
Enantioselective Total Synthesis of Multifidene, a Sex Pheromone of Brown Algae


Abstract
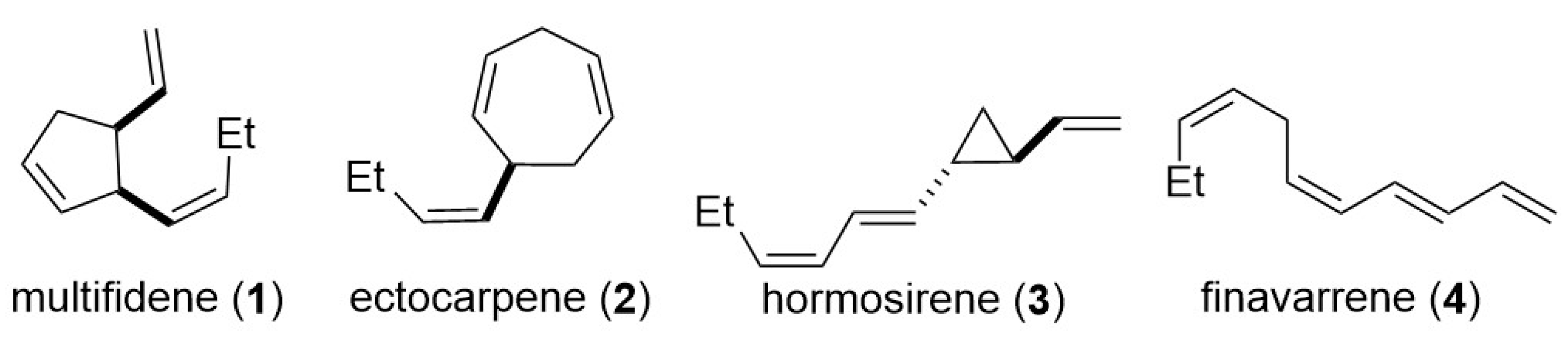
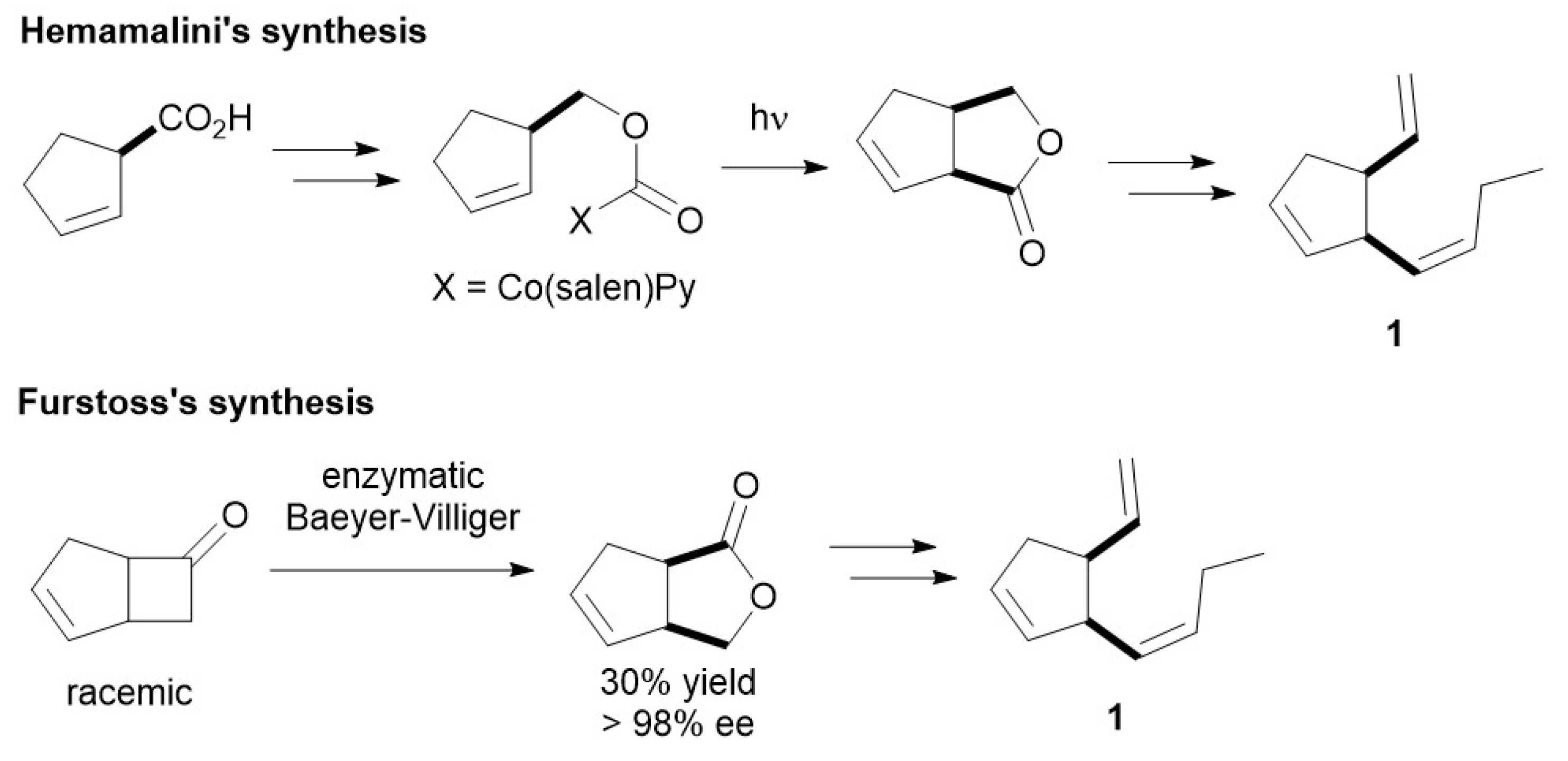
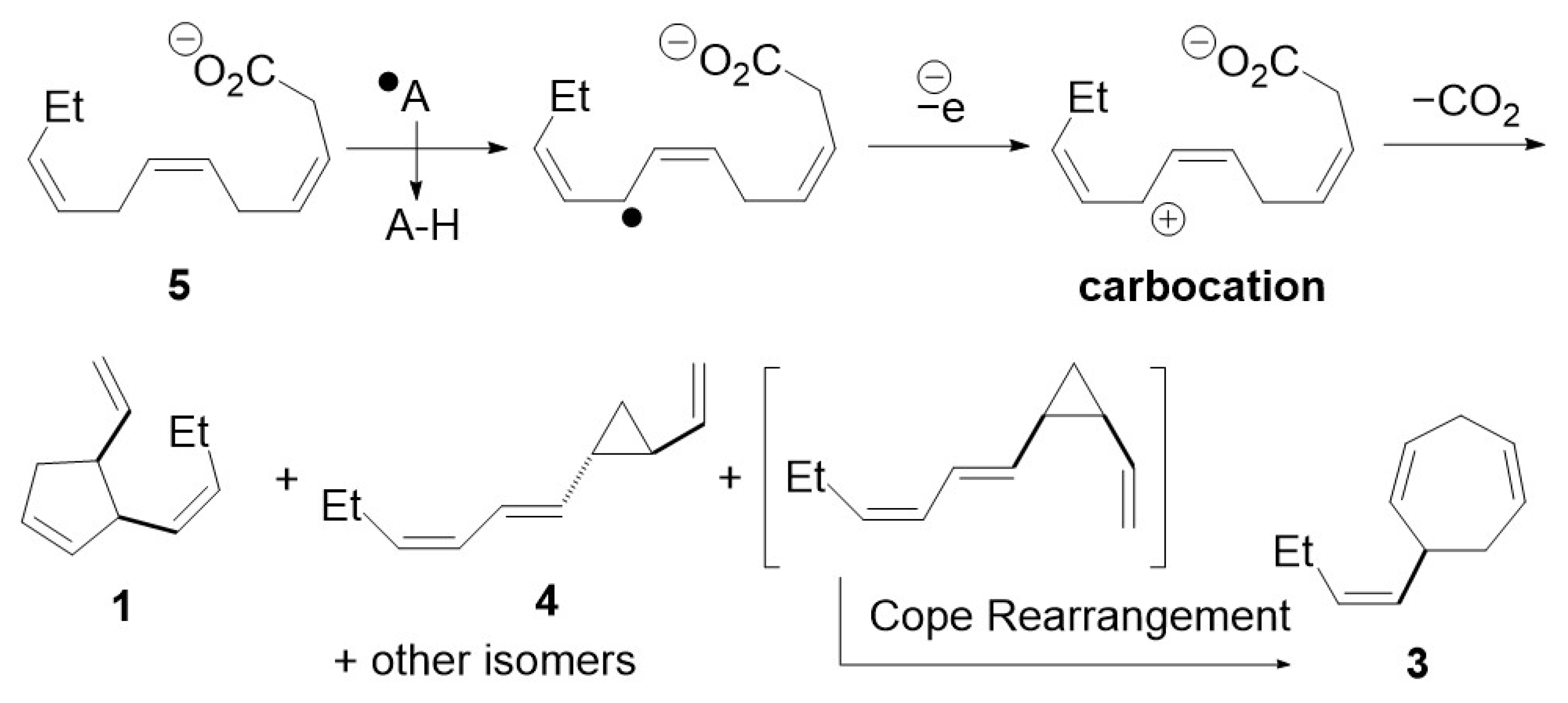
:1. Introduction
2. Materials and Methods
2.1. General Methods
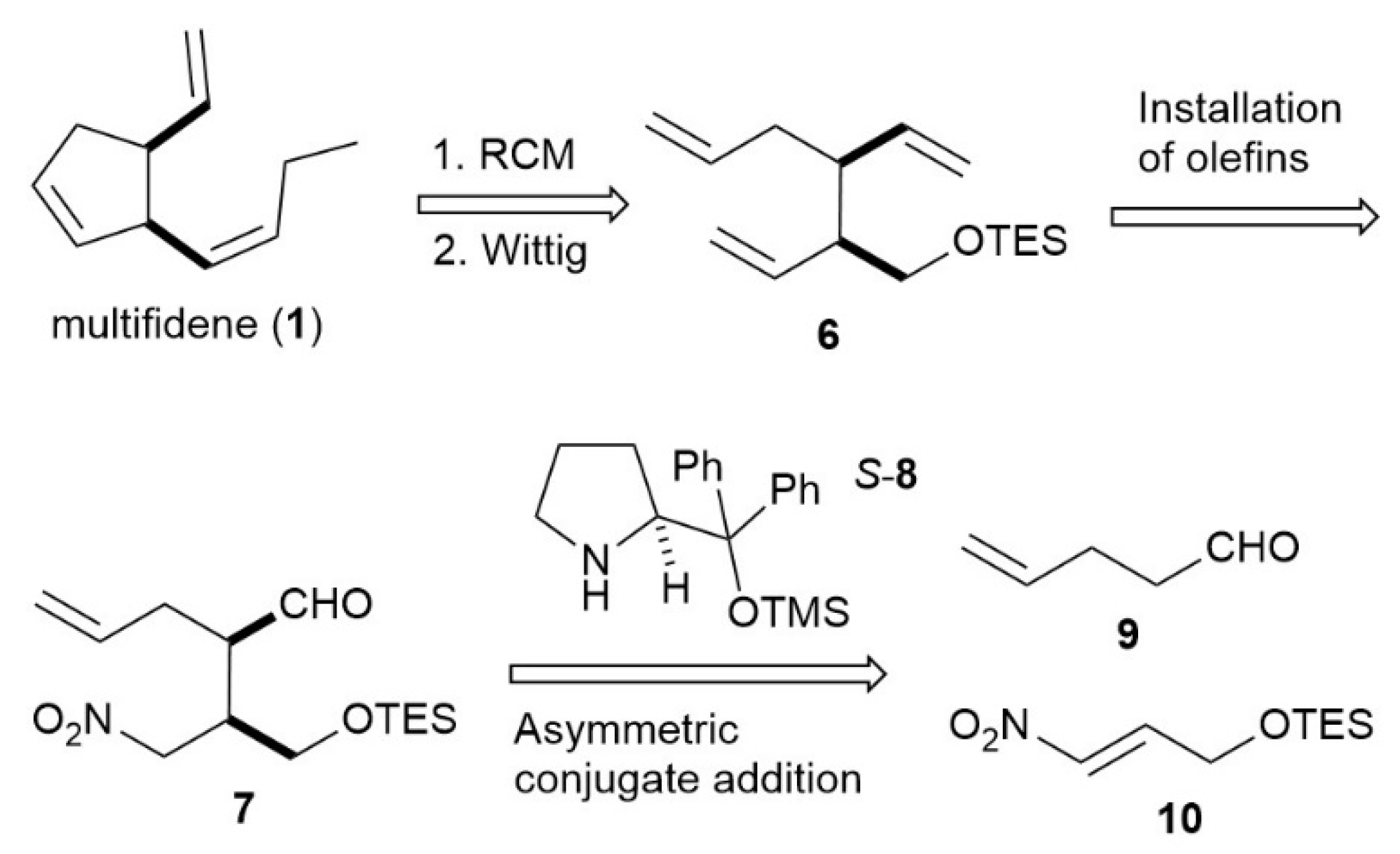
2.2. Total Synthesis of Multifidene (1)
2.2.1. Alcohol S1
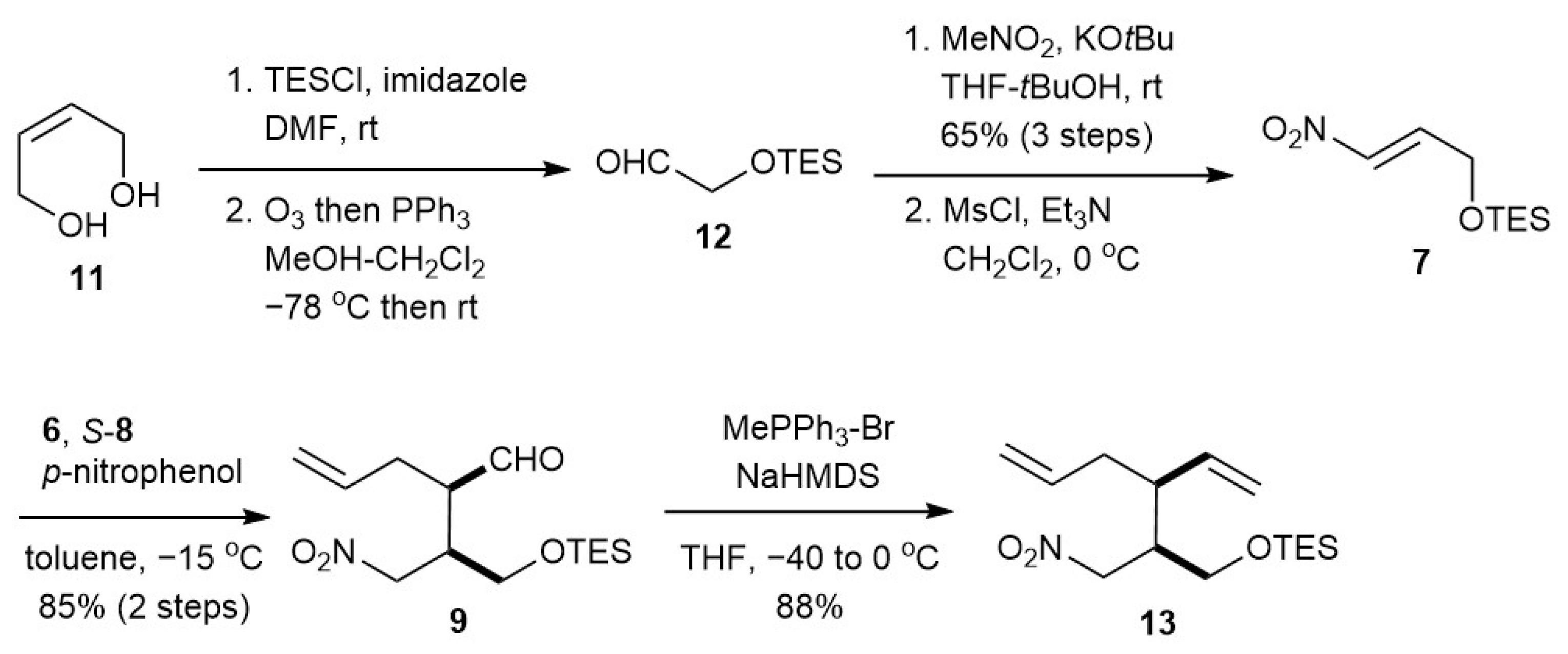
2.2.2. Adduct 7
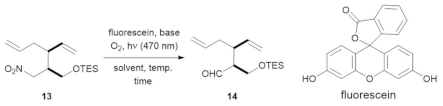
2.2.3. Diolefin 13
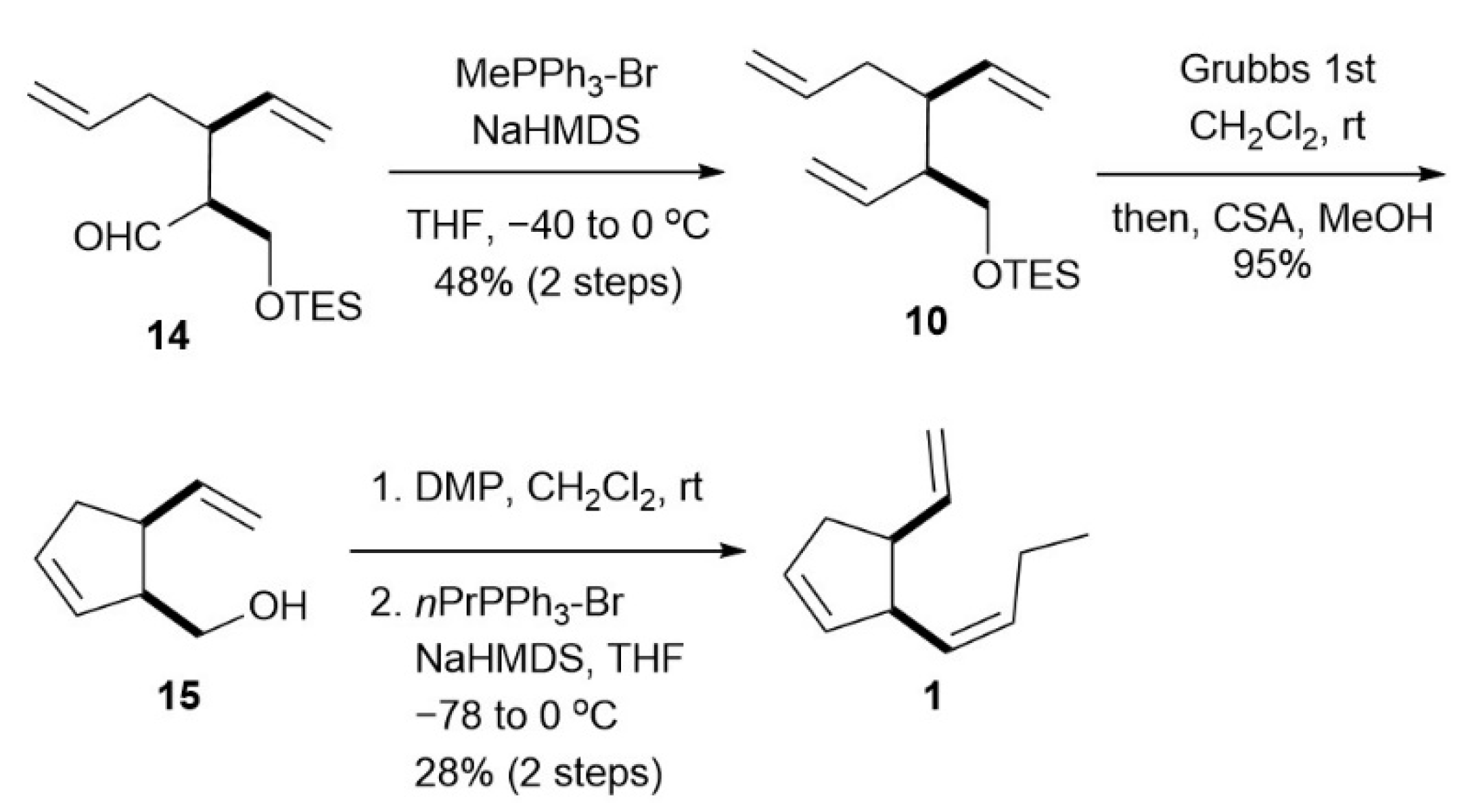
2.2.4. Triolefin 6
2.2.5. Cyclopentene 15
2.2.6. Multifidene (1)
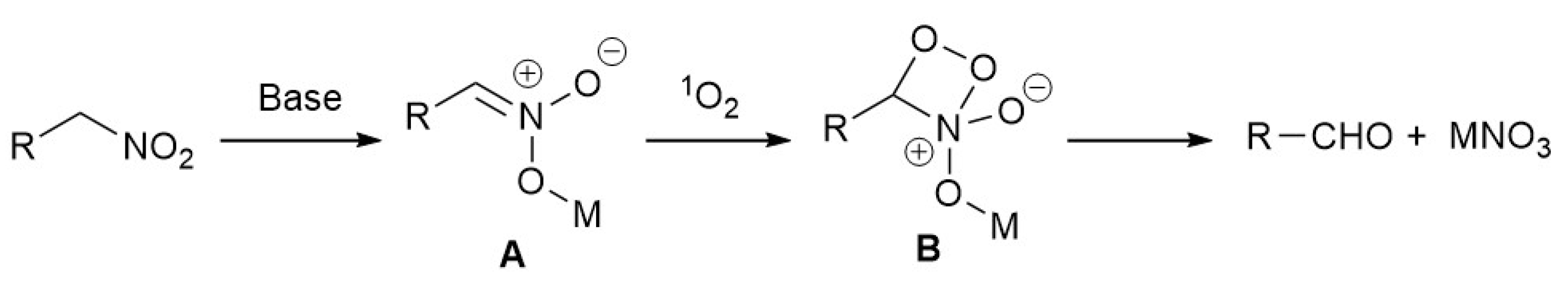
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jaenicke, L.; Müller, D.G.; Moore, R.E. Multifidene and Aucantene, C11 Hydrocarbons in the Male-Attracting Essential Oil from the Gynogametes of Cutleria multifida (Phaeophyta). J. Am. Chem. Soc. 1974, 96, 3324–3325. [Google Scholar]
- Müller, D.G.; Jaenicke, L.; Donike, M.; Akintobi, T. Sex Attractant in a Brown Alga: Chemical Structure. Science 1971, 171, 815–817. [Google Scholar]
- Moore, R.E.; Pettus, J.A., Jr. Isolation and Structure Determination of Dictyopterenes C′ and D′ from Dictyopteris. Stereospecificity in the Cope Rearrangement of Dictyopterenes A and B. J. Am. Chem. Soc. 1971, 93, 3087–3088. [Google Scholar]
- Moore, R.E.; Pettus, J.A., Jr.; Doty, M.S. Dictyopterene, A. An Odoriferous Constituent from Algae of the Genus Dictyopteris. Tetrahedron Lett. 1968, 9, 4787–4790. [Google Scholar]
- Pettus, J.A., Jr.; Moore, R.E. Isolation and Structure Determination of an Undeca-1,3,5,8-tetraene and Dictyopterene B from Algae of the Genus Dictyopteris. J. Chem. Soc. D. 1970, 1970, 1093–1094. [Google Scholar]
- Jaenicke, L.; Boland, W. Signal Substances and Their Reception in the Sexual Cycle of Marine Brown Algae. Angew. Chem. Int. Ed. Engl. 1982, 94, 643–653. [Google Scholar]
- Maier, I.; Müller, D.G. Sexual Pheromones in Algae. Bio. Bull. 1986, 170, 145–175. [Google Scholar]
- Boland, W. Chemische Kommunikation bei der Sexuellen Fortpflanzung Mariner Braunalgen. Biologie in Unserer Zeit 1987, 17, 176–185. [Google Scholar]
- Jaenicke, L. One Hundred and One Years of Chemotaxis. Pfeffer, Pheromones, and Fertilization. Bot. Acta 1988, 101, 149–159. [Google Scholar]
- Maier, I. Gamete Orientation and Induction of Gametogenesis by Pheromones in Algae and Plants. Plant Cell Environ. 1993, 16, 891–907. [Google Scholar]
- Derenbach, J.B.; Pesandeo, D. Investigations into a Small Fraction of Volatile Hydrocarbons: III. Two Diatom Cultures Produce Ectocarpene, a Pheromone of Brown Algae. Mar. Chem. 1986, 19, 337–432. [Google Scholar]
- Boland, W. The Chemistry of Gamete Attraction: Chemical Structures, Biosynthesis, and (A)biotic Degradation of Algal Pheromones. Proc. Natl. Acad. Sci. USA 1995, 92, 37–43. [Google Scholar]
- Jüttner, F.; Wurster, K. Evidence of Ectocarpene and Dictyopterenes A and C′ in the Water of a Freshwater Lake. Limnol. Oceanogr. 1984, 29, 1322–1324. [Google Scholar]
- Boland, W.; Marner, F.-J.; Jaenicke, L.; Müller, D.G.; Folster, E. Comparative Receptor Study in Gamete Chemotaxis of the Seaweeds Ectocarpus siliculosus and Cutleria multifida. Eur. J. Biochem. 1983, 134, 97–103. [Google Scholar]
- Hay, M.E.; Duffy, J.E.; Fenical, W.; Gustafson, K. Chemical Defense in the Seaweed Dictyopteris delicatula: Differential Effects against Reef Fishes and Amphipods. Mar. Ecol. Prog. Ser. 1988, 48, 185–192. [Google Scholar]
- Lebreton, J.; Alphand, V.; Furstoss, R. A Short Chemoenzymatic Synthesis of (+)-Multifidene and (+)-Viridiene. Tetrahedron Lett. 1996, 37, 1011–1014. [Google Scholar]
- Kramp, P.; Helmchen, G.; Holmes, A.B. Syntheses of Enantiomerically Pure ent-Multifidene and Related Compounds. J. Chem. Soc. Chem. Commun. 1993, 551–552. [Google Scholar]
- Wirth, D.; Fischer-Lui, I.; Boland, W.; Icheln, D.; Runge, T.; Konig, W.A.; Phillips, J.; Clayton, M. Unusual and Novel C11H16 Hydrocarbons from the Southern Australian Brown Alga Dictyopteris acuostichoides (Phaeophyceae). Helv. Chim. Acta 1992, 75, 734–744. [Google Scholar]
- Crouse, G.D.; Paquette, L.A. Total Synthesis of (±)-Multifidene, the Gamete Attractant of the Phaeophyte Cutleria multifida. J. Org. Chem. 1981, 46, 4272–4274. [Google Scholar]
- Boland, W.; Jaenicke, L.; Müller, D.G. Synthese und biologische Aktivitaten von (+)- und (−)-Multifiden. Liebigs Ann. Chem. 1981, 1981, 2266–2271. [Google Scholar]
- Boland, W.; Jaenicke, L. Stereoselective Synthesis of “Multifidenes” and Related Structures. Contribution to the Synthetic Chemistry of Cis-Disubstituted Alken-l-ylcyclopentenes. J. Org. Chem. 1979, 44, 4819–4824. [Google Scholar]
- Hemamalini, S.; Scheffold, R. Synthesis of (+)-Multifidene. Helv. Chim. Acta 1995, 78, 447–451. [Google Scholar]
- Kinoshita-Terauchi, N.; Shiba, K.; Umezawa, T.; Matsuda, F.; Motomura, T.; Inaba, K. A Brown Algal Sex Pheromone Reverses the Sign of Phototaxis by cAMP/Ca2+-Dependent Signaling in the Male Gametes of Mutimo cylindricus (Cutleriaceae). J. Photochem. Photobiol. B 2019, 192, 113–123. [Google Scholar]
- Boland, W.; Mertes, K. Biosynthesis of Algal Pheromones. Eur. J. Biochem. 1985, 147, 83–91. [Google Scholar]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar]
- Patora-Komisarska, K.; Benohoud, M.; Ishikawa, H.; Seebach, D.; Hayashi, Y. Organocatalyzed Michael Addition of Aldehydes to Nitro Alkenes—Generally Accepted Mechanism Revisited and Revised. Helv. Chim. Acta 2011, 94, 719–745. [Google Scholar]
- Humphrey, J.M.; Arnold, E.P.; Chappie, T.A.; Feltenberger, J.B.; Nagel, A.; Simon, W.; Suarez-Contreras, M.; Tom, N.J.; O’Neill, B.T. Diastereoselective Synthesis of 2,3,6-Trisubstituted Piperidines. J. Org. Chem. 2009, 74, 4525–4536. [Google Scholar]
- Umemiya, S.; Nishino, K.; Sato, I.; Hayashi, Y. Nef Reaction with Molecular Oxygen in the Absence of Metal Additives, and Mechanistic Insights. Chem. Eur. J. 2014, 20, 15753–15759. [Google Scholar]
- Vichare, P.; Chattopadhyay, A. Nitrolaldol Reaction of (R)-2,3-Cyclohexylideneglyceraldehyde: A Simple and Stereoselective Synthesis of the Cytotoxic Pachastrissamine (Jaspine B). Tetrahedron Asymmetry 2010, 21, 1983–1987. [Google Scholar]
- Muratake, H.; Natsume, N.; Nakai, H. Palladium-Catalyzed Intramolecular α-Arylation of Aliphatic Ketone, Formyl, and Nitro Groups. Tetrahedron 2004, 60, 11783–11803. [Google Scholar]
- Ghorai, S.K.; Hazra, N.K.; Mal, D. Facile Synthesis of 4-Functionalized Cyclopentenones. Synth. Commun. 2007, 37, 1949–1956. [Google Scholar]
- Balogh-Hergovich, É.; Kaizer, J.; Speier, G. Copper Mediated Conversion of Nitro Compounds to Aldehydes or Ketones by Dioxygen. Chem. Lett. 1996, 25, 573–574. [Google Scholar]
- Yamashita, M.; Nomoto, H.; Imoto, H. Preparation of Dimethyl (1-Formylalkyl)phosphonates via Singlet Oxygen Adducts. Synthesis 1987, 716–718. [Google Scholar]
- Minegishi, S.; Kobayashi, S.; Mayr, H. Solvent Nucleophilicity. J. Am. Chem. Soc. 2004, 126, 5174–5181. [Google Scholar]
- Schwab, P.; France, M.B.; Ziller, J.W.; Grubbs, R.H. A Series of Well-Defined Metathesis Catalysts-Synthesis of RuCl2(=CHR′)-(PR3)2 and Its Reactions. Angew. Chem. Int. Ed. Engl. 1995, 34, 2039–2041. [Google Scholar]
- Umezawa, T.; Mizutani, N.; Matsuo, K.; Tokunaga, Y.; Matsuda, F.; Nehira, T. Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD). Molecules 2021, 26, 1296. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Base (Equiv.) | Solvent | Time | Temp. | Ratio (13:14) 2 |
| 1 | NaOMe (1.5) | MeOH-DCE (9:1) | 8 h | 0 °C | 76:24 |
| 2 | NaOMe (1.5) | TFE-DCE (9:1) | 8 h | 0 °C | 91:9 |
| 3 | KOtBu (1.5) | MeOH-DCE (9:1) | 8 h | 0 °C | 0:100 3 |
| 4 | KOtBu (1.5) | TFE-DCE (9:1) | 8 h | 0 °C | 45:55 |
| 5 | Cs2CO3 (1.5) | MeOH-DCE (9:1) | 8 h | 0 °C | 0:100 3 |
| 6 | Cs2CO3 (1.5) | TFE-DCE (9:1) | 8 h | 0 °C | 33:67 |
| 7 | Cs2CO3 (1.5) | EtOH-DCE (9:1) | 8 h | 0 °C | 0:100 3 |
| 8 | Cs2CO3 (1.5) | iPrOH-DCE (9:1) | 8 h | 0 °C | 86:14 |
| 9 | Cs2CO3 (1.5) | MeCN-DCE (9:1) | 8 h | 0 °C | 96:4 |
| 10 | Cs2CO3 (2.0) | TFE-DCE (9:1) | 8 h | 0 °C | 0:100 4 |
| 11 | Cs2CO3 (2.0) | TFE-DCE (9:1) | 16 h | −10 °C | 0:100 4 |
| 12 | Cs2CO3 (2.0) | TFE-DCE (9:1) | 16 h | −20 °C | 44:56 |
| 13 | Cs2CO3 (2.0) | TFE-DCE (9:1) | 48 h | −20 °C | 13:87 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umezawa, T.; Hara, M.; Kinoshita-Terauchi, N.; Matsuda, F. Enantioselective Total Synthesis of Multifidene, a Sex Pheromone of Brown Algae. Organics 2022, 3, 187-195. https://doi.org/10.3390/org3030015
Umezawa T, Hara M, Kinoshita-Terauchi N, Matsuda F. Enantioselective Total Synthesis of Multifidene, a Sex Pheromone of Brown Algae. Organics. 2022; 3(3):187-195. https://doi.org/10.3390/org3030015
Chicago/Turabian StyleUmezawa, Taiki, Misaki Hara, Nana Kinoshita-Terauchi, and Fuyuhiko Matsuda. 2022. "Enantioselective Total Synthesis of Multifidene, a Sex Pheromone of Brown Algae" Organics 3, no. 3: 187-195. https://doi.org/10.3390/org3030015
APA StyleUmezawa, T., Hara, M., Kinoshita-Terauchi, N., & Matsuda, F. (2022). Enantioselective Total Synthesis of Multifidene, a Sex Pheromone of Brown Algae. Organics, 3(3), 187-195. https://doi.org/10.3390/org3030015