


Glycerol Dehydration to Acrolein Catalyzed by Silicotungstic Acid: Effect of Mesoporous Support

Abstract
:1. Introduction
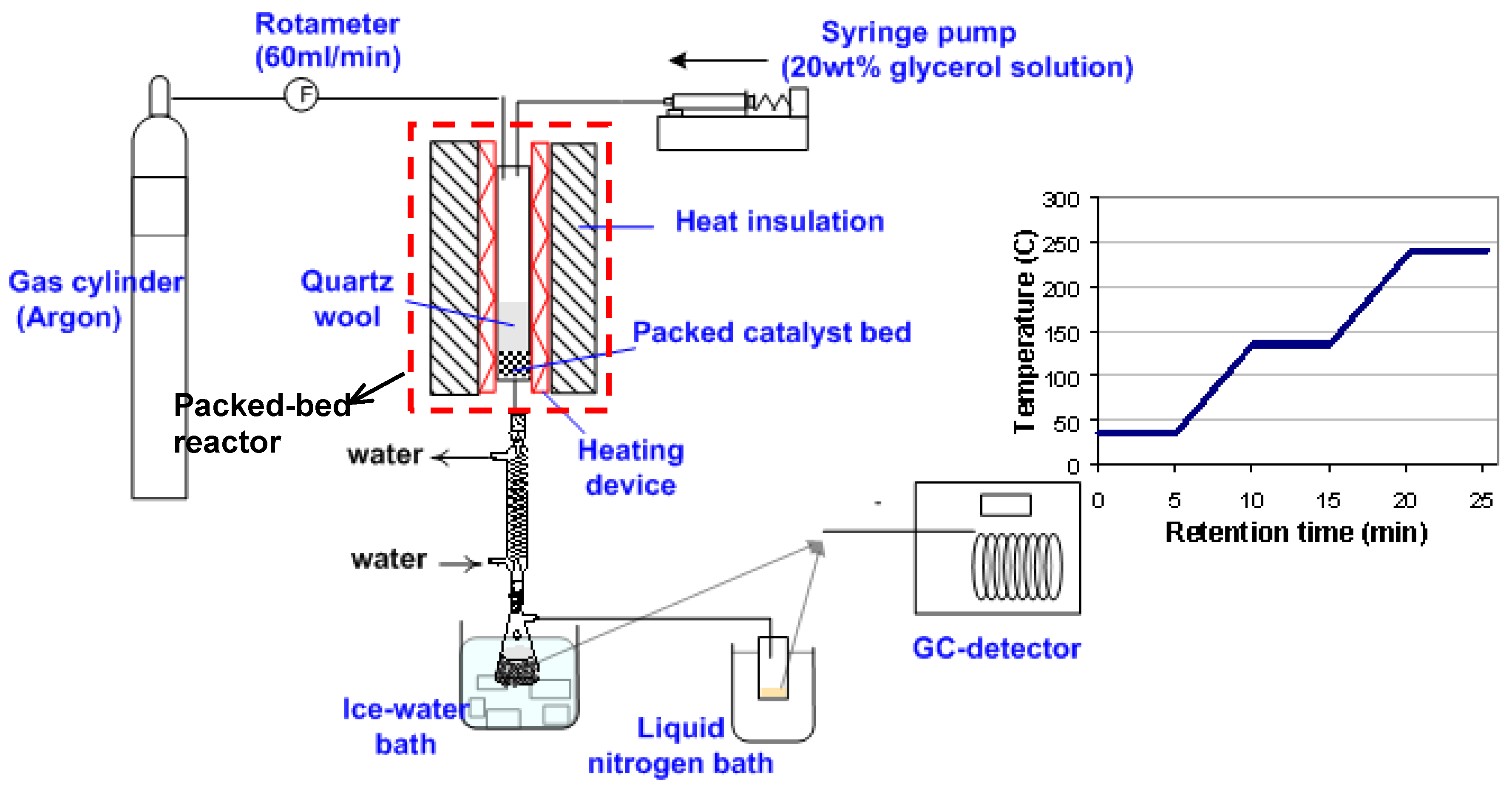
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
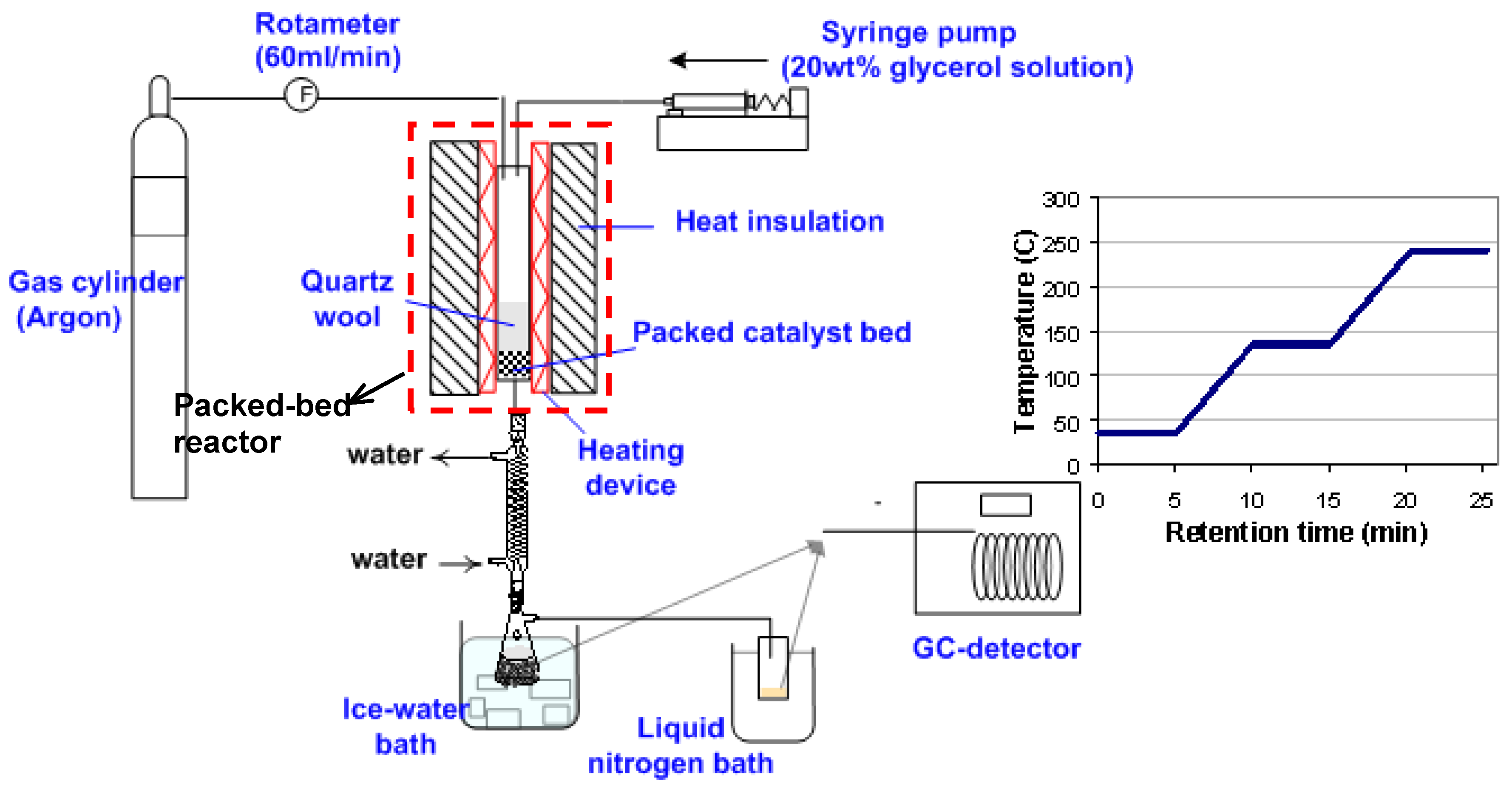
2.3. Glycerol Dehydration
3. Results and Discussion
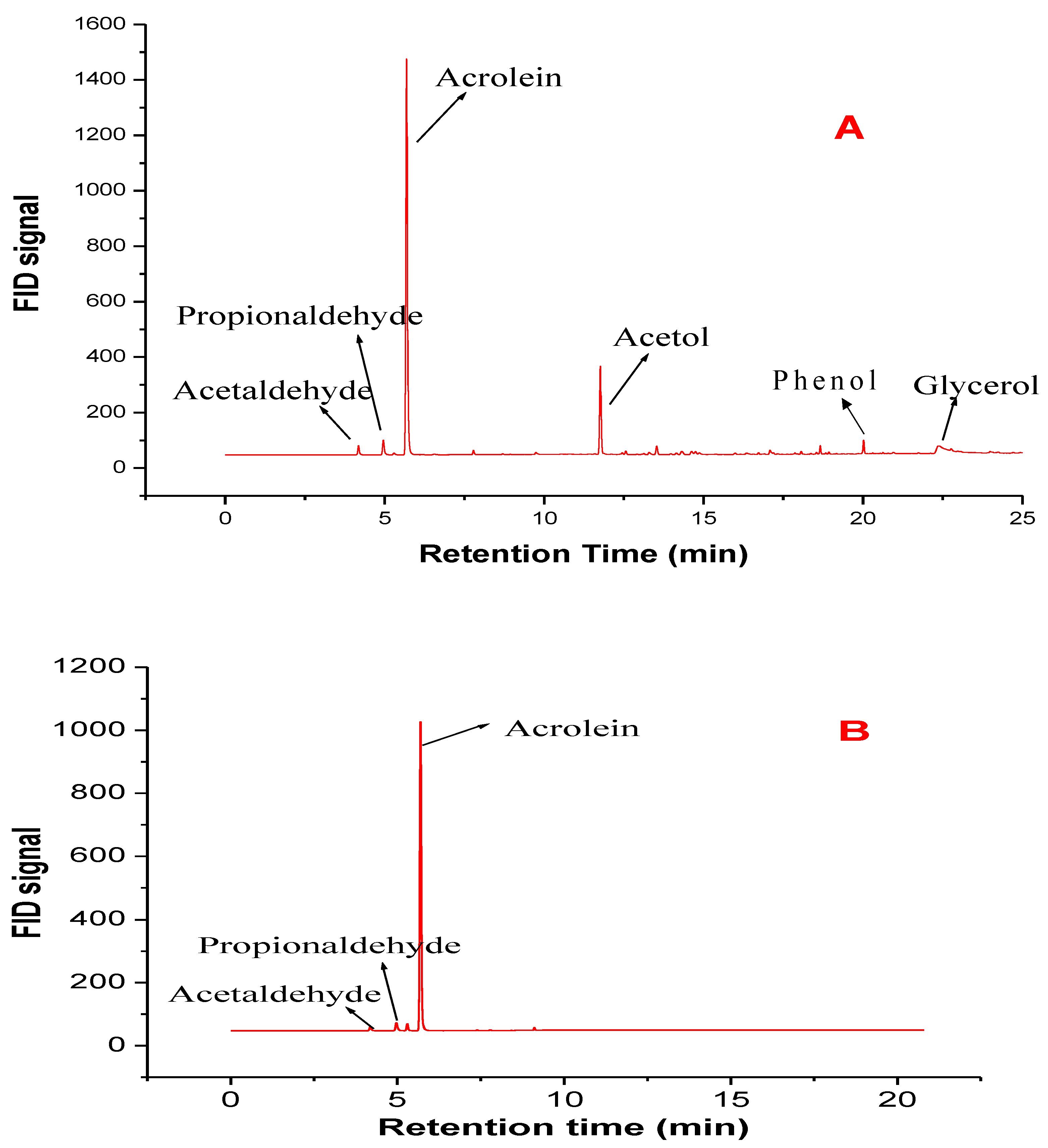
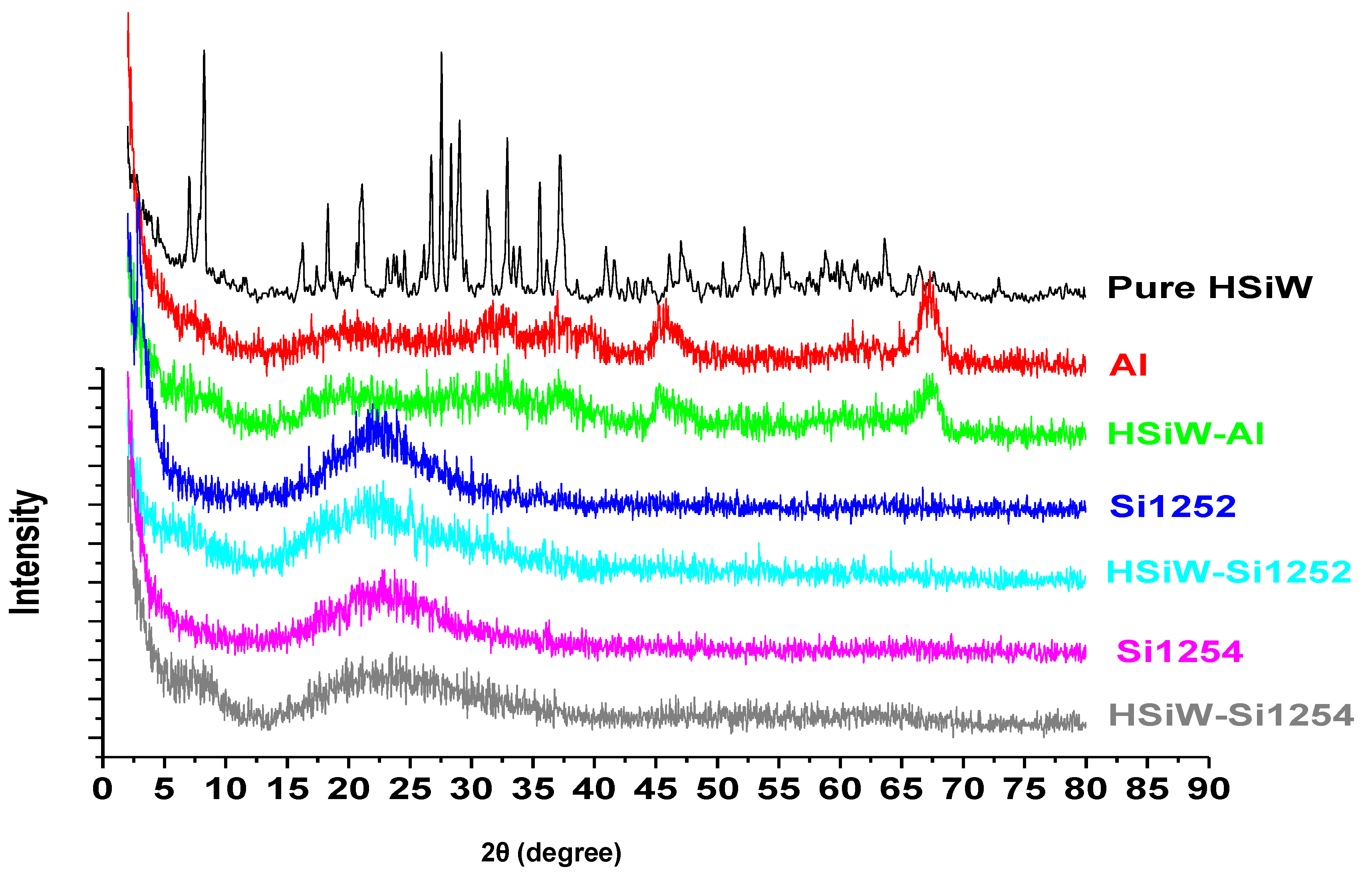
3.1. Characteristics of Fresh Catalysts
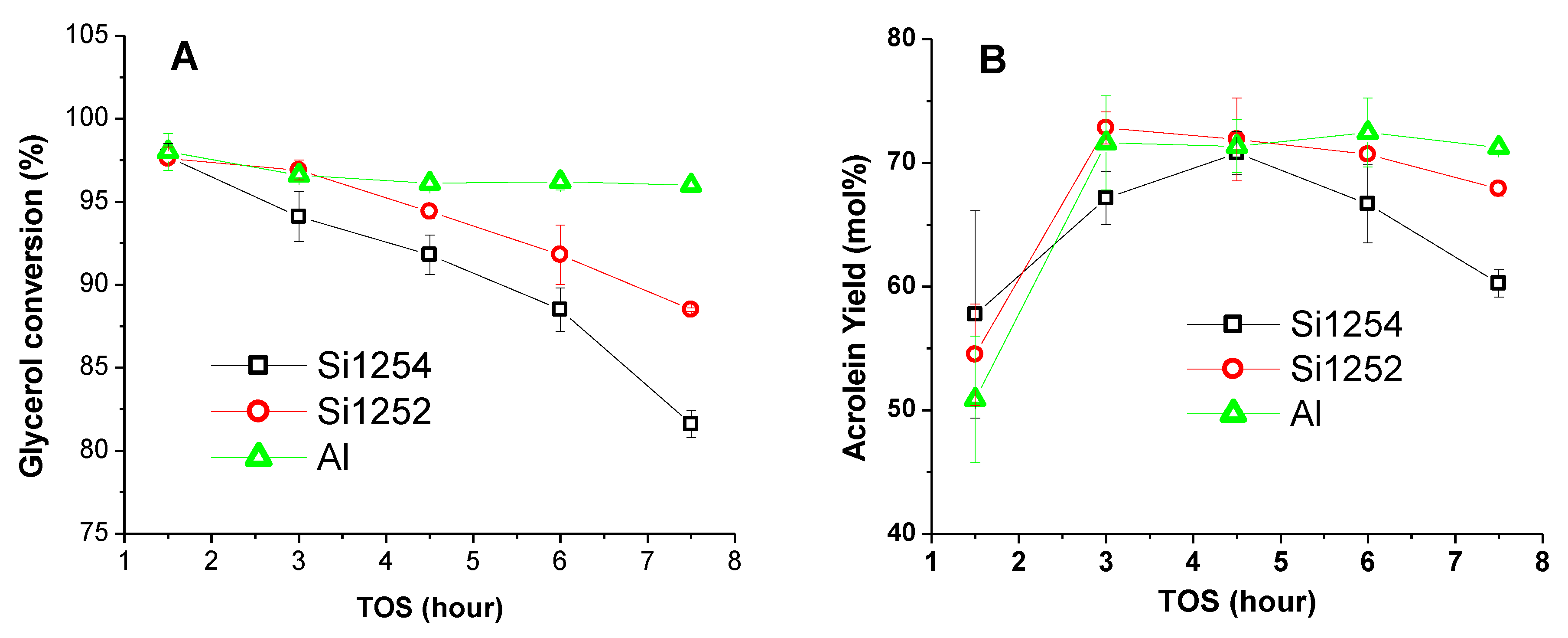
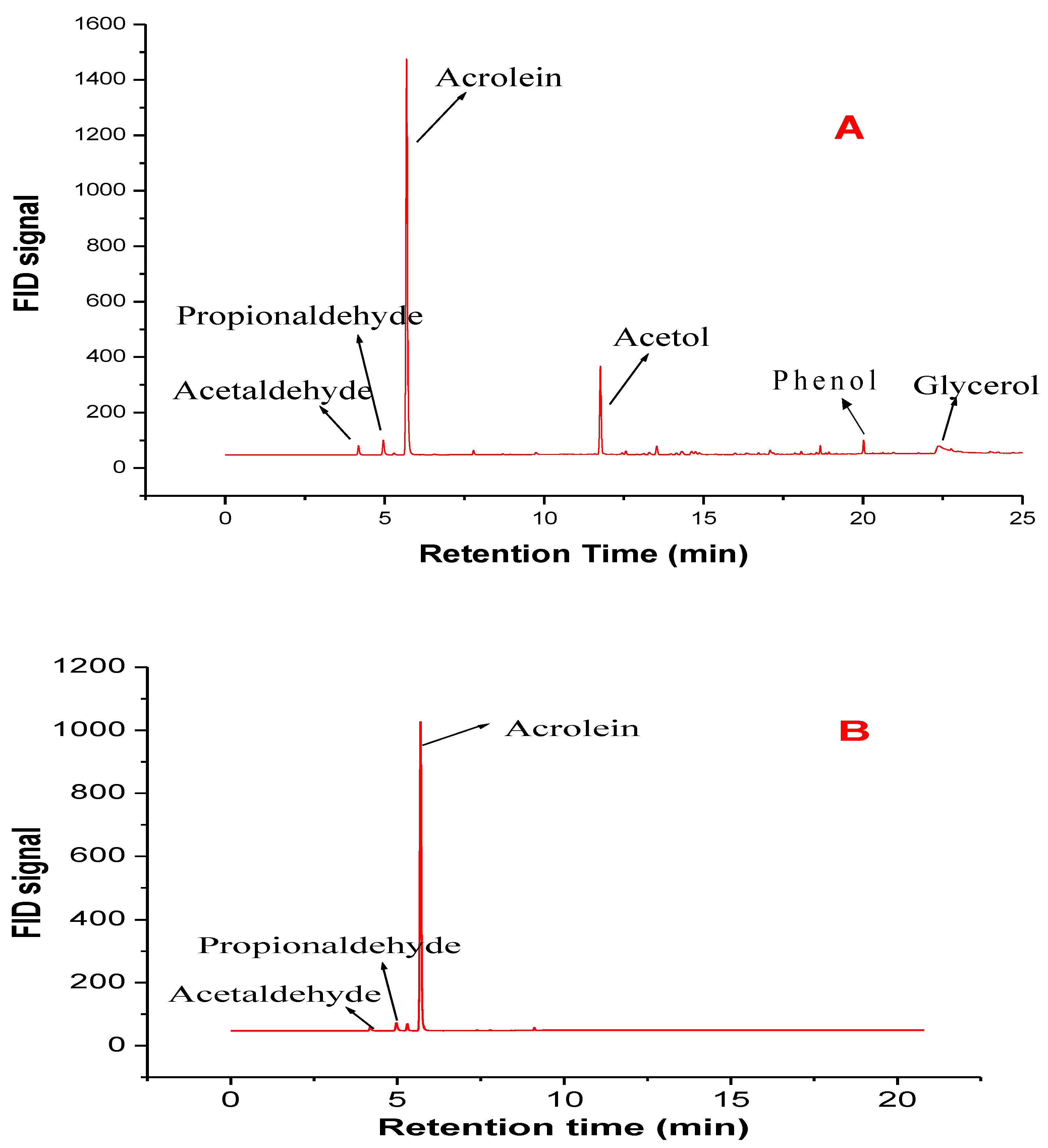
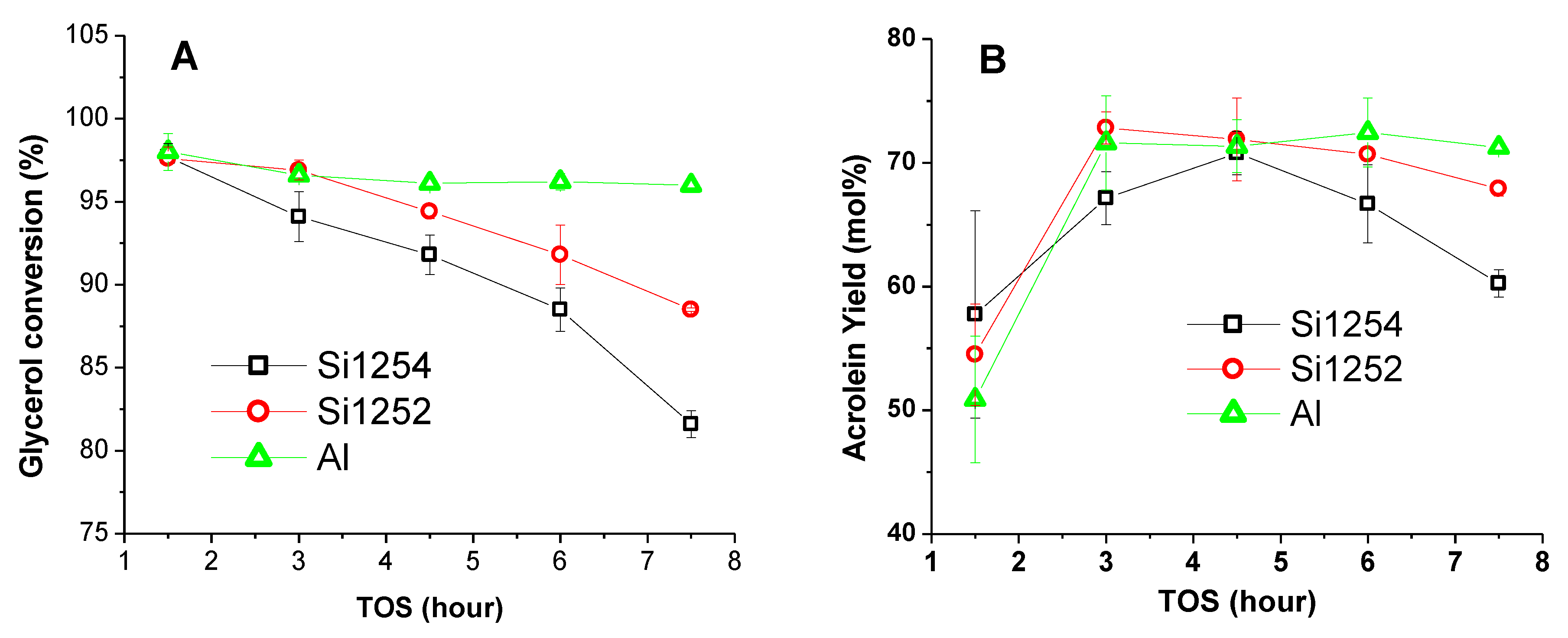
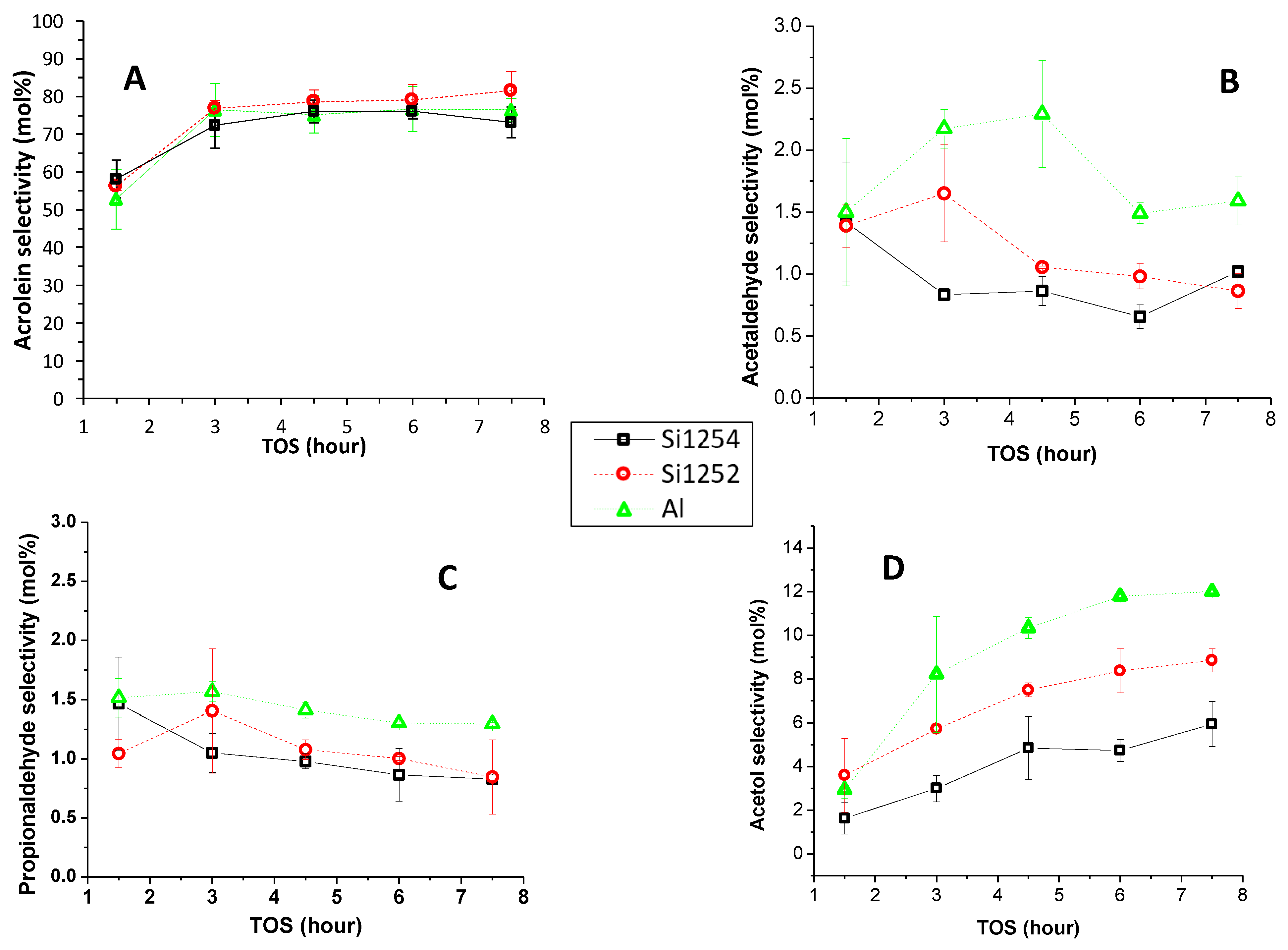
3.2. Comparative Catalyst Performance
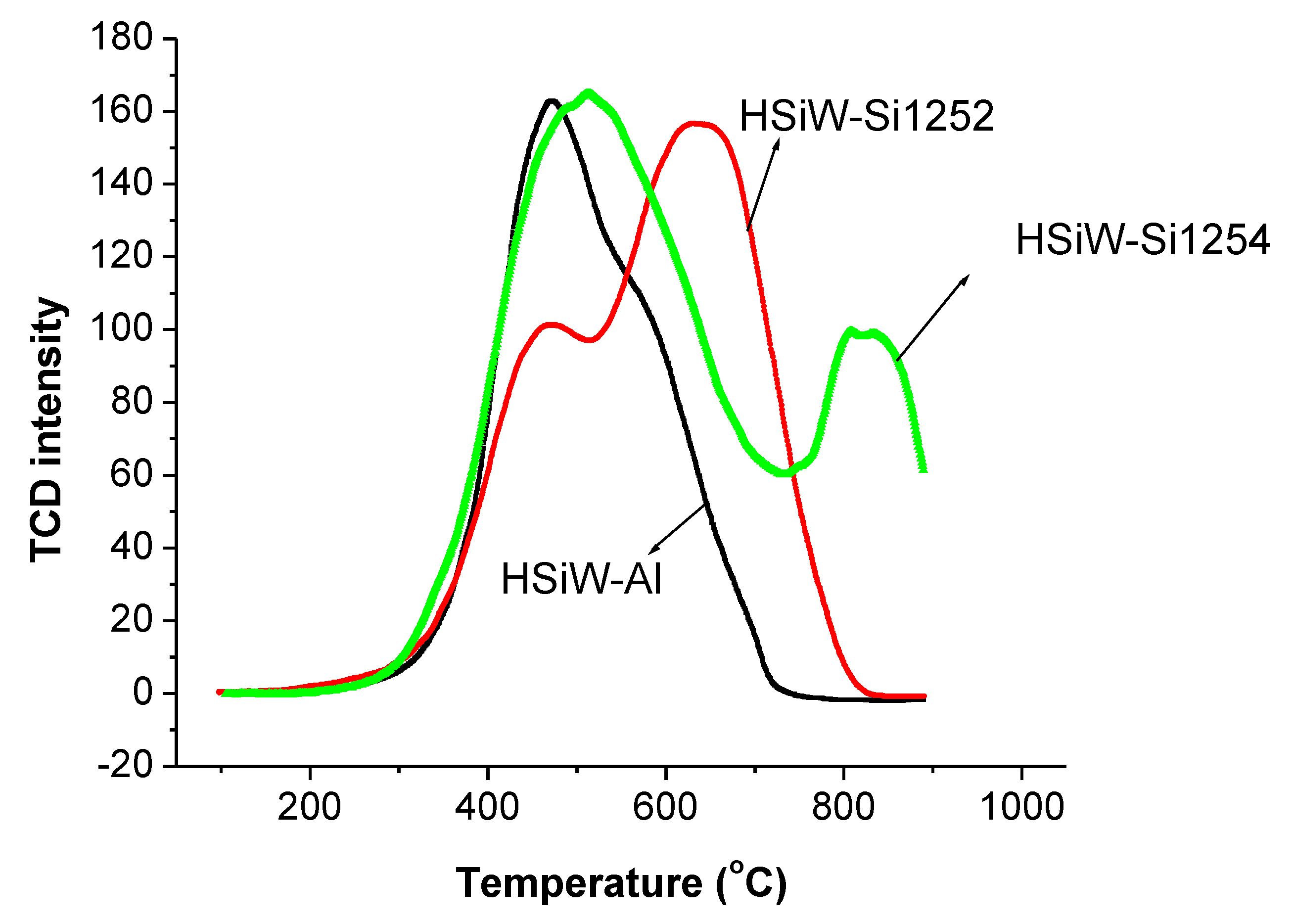
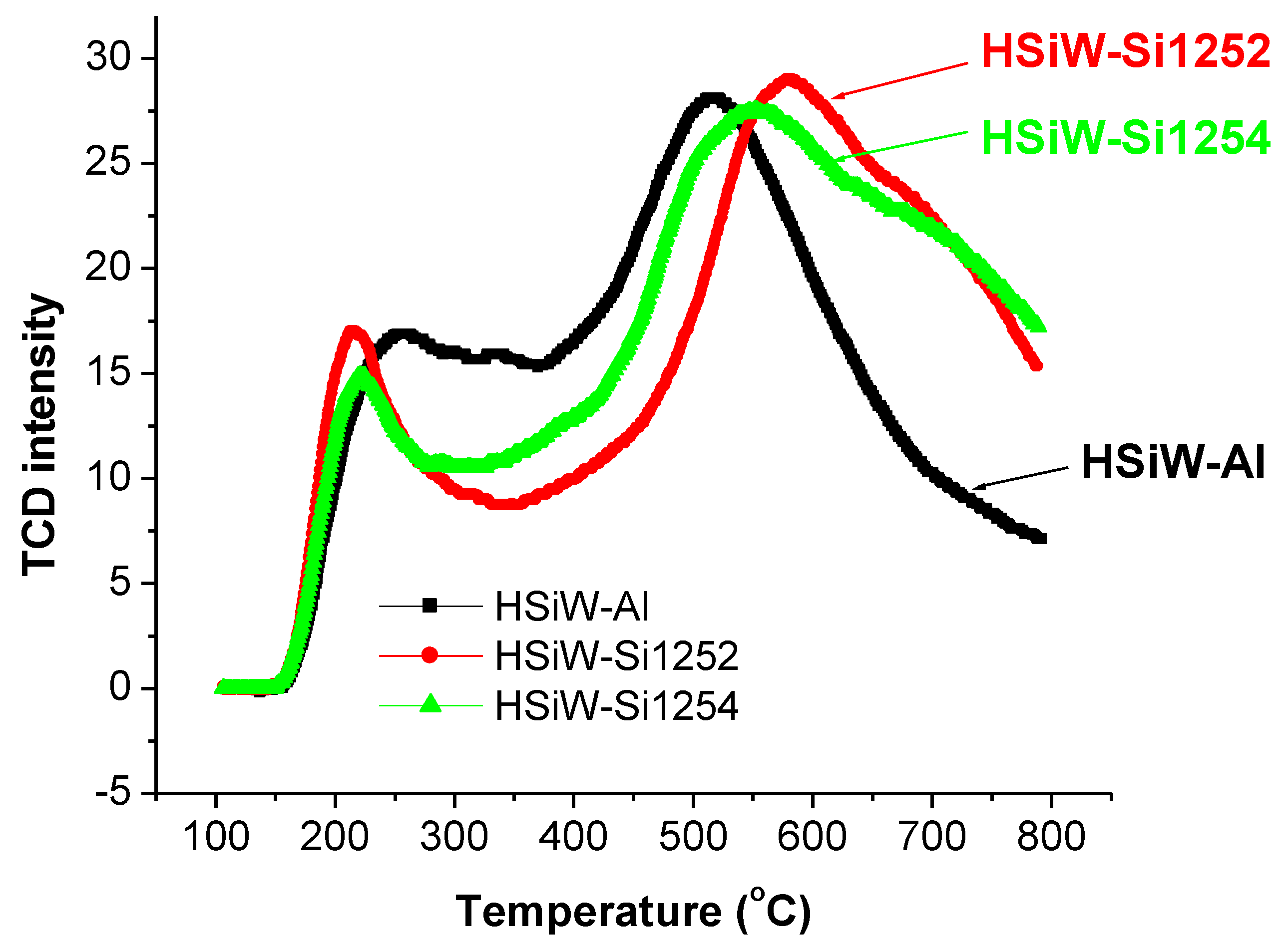
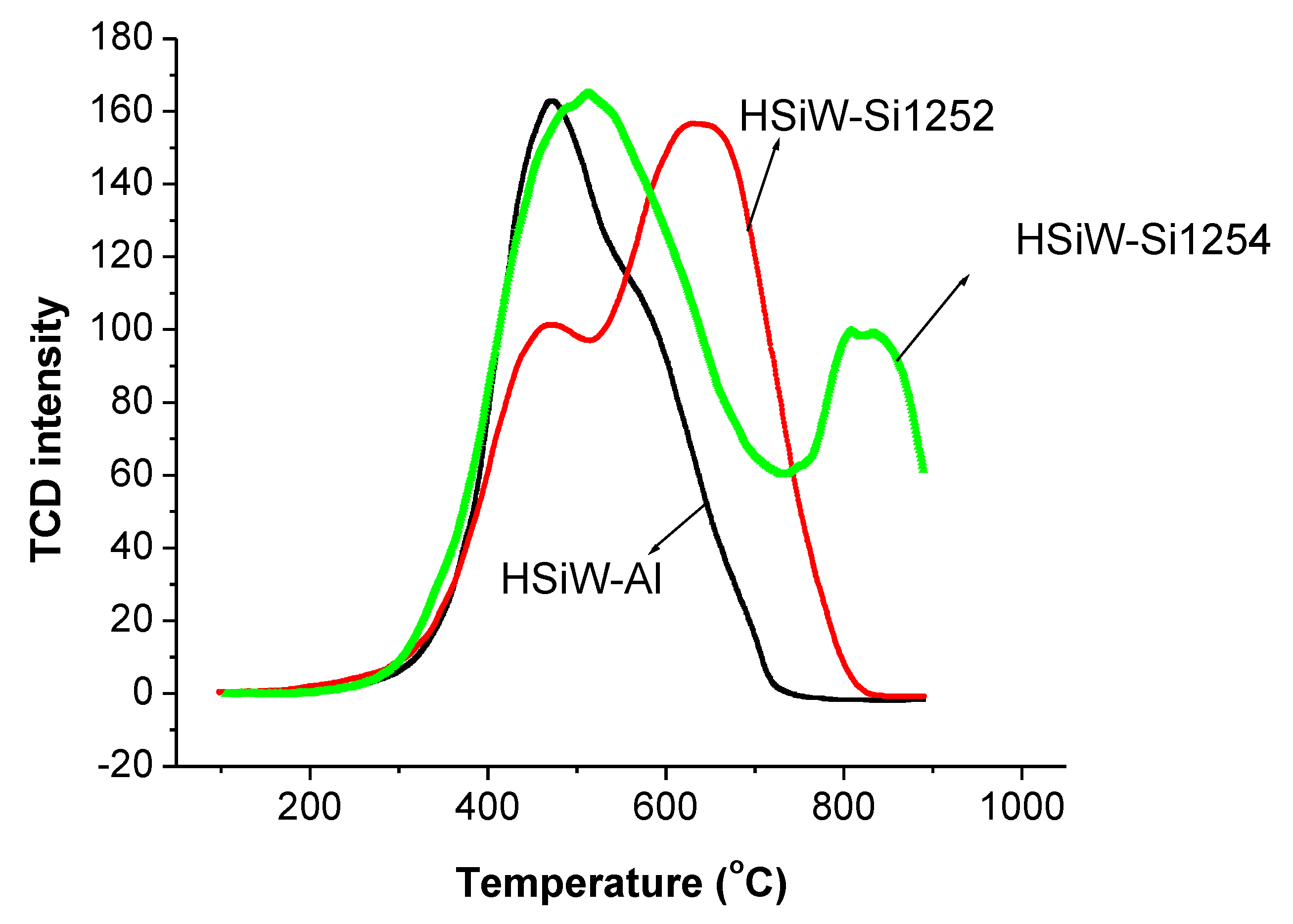
3.3. TPO Evaluation of Spent Catalysts
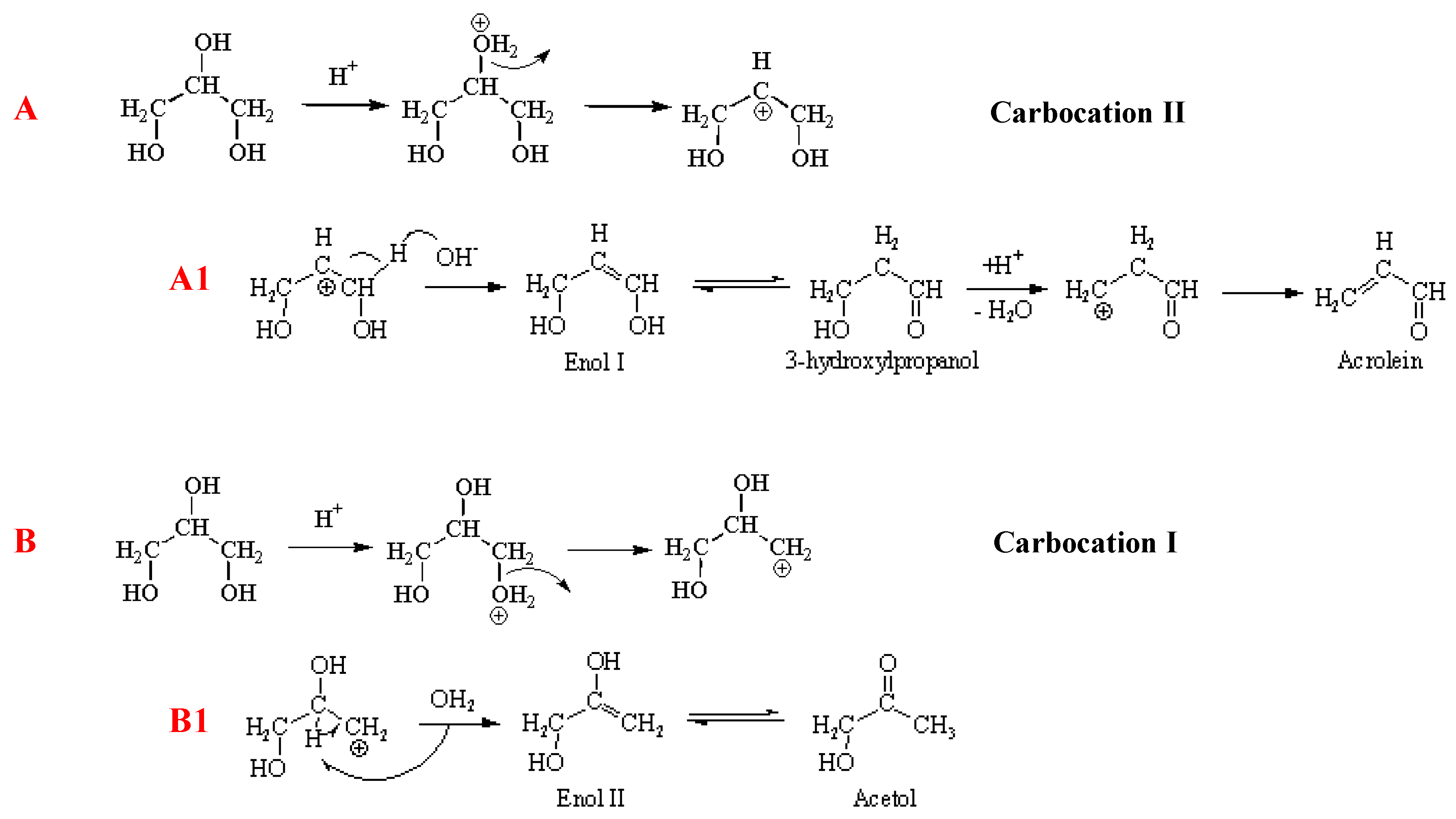
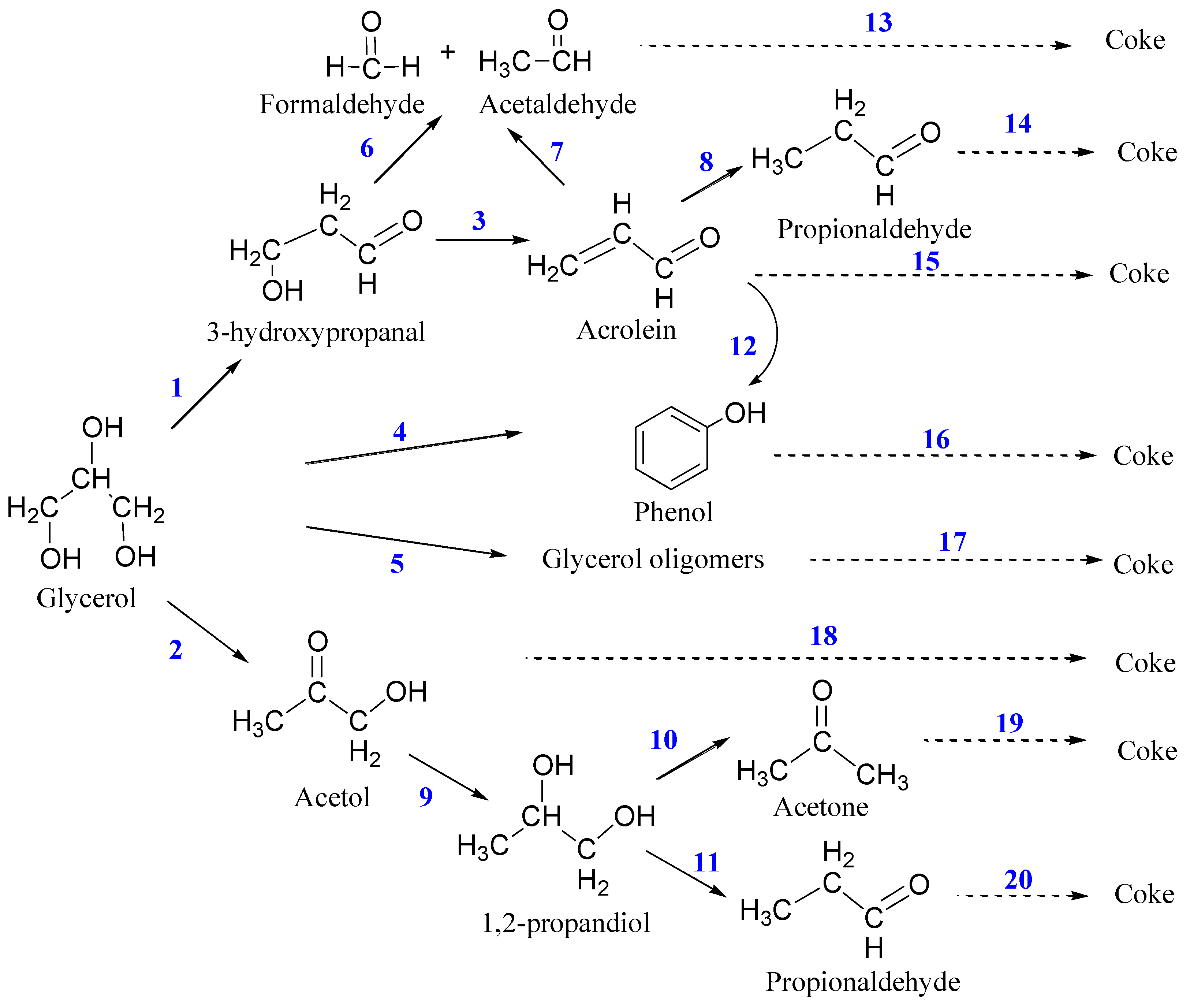
3.4. Proposed Reaction Network
4. Conclusions and Future Research
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zou, B.; Ren, S.; Ye, X. Glycerol Dehydration to Acrolein Catalyzed by ZSM-5 Zeolite in Supercritical Carbon Dioxide Medium. ChemSusChem 2016, 9, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ye, X.; Bozell, J. A Comparative Review of Petroleum-Based and Bio-Based Acrolein Production. ChemSusChem 2012, 5, 1162–1180. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.P.; Ren, S. 3-Value-Added Chemicals from Glycerol, in Soy-Based Chemicals and Materials; Brentin, R., Ed.; American Chemical Society: London, UK, 2014; pp. 43–80. [Google Scholar]
- Katryniok, B.; Paul, S.; Capron, M.; Dumeignil, F. Towards the Sustainable Production of Acrolein by Glycerol Dehydration. ChemSusChem 2009, 2, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Nimlos, M.R.; Blanksby, S.J.; Qiuan, X.; Himmel, M.E.; Johnson, D.K. Mechanisms of Glycerol Dehydration. J. Phys. Chem. A 2006, 110, 6145–6156. [Google Scholar] [CrossRef]
- Alhanash, A.; Kozhevnikova, E.; Kozhevnikov, I. Gas-phase dehydration of glycerol to acrolein catalysed by caesium heteropoly salt. Appl. Catal. A Gen. 2010, 378, 11–18. [Google Scholar] [CrossRef]
- Jia, C.J.; Liu, Y.; Schmidt, W.; Lu, A.-H.; Schüth, F. Small-sized HZSM-5 zeolite as highly active catalyst for gas phase dehydration of glycerol to acrolein. J. Catal. 2010, 269, 71–79. [Google Scholar] [CrossRef]
- Chai, S.H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Investigation of solid acid-base catalysts for gas-phase dehydration of glycerol. Green Chem. 2007, 9, 1130–1136. [Google Scholar] [CrossRef]
- Gu, Y.; Cui, N.; Yu, Q.; Li, C.; Cui, Q. Study on the influence of channel structure properties in the dehydration of glycerol to acrolein over H-zeolite catalysts. Appl. Catal. A Gen. 2012, 429, 9–16. [Google Scholar] [CrossRef]
- Kim, Y.T.; Jung, K.-D.; Park, E. Gas-phase dehydration of glycerol over ZSM-5 catalysts. Microporous Mesoporous Mater. 2010, 131, 28–36. [Google Scholar] [CrossRef]
- Ulgen, A.; Hoelderich, W. Conversion of glycerol to acrolein in the presence of WO3/ZrO2 catalysts. Catal. Lett. 2009, 131, 122–128. [Google Scholar] [CrossRef]
- Wu, Y.; Ye, X.; Yang, X.; Wang, X.; Chu, W.; Hu, Y. Heterogenization of heteropolyacids: A general discussion on the preparation of supported acid catalysts. Ind. Eng. Chem. Res. 1996, 35, 2546–2560. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Sustainable heterogeneous acid catalysis by heteropoly acids. J. Mol. Catal. A-Chem. 2007, 262, 86–92. [Google Scholar] [CrossRef]
- Timofeeva, M.N. Acid catalysis by heteropoly acids. Appl. Catal. A Gen. 2003, 256, 19–35. [Google Scholar] [CrossRef]
- Tsukuda, E.; Sato, S.; Takahashi, R.; Sodesawa, T. Production of acrolein from glycerol over silica-supported heteropoly acids. Catal. Commun. 2007, 8, 1349–1353. [Google Scholar] [CrossRef]
- Atia, H.; Armbruster, U.; Martin, A. Dehydration of glycerol in gas phase using heteropolyacid catalysts as active compounds. J. Catal. 2008, 258, 71–82. [Google Scholar] [CrossRef]
- Chai, S.-H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Gas-phase dehydration of glycerol over 12-tungstophosphoric acid supported on ZrO2 and SiO2. Green Chem. 2008, 10, 1087–1093. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicoponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Bardin, B.B.; Davis, R.J. Effect of water on silica-supported phosphotungstic acid catalysts for 1-butene double bond shift and alkane skeletal isomerization. Appl. Catal. A Gen. 2000, 200, 219–231. [Google Scholar] [CrossRef]
- Chu, W.; Yang, X.; Shan, Y.; Ye, X.; Wu, Y. Immobilization of the heteropoly acid (HPA) H4SiW12O40 (SiW12) on mesoporous molecular sieves (HMS and MCM-41) and their catalytic behavior. Catal. Lett. 1996, 42, 201–208. [Google Scholar] [CrossRef]
- Verhoef, M.J.; Kooyman, P.; Peters, J.; van Bekkum, H. A study on the stability of MCM-41-supported heteropoly acids under liquid- and gas-phase esterification conditions. Microporous Mesoporous Mater. 1999, 27, 365–371. [Google Scholar] [CrossRef]
- Izumi, Y.; Hasebe, R.; Urabe, K. Catalysis by heterogeneous supported heteropoly acid. J. Catal. 1983, 84, 402–409. [Google Scholar] [CrossRef]
- Nowinska, K.; Field, R.; Adamiec, J. Catalytic activity of supported heteropoly acids for reactions requiring strong acid centres. J. Chem. Soc. Faraday Trans. 1991, 87, 749–753. [Google Scholar] [CrossRef]
- Quantachrome. User Manual for ChemBET Pulsar TPR/TPD; Quantachrome Instrument: Boynton Beach, FL, USA, 2008; pp. 94–96. [Google Scholar]
- Lowell, S.; Shields, J.E.; Thomas, M.A. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density Particle Technology Series; Scarlett, B., Ed.; Springer: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Yuan, Q.; Yin, A.-X.; Luo, C.; Sun, L.-D.; Zhang, Y.-W.; Duan, W.-T.; Liu, H.-C.; Yan, C.-H. Facile Synthesis for Ordered Mesoporous γ-Aluminas with High Thermal Stability. J. Am. Chem. Soc. 2008, 130, 3465–3472. [Google Scholar] [CrossRef]
- Tsyganenko, A.; Storozheva, E.; Manoilova, O.; Lesage, T.; Daturi, M.; LaValley, J.-C. Brønsted acidity of silica silanol groups induced by adsorption of acids. Catal. Lett. 2000, 70, 159–163. [Google Scholar] [CrossRef]
- Levchenko, A.A.; Jain, P.; Trofymluk, O.; Yu, P.; Navrotsky, A.; Sen, S. Nature of molecular rotation in supercooled glycerol under nanoconfinement. J. Phys. Chem. 2010, 114, 3070–3074. [Google Scholar] [CrossRef]
- Tomin, V.I.; Hubis, K. Instantaneous emission spectra and molecular rotation of n-dimethylaminobenzonitrile fluorescing in the long-wavelength spectral range. Opt. Spectrosc. 2005, 100, 65–74. [Google Scholar]
- Hao, L.; Leaist, D.G. Large soret effect for silicotungstic acid in a supporting electrolyte: 9% Change in concentration per degree. J. Phys. Chem. 1994, 98, 13741–13744. [Google Scholar] [CrossRef]
- Suprun, W.; Lutecki, M.; Haber, T.; Papp, H. Acidic catalysts for the dehydration of glycerol: Activity and deactivation. J. Mol. Catal. A Chem. 2009, 309, 71–78. [Google Scholar] [CrossRef]
- Corma, A.; Huber, G.W.; Sauvanaud, L.; O’Connor, P. Biomass to chemicals: Catalytic conversion of glycerol/water mixtures into acrolein, reaction network. J. Catal. 2008, 257, 163–171. [Google Scholar] [CrossRef]
- Wolf, E.E.; Petersen, E.E. Catalysts deactivation by coking. Catal. Rev. Sci. Eng. 2007, 24, 329–371. [Google Scholar] [CrossRef]
- Tanabe, K. Solid Acids and Bases: Their Catalytic Properties. Studies in Surface Sciences and Catalysis; Elsevier: Tokyo, Japan, 1989. [Google Scholar]
- Liu, L. Roles of Non-thermal Plasma in Gas-phase Glycerol Dehydration Catalyzed by Supported Silicotungstic Acid. Ph.D. Thesis, The University of Tennessee, Knoxville, TN, USA, 2011. [Google Scholar]
- NIH. National Library of Medicine, Material Data Sheet. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Glycerol (accessed on 31 December 2022).
- Yaws, C.L.; Narasimhan, P.; Gabbula, C. Yaws’ Handbook of Antoine Coefficients for Vapor Pressure, 2nd ed.; Knovel: New York, NY, USA, 2009. [Google Scholar]
- The Dortmund Data Bank. Available online: http://www.ddbst.com/en/online/Online_Calc_vap_Form.php?component=Water (accessed on 31 December 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mesoporous Support | Si1252 | Si1254 | Al2700 |
|---|---|---|---|
| Shape | granules | granules | beads |
| Particle size (mm) | 1–3 | 1–3 | 1.2–2.4 |
| Average pore diameter (nm) | 11 | 6 | 30 |
| Pore volume (cc/g) | 1.02 | 0.81 | 1.10 |
| Surface area (m2/g) | 390 | 540 | 150 |
| Density (g/cm3) | 0.43 | 0.38 | 0.40 |
| Glycerol Conversion (mol%) | Selectivity (mol%) | Coke (wt.%) 4 | Acrolein Yield (mol%) | ||||
|---|---|---|---|---|---|---|---|
| Acrolein | Acetaldehyde | Propion-aldehyde | Acetol | ||||
| HSiW-Si1252 | 92.9 1 ± 0.6 2 (98.7 ± 0.7) 3 | 79.5 ± 0.2 | 1.1± 0.2 | 1.1± 0.0 | 7.6 ± 0.3 | 14.3 ± 0.5 | 73.9 |
| HSiW-Si1254 | 89.0± 1.2 (98.0 ± 0.8) | 74.4 ± 0.7 | 0.8± 0.1 | 0.9± 0.1 | 4.2 ± 0.9 | 19.4 ± 0.9 | 66.2 |
| HSiW-Al | 96.2± 0.3 (98.0 ± 1.1) | 77.0 ± 0.9 | 1.9± 0.1 | 1.4± 0.1 | 11.6 ± 0.9 | 8.7 ± 0.9 | 74.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Yu, F.; Wang, S.; Ye, X.P. Glycerol Dehydration to Acrolein Catalyzed by Silicotungstic Acid: Effect of Mesoporous Support. Eng 2023, 4, 206-222. https://doi.org/10.3390/eng4010012
Liu L, Yu F, Wang S, Ye XP. Glycerol Dehydration to Acrolein Catalyzed by Silicotungstic Acid: Effect of Mesoporous Support. Eng. 2023; 4(1):206-222. https://doi.org/10.3390/eng4010012
Chicago/Turabian StyleLiu, Lu, Fei Yu, Siqun Wang, and Xiaofei Philip Ye. 2023. "Glycerol Dehydration to Acrolein Catalyzed by Silicotungstic Acid: Effect of Mesoporous Support" Eng 4, no. 1: 206-222. https://doi.org/10.3390/eng4010012
APA StyleLiu, L., Yu, F., Wang, S., & Ye, X. P. (2023). Glycerol Dehydration to Acrolein Catalyzed by Silicotungstic Acid: Effect of Mesoporous Support. Eng, 4(1), 206-222. https://doi.org/10.3390/eng4010012