1. Introduction
Quantum computers offer the ability to address problems in quantum many-body chemistry and physics by quantum simulation or in a hybrid quantum-classical approach. The latter method is considered the most promising approach for noisy-intermediate scale quantum (NISQ) devices [
1]. The prospect and benefits of quantum algorithms, along with suitable hardware, is in overcoming the complexity of the wave-function of a quantum system as it scales exponentially with system size [
2]. Therefore, developing techniques and algorithms for NISQ era devices that may prove to have some computational advantage themselves, or establish a path towards ideas and foundations that provide advantage for future error-corrected quantum devices, is a worthwhile pursuit.
The leading algorithms intended to be executed on NISQ devices, which aim to determine solutions to an electronic Hamiltonian, are variational in nature [
3]. One specific algorithm is the variational quantum eigensolver (VQE), which has been tremendously successful in addressing chemistry and physics problems on quantum hardware and NISQ devices [
4,
5,
6,
7,
8,
9,
10,
11,
12]. However, the restriction or challenge that exist with VQE is the need for prior insight with regard to selecting the trial quantum state, i.e., ansatz circuit. Furthermore, the classical optimization of the ansatz parameters may be a poorly converging problem [
13,
14], therefore limiting the applicability of VQE for obtaining results accurate enough for chemical or physical interpretation. Finally, the realization of ansatz circuits that are motivated by domain knowledge, for example, the unitary coupled cluster ansatz for chemistry problems [
15], may not be directly applicable on NISQ hardware, therefore requiring clever modification to obtain hardware efficient ansätze [
6,
7,
16].
In this work, we present a pragmatic hybrid quantum-classical approach for calculating the eigenenergy spectrum of a quantum system within an effective model. Firstly, an effective Hamiltonian is obtained through measurement of short-depth quantum circuits. The effective Hamiltonian is essentially the projection of the quantum system Hamiltonian onto a limited set of computational bases. The basis set is prepared with the intent of ensuring the dimensions of the corresponding matrix does not grow exponentially with the system size. In order to evaluate the matrix elements of the effective Hamiltonian, suitable non-parametric quantum circuits are specified. The quantum circuits are designed, executed, and measured. From the result of the measurements, the diagonal and off-diagonal terms of the effective Hamiltonian matrix are obtained. On the classical side, the effective Hamiltonian matrix, with suitable dimensions, is diagonalized numerically using a classical computer.
The paper is organized as follows: A short background is presented in
Section 2. In
Section 3, the steps taken in our hybrid quantum-classical approach are explained in detail. In
Section 4, we demonstrate the application of this hybrid approach on simple chemical molecules BeH
and LiH. In
Section 5, the approach is demonstrated on the IBMQ 5-qubit Valencia quantum processor [
17] for H
molecule. Finally, we discuss the integration of VQE and the proposed approach in
Section 6.
2. Background
Consider a quantum many-body system of electrons with the second quantized Hamiltonian:
and
are the creation and annihilation operators, respectively. The anticommutator for the creation and annihilation are given by:
and
. These rules enforce the non-abelian group statistics for fermions, that is, under exchange of two fermions the wave-function yields a minus sign.
The indices in Equation (
1) refer to single-electron states. The coefficients
and
are the matrix integrals
and
where
and
operators correspond to one- and two-body interactions respectively. Since
and
can depend on other parameters, such as the distance between nuclei, the Hamiltonian in Equation (
1) represents a class of problems. However, this class of problems has the common property that the number of fermions is a conserved value. Strictly speaking, the terms in the Hamiltonian act on fixed-particle-number Hilbert spaces,
, that have the correct fermionic antisymmetry, with
denoting the number of electrons. In this paper, we consider this class of electronic systems where the Hamiltonian is assumed to be in the form of Equation (
1). The coefficients expressed in Equations (
2) and (
3) can be obtained using software packages developed for quantum chemistry calculations that perform efficient numerical integration [
18].
Mapping to Qubits & Computational Basis
The Hamiltonian as written in Equation (
1) can be expressed in the form of qubit operations (i.e., Pauli matrices). This requires a transformation that preserves the anticommutation of the annihilation and creation operators. One transformation that satisfies the criteria, and is based on the physics of spin-lattice models, is the Jordan-Wigner (JW) transformation [
19]. The JW-transformed Hamiltonian takes the form
in which
’s are scalar and a
Pauli string operator
is defined as
acts on the
i-th qubit,
are the three Pauli matrices [
20], and
is the identity matrix with the number of qubits denoted as
N.
If the number of
operators in the tensor product of
is
, we call
a
k-local Pauli string operator. Upon the JW transformation of the Hamiltonian, a Fock basis of the second quantization representation is in one-to-one correspondence with a computational basis of the qubits [
21]. In other words, a computational basis of
with
N qubits, where
, is equivalent to a an antisymmetric Fock basis.
Within the finite, but exponentially large, Hilbert space spanned by computational basis set, an effective matrix representation of the Hamiltonian may be possible, specifically, if one can efficiently evaluate the matrix elements for an arbitrary computational basis and . Furthermore, assuming that the dimensions of the resulting effective matrix are relatively small, the matrix can be diagonalized on a classical computer, where its eigenvalues approximate the spectrum of the original Hamiltonian.
In this paper, we show how to evaluate a matrix element
for arbitrary computational basis
and
, using a quantum circuit that has a circuit depth
. We do so by using ancilla qubits; thus,
physical resources are needed. In addition, we discuss how to choose an effective subspace for a given electronic Hamiltonian, with a dimension
, based on physical motivations (see
Section 3.3). The condition
makes it possible to diagonalize the Hamiltonian on a classical computer. In
Section 4, we numerically demonstrate this method for simple quantum chemistry systems, focusing on ground state energy and the density of state calculations of the low-energy spectrum. Furthermore, in
Section 5 we use the IBMQ 5-qubit Valencia device to measure the terms for H
in the complete computational basis of 4 qubits.
4. Numerical Demonstration: LiH and BeH
The application of the methodology discussed in
Section 3 is focused on BeH
and LiH molecules due to thier relatively small number of electrons and molecular orbital footprint. The number of electrons in BeH
and LiH is six and four, respectively. The single-electron molecular spin-orbitals in the second quantized Hamiltonian are constructed using the minimal atomic STO-3G basis set [
2,
6]. For BeH
, there are a total of 14 spin-orbital, thus corresponding to 14 qubits; LiH contains 12 spin orbitals and, hence, 12 qubits. For the purpose of measuring the matrix elements of the effective Hamiltonian, Equation (
7), one additional ancillary qubit is required, as illustrated in the quantum circuits shown in
Figure 1. Therefore, the total number of qubits for the simulation of BeH
and LiH is 15 and 13, respectively.
To obtain the coefficients in the Hamiltonian of Equation (
1)—more specifically as defined in Equations (
2) and (
3)—we make use of the Psi4 quantum chemistry package [
30]. The second quantized Hamiltonian is further transfomed onto a set of Pauli strings and their corresponding weights, Equation (
1), via JW transform using OpenFermion package [
31].
In order to construct the potential energy surface for each inter-nuclear distance,
R, the steps indicated in the previous paragraph are repeated. The distance
R corresponds to the bond length between Be–H or Li–H in a given molecule; both LiH and BeH
are linear molecules. We note that these calculations are assuming the total Hamiltonian can be represented using the Born-Oppenheimer approximation, where the dynamics of the core nuclei are neglected. This is standard practice in quantum chemistry calculations [
26]; thus, at every distance
R, the nuclear-nuclear repulsion energy is treated classically and is added to the Hamiltonian as a constant.
For every distance
R, a set of computational bases is chosen. This set includes the basis with the lowest energy expectation (the reference configuration) and computational bases that correspond to the low-order excitations (i.e.,
S,
D, etc.) with respect to the reference configuration. In the case of BeH
, we construct the effective Hamiltonian matrix by keeping the
S,
D, and
T excitations; thus, the total number of computational bases for BeH
is
. The low dimension of the effective Hamiltonian matrix makes it possible to diagonalize the matrix on a classical computer using standard numerical techniques [
32]. Thus, the lowest energies, including the ground state energy, are obtained at every
R. The results for this process are shown in
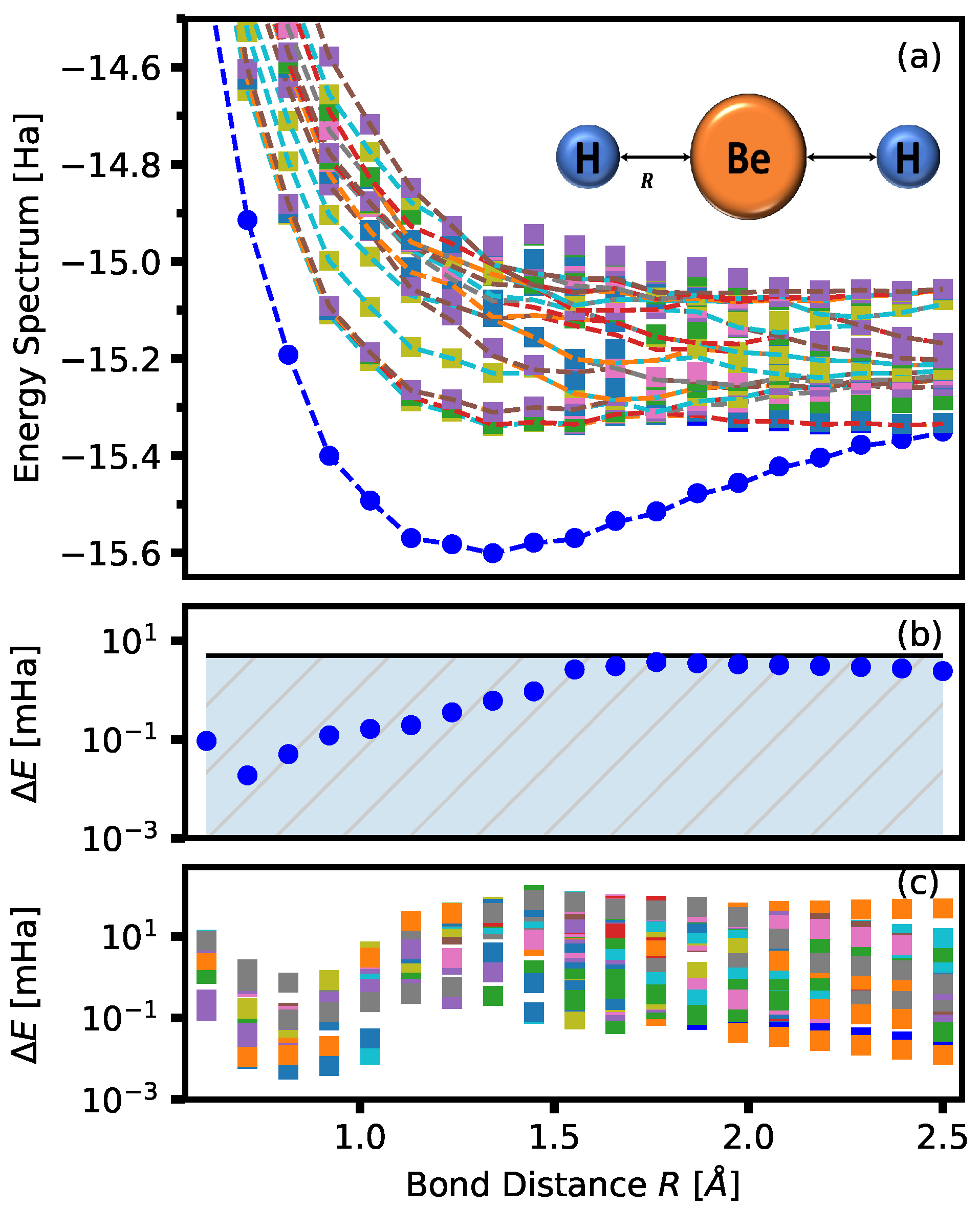
Figure 3.
Figure 3a shows the ground state energy, as well as a few low-lying excited states, for BeH
of the effective Hamiltonian. The exact energies are given by the dashed curves in
Figure 3a. In
Figure 3b, the difference between the exact and obtained ground state energy is shown. An error within the hatched area indicates results that are within chemical accuracy. Chemical accuracy is typically identified as
Hatrees.
Figure 3c shows the same energy difference for all other excited-state energies. The same process is done for LiH where only
S and
D configurations are used. The total number of permutations of 4 fermions (495) is reduced to
, and the results are shown in
Figure 4.
The total number of configurations can be approximated by the highest excitation considered. For n-electron excitation, the number of configurations is . Thus, the total number of configurations is less than when . Of course, this argument is valid as long as n can be assumed to be finite and independent of the size of the system N or the number of electrons . Under these assumptions, the dimensions of the effective Hamiltonian, , remains polynomial if one replaces the minimal STO-3G with the extended atomic basis.
Density of States
Knowledge of density of states (DOS) can be important in analyzing the thermodynamic behaviour of a system at finite temperature and in the analysis of transition states important in chemical reactions [
33]. One advantage of the method proposed in this paper is the insights provided into the low-energy spectrum of the quantum system, more precisely, the degeneracy of the energy levels. To illustrate this, in
Figure 5, the unnormalized DOS of the LiH obtained via the effective Hamiltonian is compared with the exact density, which shows qualitatively good agreement.
5. Hardware Demonstration: H
In this section, we demonstrate the feasibility of the hybrid quantum-classical approach discussed in this paper on a IBMQ hardware device. Specifically, we calculate the eigenenergy spectrum of H molecule using STO-3G minimal basis-set.
Within STO-3G basis set, the total number of spin-orbitals for H
is four. In contrast to the numerical demonstration, where the computational basis was prepared using the UCC ansatz, here, we employ the complete computational basis with conserved number of electrons for H
(
). The number of computational basis set with conserved electron number, is
, and the bases are:
The number of bases is not further reduced; that is, the effective Hamiltonian constructed here is equal to the full Hamiltonian within the
electron number. The above set can be interpreted as the collection of all the
S and
D exciations, as introduced in
Section 3.3, above a classical configuration, such as
.
The Hamiltonian of the problem, stated as a weighted sum of Pauli string operators, as in Equation (
4), has a total of fifteen operators, which can be grouped as:
and
Here, a Pauli string operator, such as
, is a shorthand for
, as introduced in Equation (
5).
Our goal is to use the quantum hardware to evaluate the values for
, where
belongs to
or
, and
and
can be any of the bases listed in Equation (
19).
Note that this task is not a difficult one for a classical computer. However, the objective of the paper and hardware demonstration is to establish a hybrid quantum-classical computational scheme in which the quantum processor performs some of the computational tasks. In the simulations of this paper, the task is the evaluation of the phase associated with the exchange of fermions; since can be understood as exchanging fermions on via the operator , and to arrive at . The output can only take discrete values of , , and 0.
In each group
and
, Pauli string operators commute and, thus, theoretically, can be measured simultaneously. However, in order to perform simultaneous measurement, one needs to adjust the computational bases, by applying further quantum gates on the target qubits. The appended measurement circuit further increases the circuit depth [
34]. Thus, practically, the simultaneous measurement may not be advantageous, and, in contrast, induces further noise and decoherence on NISQ devices. Nevertheless, for
, no further circuit depth is required, as they are all measured in the
z-basis.
We can further reduce the quantum coprocessor computational load by excluding circuits containing terms that are classically efficient to compute. In Equations (
20) and (
21), the operators in
do not contribute to off-diagonal matrix element of the Hamiltonian, while operators in
contribute
only to the off-diagonal matrix elements. This information is used to reduce the essential number of circuits to be run on the hardware.
The evaluation of an element
is performed by assigning an appropriate circuit, as introduced in
Section 3.1, to a quantum coprocessor. In this work, we make use of the IBMQ cloud open devices.
Once a circuit is loaded onto a quantum coprocessor, each circuit is executed a several times. The number of times a circuit is run is referred to as the number of shots. In this work, each circuit was executed for a total of 8000 shots. At each run, the qubits are measured, and the collection of the realized configurations is used to evaluate value of .
The preparation of the circuits is performed using the transpile and assemble routines available in the IBM Qiskit API [
35], which enables collating individual circuits, corresponding to the diagonal and off-diagonal terms, to prepare each circuit to run as a single job on a target IBMQ device. In this work, we make use of the IBMQ 5-qubit Valencia hardware [
17]. The number of circuits—for both real and imaginary parts separately—after the transpile and assemble routines for the diagonal and off-diagonal terms corresponds to 66 and 60 circuits, respectively. The execution timings for the IBMQ QASM simulator range between 0.21–1.5 s and for the IBMQ Valencia 5 qubit device 274–298 s with 8000 shots per circuit. Due to the limited number of qubits available on this processor, direct measurement is used, that is by measuring all the qubits at the end of the circuit.
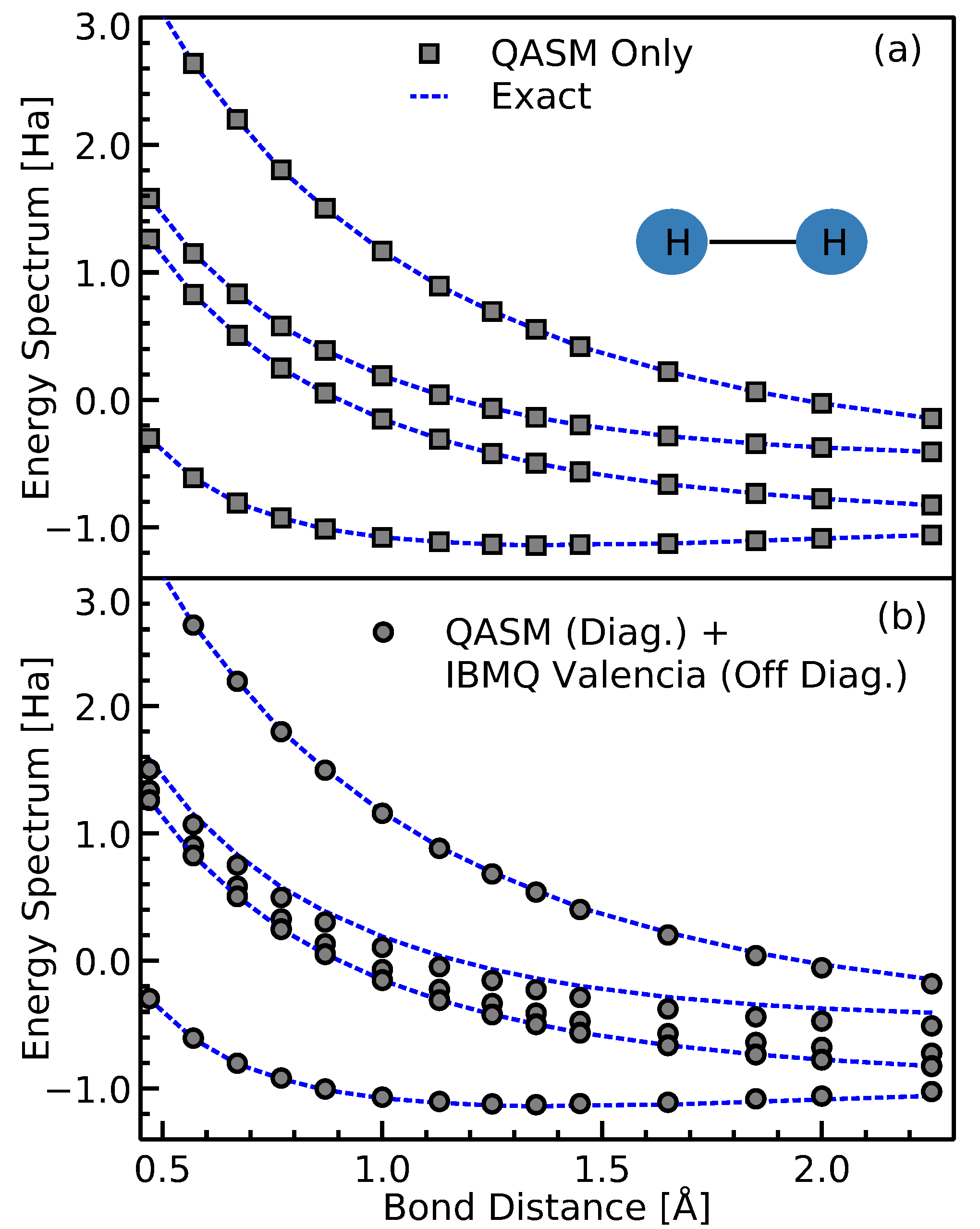
As a sanity check, we first use IBMQ QASM simulator [
36] to perform all steps involved as outlined in
Section 3. The calculations are shown in
Figure 6a. The results perfectly match the exact values as expected. Furthermore, in
Figure 6b the off-diagonal terms in the Hamiltonian are evaluated using IBMQ Valencia device, while the diagonal elements are evaluated using the IBMQ QASM simulator. In both cases, upon diagonalization of the obtained Hamiltonian, the energy spectrum is in good agreement with the exact values. We observe in
Figure 6b that there is a slight lifting of the double degeneracy of H
, which we associate to the device noise. This is due to the occurance of non-zero values in the measured off-diagonal terms when in fact the exact value should be zero.
Finally, all the matrix elements, that is, diagonal and off-diagonal elements, are evaluated using IBMQ Valencia device. The results are shown in
Figure 7a, which was done with and without measurement error mitigation. The use of measurement error mitigation marginally improves the results. The discrepancy between the exact and hardware values, without and with measurement error mitigation, are shown in
Figure 7b,c.
The effect of noise is ubiquitous in current quantum hardware and is inherently complex and difficult to characterize. However, the result of noise due to the measurement process can be mitigated to some degree by using a calibration matrix [
35]. The resulting calibration matrix is then used to reduce errors of subsequent circuit measurements. The application of measurement error mitigation is demonstrated in
Figure 7a with beneficial impact on error shown in
Figure 7c.
The calculations in
Figure 7, that is construction of the effective Hamiltonian,
, are performed through five independent IBMQ job submissions. Each time the energy-surface is slightly different, which can be associated to the inherent device errors and perhaps low number of shots used. The error bars in
Figure 7b,c indicate the min/max range in errors of the five different calculation.
6. Discussion and Summary
Recently an abundance of Variational Quantum Algorithms (VQA) have been introduced to solve electronic quantum many-body problems on NISQ devices [
3,
4,
5,
8,
9,
10,
11,
28]. These VQA proposals mostly rely on optimization of parametric circuits. In this paper, we demonstrate that an alternative approach exists, specifically for quantum chemistry problems, which does not require an optimization procedure and parametric ansatz. In this approach, the quantum hardware is used only to prepare a many-body quantum state and to efficiently measure the expectation values of certain target observables with respect to this quantum state. From the output measurements, one can construct what we refer to as an effective Hamiltonian matrix,
. Upon diagonalization of
using classical eigen-decomposition numerical methods one obtains the ground state and low-lying excited states of the system.
The approach introduced in this paper is particularly suitable for NISQ devices where short quantum circuit depth is essential due to lack of error-correction protocols on these devices. An additional important aspect of this approach is that it provides access to the low-lying energy spectra of the system and not just the ground state in comparison with the original VQE process.
In the context of VQE and VQE-type algorithms, several attempts have been made to extend the variational approach to excited states [
10,
11,
37,
38,
39,
40]. Quantum subspace expansion [
41,
42], for example, constructs a set of non-orthogonal bases out of an optimized ansatz, and performs post-processing to obtain excited states. Deflation techniques as described in ref. [
11], constructs a pseudo-Hamiltonian in which the ground state is excluded and orthogonality is enforced through regularization. Successful examples are introduced for some low-lying excited states of LiH [
8]. In all these previous works, optimization of a parametric ansatz is required and, therefore, necessitates an enormous number of quantum circuit executions and sampling.
The approach introduced in this paper is similar to multistate contracted VQE (MC-VQE) in ref. [
10]. The main difference is the application of VQE: MC-VQE is obviously a VQE-type algorithm, our approach is distinct in that it does not require a parametric circuit or variational procedure for optimization. In addition, our method differs since it uses supporting quantum circuit resources, i.e., ancilla qubits, in order to perform interference and measurement that is different from the quantum circuit in ref. [
43] and its generalization in ref. [
10].
An extension of our approach to VQE type algorithm is possible. This can be done by appending a set of parametric gates that act only on the target qubits to the circuits in
Figure 1. Let us denote this part of the circuit with
, where
stands for a set of parameters. Then, it can be verified that, given
, the final matrix element obtained from the circuit after measurement (see Equation (
2), for example) becomes
, compared to
. The appended parametric circuit
allows one to project the Hamiltonian onto
, for a given
. This means the
effective Hamiltonian is now parametric and depends on the value(s) of
. The optimal parameter(s) are then obtained by minimizing the ground-state energy of the effective Hamiltonian matrix. The reference state
can be regarded as the contracted reference states introduced in ref. [
10].
One possible limitation of the circuits shown in
Figure 1 is the execution of two-qubit gates corresponding to control operations over
n-qubits (i.e., series of
CNOT gates). For NISQ devices with hardware-restricted qubit connectivity, this may require a number of
SWAP gate operations and, therefore, can increase the circuit depth and subsequent error rates [
44,
45]. In essence, the implementation of the circuits described in this paper will depend on the ability to limit circuit depth and associated error rates by NISQ hardware circuit optimization (i.e., scheduling). However, significant improvements in qubit connectivity of various modalities (e.g., ion traps) [
22,
46] or optimizing quantum circuits against decoherence [
47,
48] may blunt this concern.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






