mao, H.-K.; Hemley, R.J. Novel Broken Symmetry Phase from N2O at High Pressures and High Temperatures. Phys. Rev. Lett. 2001, 87, 135504. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.D.; Pennington, W.T. Tetramethylpyrazinium polyiodides. Acta Crystallogr. B 1995, 51, 810–815. [Google Scholar] [CrossRef]
- Kumar, V.; Scilabra, P.; Politzer, P.; Terraneo, G.; Daolio, A.; Fernandez-Palacio, F.; Murray, J.S.; Resnati, G. Tetrel and Pnictogen Bonds Complement Hydrogen and Halogen Bonds in Framing the Interactional Landscape of Barbituric Acids. Cryst. Growth Des. 2021, 21, 642–652. [Google Scholar] [CrossRef]
- Butcher, R.J.; Bottaro, J.C.; Gilardi, R. Ammonium 1,3,4,6-tetranitro-2,5-diazapentalene. Acta Crystallogr. E 2003, 59, o1149–o1150. [Google Scholar] [CrossRef]
- Butcher, R.J.; Bottaro, J.C.; Gilardi, R. 1,3,4-Trinitro-7,8-diazapentalene. Acta Crystallogr. E 2003, 59, o1780–o1782. [Google Scholar] [CrossRef]
- Gilardi, R.; Evans, R.N.; Manser, G.E. 3,3-Bis(difluoroaminomethyl)oxetane, a promising new energetic material. Acta Crystallogr. E 2003, 59, o2032–o2034. [Google Scholar] [CrossRef]
- Molchanov, A.P.; Efremova, M.M.; Kryukova, M.A.; Kuznetsov, M.A. Selective and reversible 1,3-dipolar cycloaddition of 6-aryl-1,5-diazabicyclo[3.1.0]hexanes with 1,3-diphenylprop-2-en-1-ones under microwave irradiation. Beilstein J. Org. Chem. 2020, 16, 2679–2686. [Google Scholar] [CrossRef]
- Olejniczak, A.; Katrusiak, A.; Podsiadlo, M.; Katrusiak, A. Crystal design by CH…N and N…N interactions: High-pressure structures of high-nitrogen-content azido-triazolopyridazines compounds. Acta Crystallogr. B 2020, 76, 1136–1142. [Google Scholar] [CrossRef]
- Allcock, H.R.; Ngo, D.C.; Parvez, M.; Visscher, K. Cyclic and short-chain linear phosphazenes with hindered aryloxy side groups. J. Chem. Soc. Dalton Trans. 1992, 10, 1687–1699. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | NF3 | NCl3 | NBr3 | NI3 | ||||
|---|---|---|---|---|---|---|---|---|
| Method | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) |
| Extrema of potential | Vs,max | |||||||
| N–X (one on each X) | 4.7 | 5.2 | 25.0 | 24.5 | 29.7 | 28.2 | 36.3 | 32.1 |
| X–N (one on each extension) | 16.5 | 16.5 | 5.2 | 4.6 | 3.5 | 3.5 | 2.1 | 2.3 |
| Center of the triangular X3 face | 8.8 | 7.5 | 5.6 | 4.9 | 5.4 | 4.3 | 6.4 | 4.9 |
| VS,min | ||||||||
| On lone-pair region on N | −4.1 | −3.7 | −11.9 | −12.6 | −14.1 | −13.8 | −17.2 | −15.2 |
| On F (within the X3 face) | −1.8 | −1.9 | −3.7 | −3.4 | −4.3 | −3.9 | −4.9 | −4.0 |
| On X (opposite of the X3 face) | −1.3 | −1.3 | −4.0 | −4.1 | −4.8 | −4.6 | −5.3 | −4.8 |
| Species | FN3 | ClN3 | BrN3 | IN3 | ||||
|---|---|---|---|---|---|---|---|---|
| Method | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) | ωB97XD | MP2 (Full) |
| Extrema | VS,min | |||||||
| On N3 of N2=N3 | −4.5 | −4.3 | −8.4 | −9.1 | −10.3 | −11.2 | −13.8 | −13.8 |
| On N1 of N2=N1 | −12.5 | −11.6 | −14.6 | −13.4 | −15.0 | −13.4 | −15.9 | −14.5 |
| On X of N1–X | −14.5 | −13.4 | −5.6 | −4.1 | −3.7 | −2.1 | −1.1 | 0.02 |
| VS,max | ||||||||
| On X of N–X | −3.4 | −1.7 | 23.5 | 25.3 | 31.7 | 33.1 | 42.8 | 41.2 |
| On N1 of N2=N1 | −6.4 | −5.3 | −2.2 | −0.8 | −0.5 | 1.0 | 2.5 | 3.4 |
| On N2 | 25.8 | 24.6 | 23.7 | 21.5 | 22.4 | 20.1 | 21.0 | 18.3 |
| On N2 | 30.2 | 30.8 | 24.6 | 23.4 | 22.3 | 20.7 | 17.8 | 16.4 |
 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Compounds 2022, 2, 80-110. https://doi.org/10.3390/compounds2010007
Varadwaj PR, Varadwaj A, Marques HM, Yamashita K. The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Compounds. 2022; 2(1):80-110. https://doi.org/10.3390/compounds2010007
Chicago/Turabian StyleVaradwaj, Pradeep R., Arpita Varadwaj, Helder M. Marques, and Koichi Yamashita. 2022. "The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor" Compounds 2, no. 1: 80-110. https://doi.org/10.3390/compounds2010007
APA StyleVaradwaj, P. R., Varadwaj, A., Marques, H. M., & Yamashita, K. (2022). The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Compounds, 2(1), 80-110. https://doi.org/10.3390/compounds2010007