
Synthesis and Theoretical Studies of Aromatic Azaborines




Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Modeling
2.2. Syntheses
2.3. HPCCC Purifications
3. Results
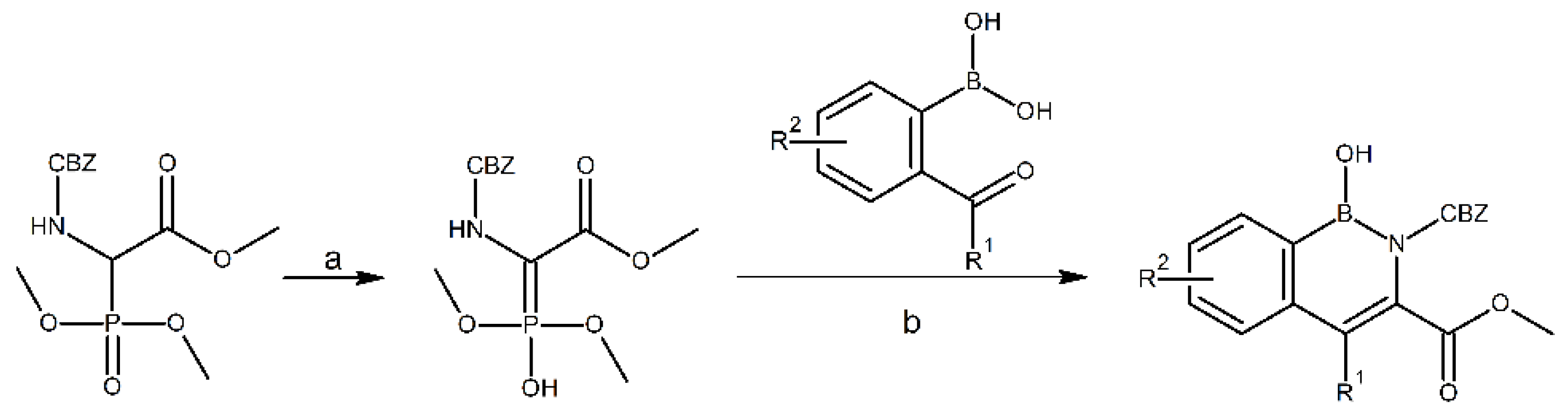
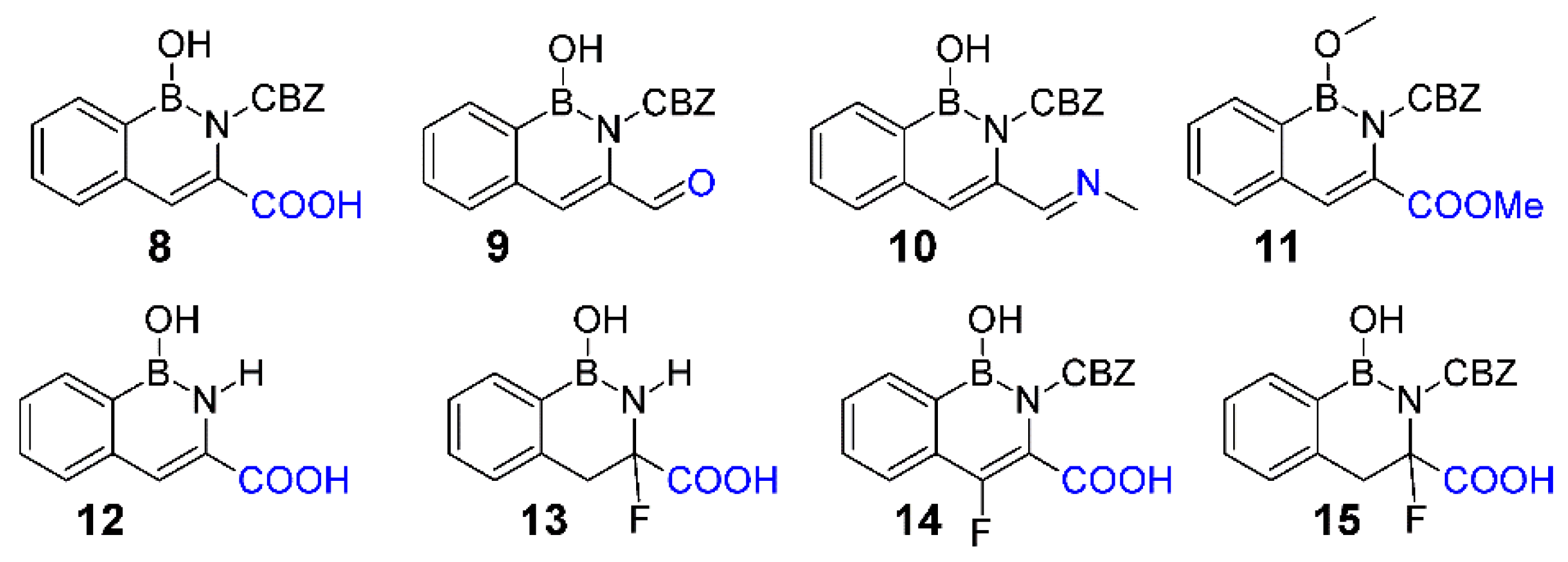
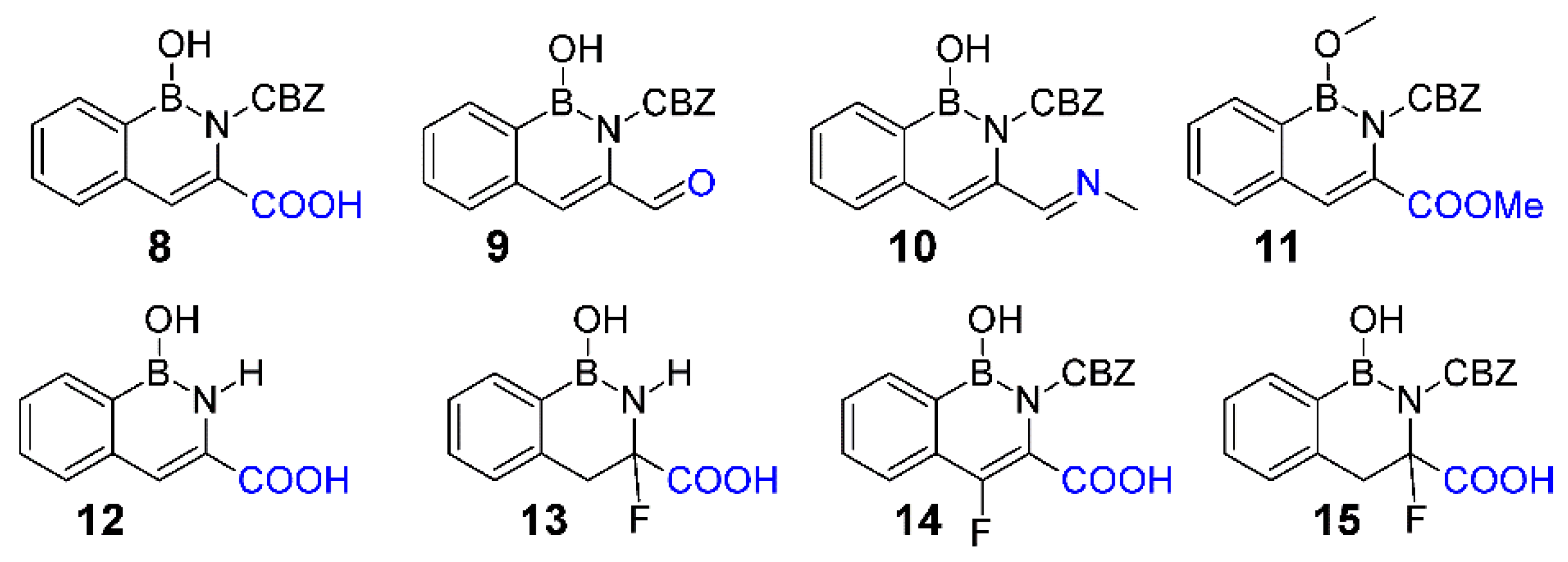
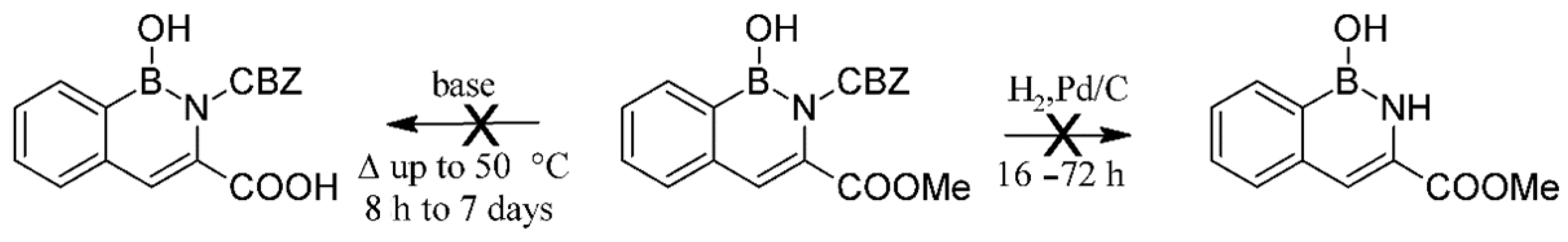
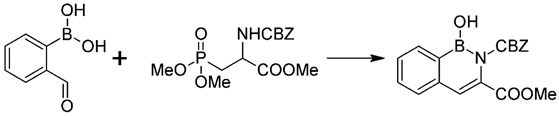
3.1. Syntheses
3.2. Molecular Stability
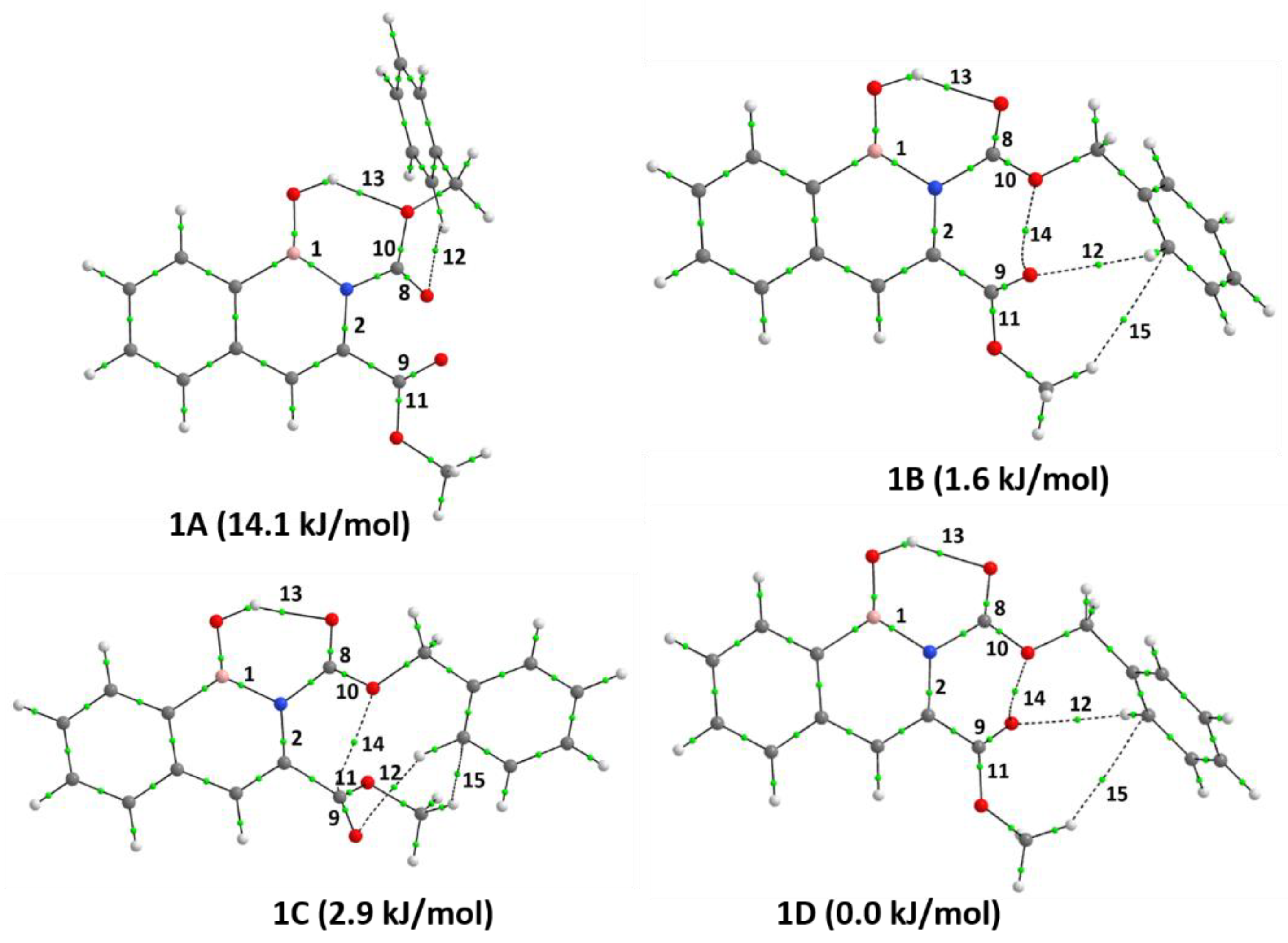
3.3. Intramolecular Interactions
4. Discussion
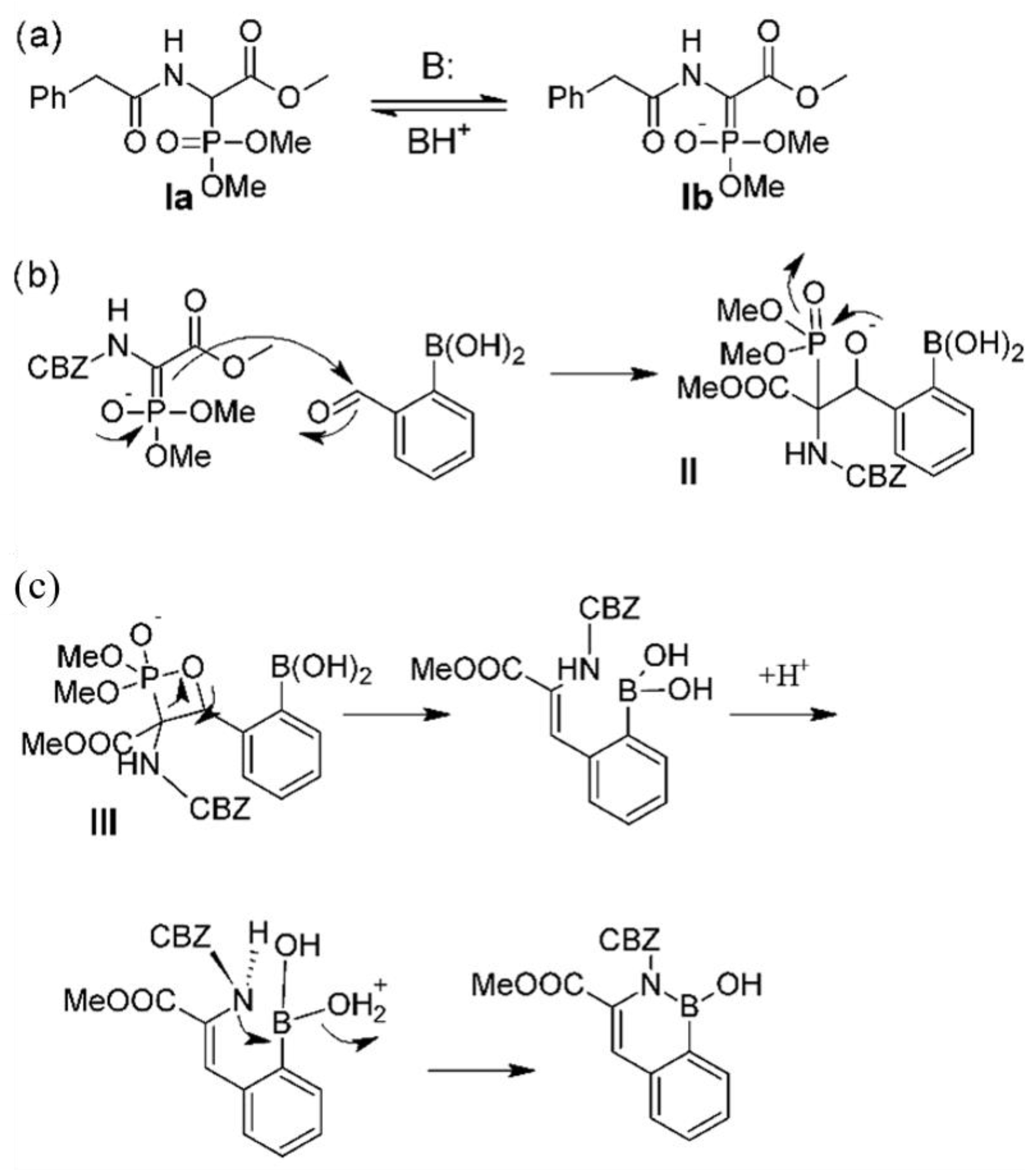
4.1. Proposed Reaction Mechanism
4.2. Relative Stability and Nature of Bonding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef]
- von Grotthuss, E.; John, A.; Kaese, T.; Wagner, M. Doping polycyclic aromatics with boron for superior performance in materials science and catalysis. Asian J. Org. Chem. 2018, 7, 37–53. [Google Scholar] [CrossRef]
- Saint-Louis, C.J.; Magill, L.L.; Wilson, J.A.; Schroeder, A.R.; Harrell, S.E.; Jackson, N.S.; Trindell, J.A.; Kim, S.; Fisch, A.R.; Munro, L.; et al. The synthesis and characterization of highly fluorescent polycyclic azaborine chromophores. J. Org. Chem 2016, 81, 10955–10963. [Google Scholar] [CrossRef]
- Noda, H.; Furutachi, M.; Asada, Y.; Shibasaki, M.; Kumagai, N. Unique physicochemical and catalytic properties dictated by the B3NO2 ring system. Nat. Chem. 2017, 9, 571–577. [Google Scholar] [CrossRef]
- Saint-Louis, C.J.; Shavnore, R.N.; McClinton, C.D.C.; Wilson, J.A.; Magill, L.L.; Brown, B.M.; Lamb, R.W.; Webster, C.E.; Schrock, A.K.; Huggins, M.T. Synthesis, computational, and spectroscopic analysis of tunable highly fluorescent BN-1,2-azaborine derivatives containing the N-BOH moiety. Org. Biomol. Chem 2017, 15, 10172–10183. [Google Scholar] [CrossRef]
- Choi, J.H.; Lim, H.J. Mild one-pot Horner-Wadsworth-Emmons olefination and intramolecular N-arylation for the syntheses of indoles, all regio-isomeric azaindoles, and thienopyrroles. Org. Biomol. Chem. 2015, 13, 5131–5138. [Google Scholar] [CrossRef] [PubMed]
- Lescure, L.R.; Jesse, T.; Groziak, M.P.; Powers, X.B.; Olmstead, M.M. Polycyclic aromatic heterocycles with a Benzo[c][1,2]azaborinine core. J. Heterocycl. Chem. 2019, 56, 2960–2965. [Google Scholar] [CrossRef]
- Gwynne, E.; Holt, J.; Dwan, J.; Appoh, F.; Vogels, C.; Decken, A.; Westcott, S. Reaction of hydantoin with boronic acids. Helv. Chim. Acta 2010, 93, 1093–1100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll, version 19.2.13; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Puljula, E.; Vepsäläinen, J.; Turhanen, P.A. Synthesis of medronic acid monoesters and their purification by high-performance countercurrent chromatography or by hydroxyapatite. Beilstein J. Org. Chem. 2016, 12, 2145–2149. [Google Scholar] [CrossRef] [Green Version]
- Puljula, E.; Turhanen, P.A. Semi-preparative high-performance countercurrent chromatography method for the purification of chemically synthesized ATP analogue, ApppI. J. Chromatogr. B 2017, 1063, 180–182. [Google Scholar] [CrossRef]
- Turhanen, P.A. Synthesis of a biologically important adenosine triphosphate analogue, ApppD. ACS Omega 2017, 2, 2835–2838. [Google Scholar] [CrossRef] [Green Version]
- Park, K.C. A high-yield synthesis of 4-Borono-dl-phenylalanine. Synthesis 1999, 1999, 2041–2044. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Time | Temperature | Equiv (Boronic Acid) | Equiv (Glynate) | Equiv (Base) | Yield/Conversion |
| 1 | DBU | DCM | 16 h | rt | 1.0 | 1.05 | 1.1 | 136 mg/25% a |
| 2 | TEA | DCM | 16 h | rt | 1.0 | 1.05 | 1.1 | n.d. a |
| 3 | DIPEA | DCM | 16 h | rt | 1.0 | 1.05 | 1.1 | n.d. a |
| 4 | NMM | DCM | 16 h | rt | 1.0 | 1.05 | 1.1 | n.d. a |
| 5 | DBU | THF | 16 h | rt | 1.0 | 1.05 | 1.1 | 102 mg/39% a |
| 6 | DBU | MeCN | 16 h | rt | 1.0 | 1.05 | 1.1 | 132 mg/31% a |
| 7 | DBU | MeCN | 8 h | rt | 1.0 | 1.05 | 1.1 | 32% a, b |
| 8 | DBU | MeCN | 4 h | rt | 1.0 | 1.05 | 1.1 | 31% a, b |
| 9 | DBU | MeCN | 3 h | rt | 1.0 | 1.05 | 1.1 | 32% a, b |
| 10 | DBU | MeCN | 2 h | rt | 1.0 | 1.05 | 1.1 | 5% a, b |
| 11 | DBU | MeCN | 1 h | rt | 1.0 | 1.05 | 1.1 | n.d. a, b |
| 12 | DBU | MeCN | 4 h | rt | 1.0 | 1.0 | 1.0 | 24% c |
| 13 | DBU | MeCN | 4 h | rt | 1.0 | 1.0 | 2.0 | 49% c |
| 14 | DBU | MeCN | 4 h | rt | 1.0 | 1.0 | 3.0 | 46% c |
| 15 | DBU | MeCN | 4 h | rt | 2.0 | 1.0 | 1.0 | 36% c |
| 16 | DBU | MeCN | 4 h | rt | 2.0 | 1.0 | 2.0 | 45% c |
| 17 | DBU | MeCN | 4 h | rt | 2.0 | 1.0 | 3.0 | 35% c |
| 18 | NaH + DBU | ACNe | 4 h | rt | 1.2 + 1.0 | 1.0 | 1.0 | 114 mg/29% a |
| Boronic Acid | Yield | Best Conditions | Boronic Acid | Yield | Best Conditions |
|---|---|---|---|---|---|
 | n.d. | 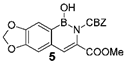 | 10% | 100 mg (2 equiv) boronic acid, 86 mg (1 eq) trimethyl phosphonoglycinate, 116 µL (3 equiv) DBU. | |
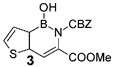 | n.d. | 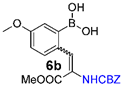 | 7% | 50 mg (1 equiv) boronic acid, 85 mg (1 eq) trimethyl phosphonoglycinate, 116 µL (3 equiv) DBU. | |
 | 30% | 100 mg (2 equiv) boronic acid, 90 mg (1 eq) trimethyl phosphonoglycinate, 81 µL (2 equiv) DBU. | 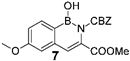 | 22% | 100 mg (2 equiv) boronic acid, 92 mg (1 eq) phosphonoglycine trimethyl ester, 125 µL (3 equiv) DBU. |
| 1 | 3 | 4 | 7 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ρ | 1.19 | 1.17 | 1.20 | 1.18 | 1.20 | 1.20 | 1.24 | 1.31 | 1.33 | 1.19 | 1.19 |
| |V|/G | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.49 | 1.51 | 1.51 | 1.53 |
| DI | 0.41 | 0.41 | 0.41 | 0.40 | 0.41 | 0.41 | 0.42 | 0.44 | 0.44 | 0.41 | 0.40 |
| EBCP | −532 | −523 | −537 | −527 | −538 | −538 | −566 | −610 | −618 | −533 | −524 |
| BCP | Type | d [Å] | ρ [e/Å3] | E [kJmol−1] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A | 1B | 1C | 1D | 1A | 1B | 1C | 1D | 1A | 1B | 1C | 1D | ||
| 1 | B-N | 1.472 | 1.473 | 1.481 | 1.475 | 1.191 | 1.189 | 1.169 | 1.186 | −532 | −532 | −519 | −530 |
| 2 | N-C | 1.405 | 1.401 | 1.407 | 1.403 | 1.973 | 1.978 | 1.954 | 1.973 | −839 | −851 | −829 | −838 |
| 8 | C=O | 1.205 | 1.218 | 1.220 | 1.219 | 2.840 | 2.771 | 2.764 | 2.765 | −1973 | −1877 | −1864 | −1869 |
| 9 | C=O | 1.209 | 1.212 | 1.210 | 1.212 | 2.801 | 2.784 | 2.800 | 2.785 | −1940 | −1917 | −1933 | −1919 |
| 10 | C-O | 1.349 | 1.329 | 1.332 | 1.329 | 2.053 | 2.155 | 2.130 | 2.157 | −1104 | −1209 | −1194 | −1212 |
| 11 | C-O | 1.340 | 1.338 | 1.340 | 1.339 | 2.076 | 2.084 | 2.077 | 2.079 | −1158 | −1168 | −1158 | −1162 |
| 12 | O…H | 2.298 | 2.660 | 2.705 | 2.598 | 0.051 | 0.042 | 0.039 | 0.048 | −6 | −5 | −5 | −6 |
| 13 | O…H | 1.876 | 1.845 | 1.810 | 1.826 | 0.216 | 0.232 | 0.252 | 0.242 | −38 | −40 | −44 | −42 |
| 14 | O…O | - | 2.760 | 2.645 | 2.733 | - | 0.102 | 0.111 | 0.105 | - | −16 | −17 | −17 |
| 15 | H…C | - | 2.999 | 3.044 | 3.083 | - | 0.028 | 0.026 | 0.024 | - | −2 | −2 | −2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirva, P.; Turhanen, P.; Timonen, J.M. Synthesis and Theoretical Studies of Aromatic Azaborines. Organics 2022, 3, 196-209. https://doi.org/10.3390/org3030016
Hirva P, Turhanen P, Timonen JM. Synthesis and Theoretical Studies of Aromatic Azaborines. Organics. 2022; 3(3):196-209. https://doi.org/10.3390/org3030016
Chicago/Turabian StyleHirva, Pipsa, Petri Turhanen, and Juri M. Timonen. 2022. "Synthesis and Theoretical Studies of Aromatic Azaborines" Organics 3, no. 3: 196-209. https://doi.org/10.3390/org3030016
APA StyleHirva, P., Turhanen, P., & Timonen, J. M. (2022). Synthesis and Theoretical Studies of Aromatic Azaborines. Organics, 3(3), 196-209. https://doi.org/10.3390/org3030016