
Solvent-Free 1,3-Dipolar Cycloadditions of Nitrones for a More Sustainable Synthesis of Glycomimetics





Abstract

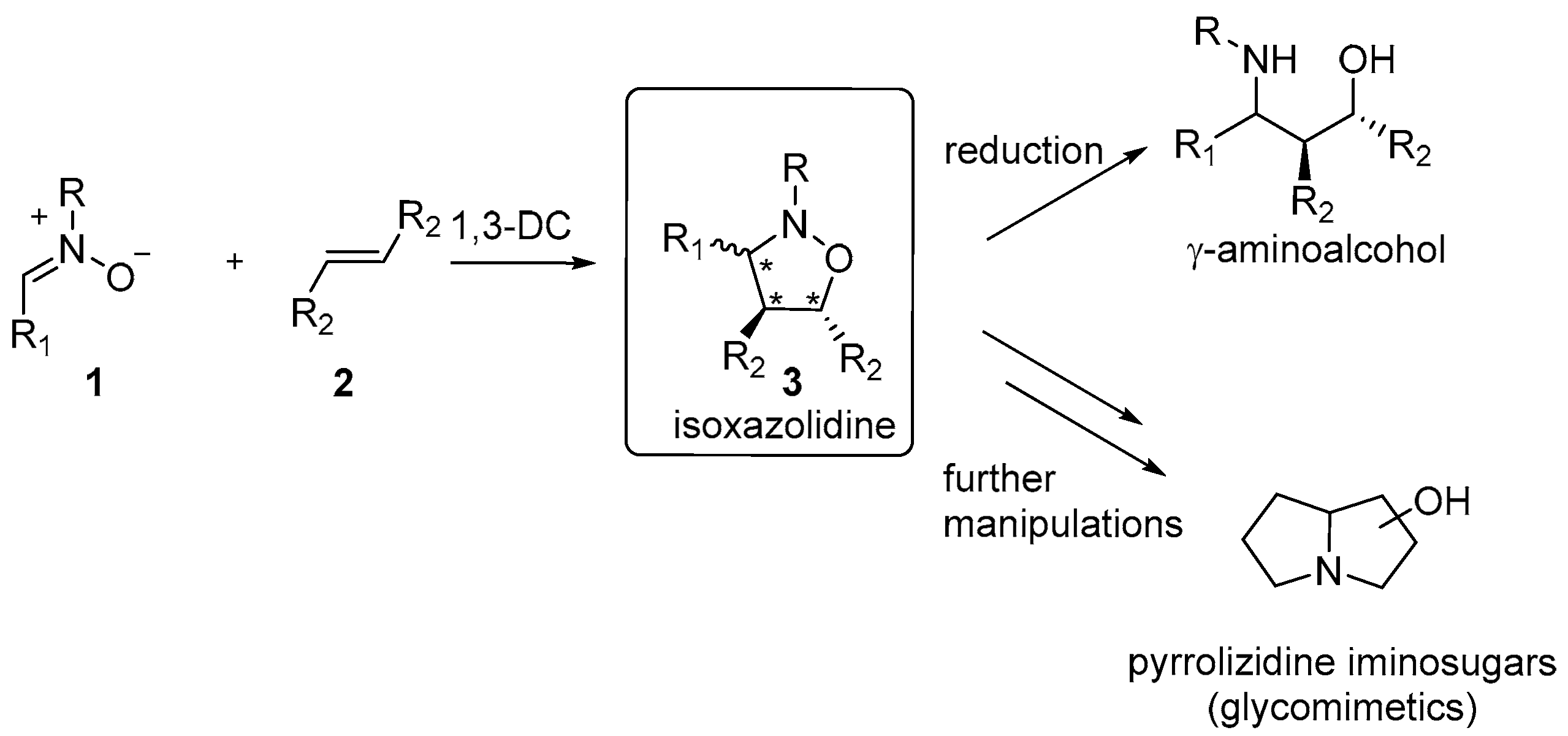
1. Introduction
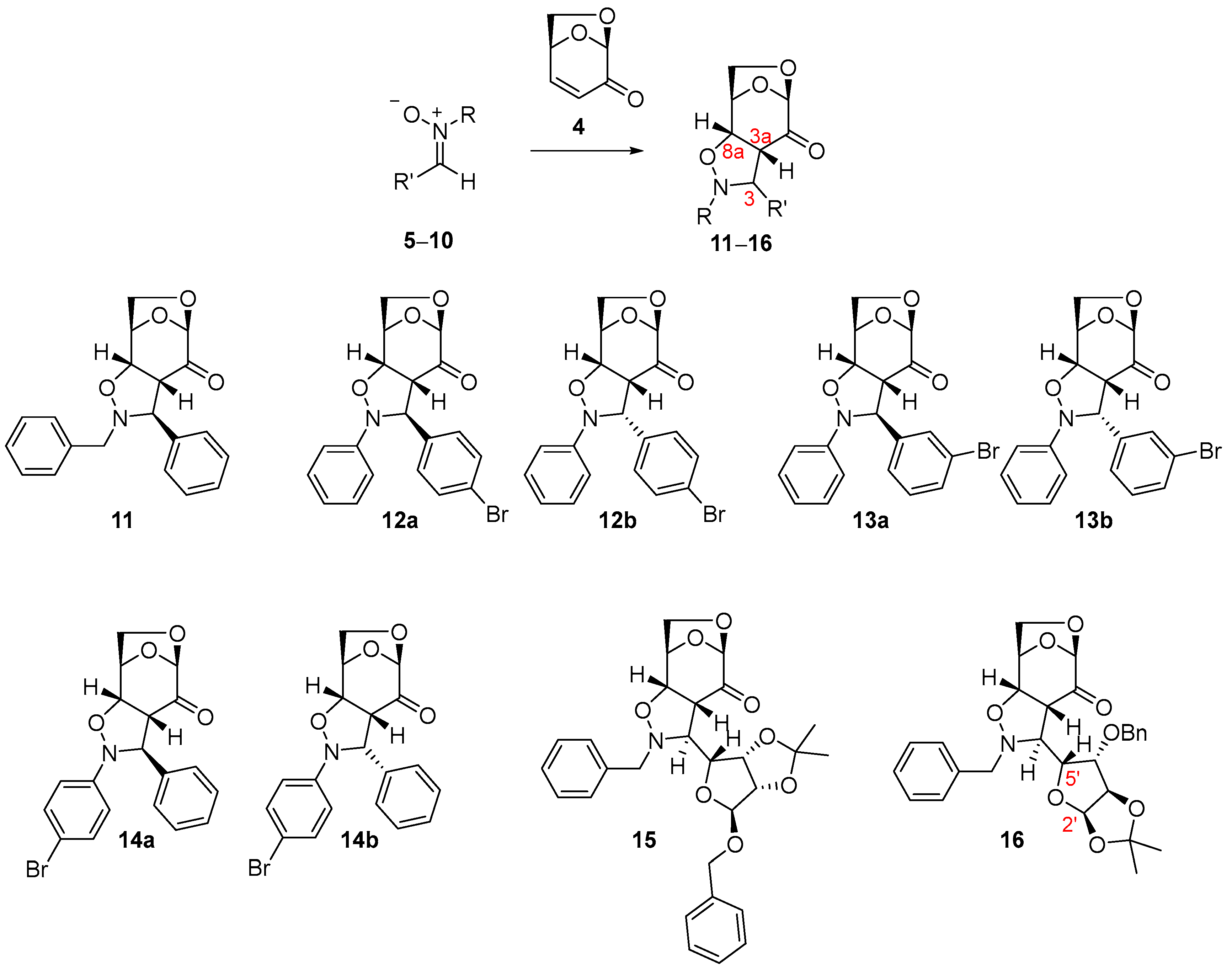
2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Nitrone | R | R′ | Reaction Conditions | Heating | Temperature | Time | Product (Yield) a |
|---|---|---|---|---|---|---|---|---|
| 1 b | 5 | -benzyl | -phenyl | dry toluene | standard | 110 °C | 48 h | 11 (87%) |
| 2 | 5 | -benzyl | -phenyl | solvent-free | standard | 40 °C | 96 h | 11 (57%) |
| 3 | 5 | -benzyl | -phenyl | solvent-free | standard | 60 °C | 17 h | 11 (58%) |
| 4 | 5 | -benzyl | -phenyl | solvent free | standard | 110 °C | 3 h | 11 (63%) |
| 5 | 5 | -benzyl | -phenyl | solvent-free | microwaves (150 W) | 110 °C | 1.5 h | 11 (63%) |
| 6 | 5 | -benzyl | -phenyl | solvent-free | microwaves (300 W) | 110 °C | 1 h | 11 (68%) |
| 7 c | 6 | -phenyl | -4-bromophenyl | dry CH2Cl2ZnCl2 · Et2O | - | r.t. | 8 h | 12 (77%) |
| 8 | 6 | -phenyl | -4-bromophenyl | solvent-free | standard | 80 °C | 2.5 h | 12 (81%) |
| 9 | 6 | -phenyl | -4-bromophenyl | solvent-free | microwaves (150 W) | 80 °C | 45 min | 12 (79%) |
| 10 c | 7 | -phenyl | -3-bromophenyl | dry CH2Cl2ZnCl2 · Et2O | - | r.t. | 8 h | 13 (69%) |
| 11 | 7 | -phenyl | -3-bromophenyl | solvent-free | standard | 80 °C | 3 h | 13 (69%) |
| 12 | 7 | -phenyl | -3-bromophenyl | solvent-free | microwaves (150 W) | 80 °C | 45 min | 13 (72%) |
| 13 c | 8 | -4-Br-phenyl | -phenyl | dry CH2Cl2ZnCl2 · Et2O | - | r.t. | 8 h | 14 (84%) |
| 14 | 8 | -4-Br-phenyl | -phenyl | solvent-free | standard | 80 °C | 3 h | 14 (75%) |
| 15 | 8 | -4-Br-phenyl | -phenyl | solvent-free | microwaves (150 W) | 80 °C | 45 min | 14 (81%) |
| 16 d | 9 | -benzyl |  | dry toluene | standard | 60 °C | 24 h | 15 (78%) |
| 17 e | 9 | -benzyl | 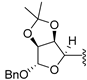 | solvent-free | standard | 60 °C | 6 h | 15 (61%) |
| 18 e | 9 | -benzyl |  | solvent-free | microwaves (150 W) | 60 °C | 4 h | 15 (62%) |
| 19 | 10 | -benzyl | 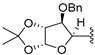 | dry toluene | standard | 60 °C | 18 h | 16 (60%) |
| 20 | 10 | -benzyl | 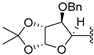 | solvent-free | standard | 60 °C | 6 h | 16 (62%) |
| 21 | AG | -benzyl | 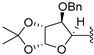 | solvent-free | microwaves (150 W) | 60 °C | 3 h | 16 (57%) |
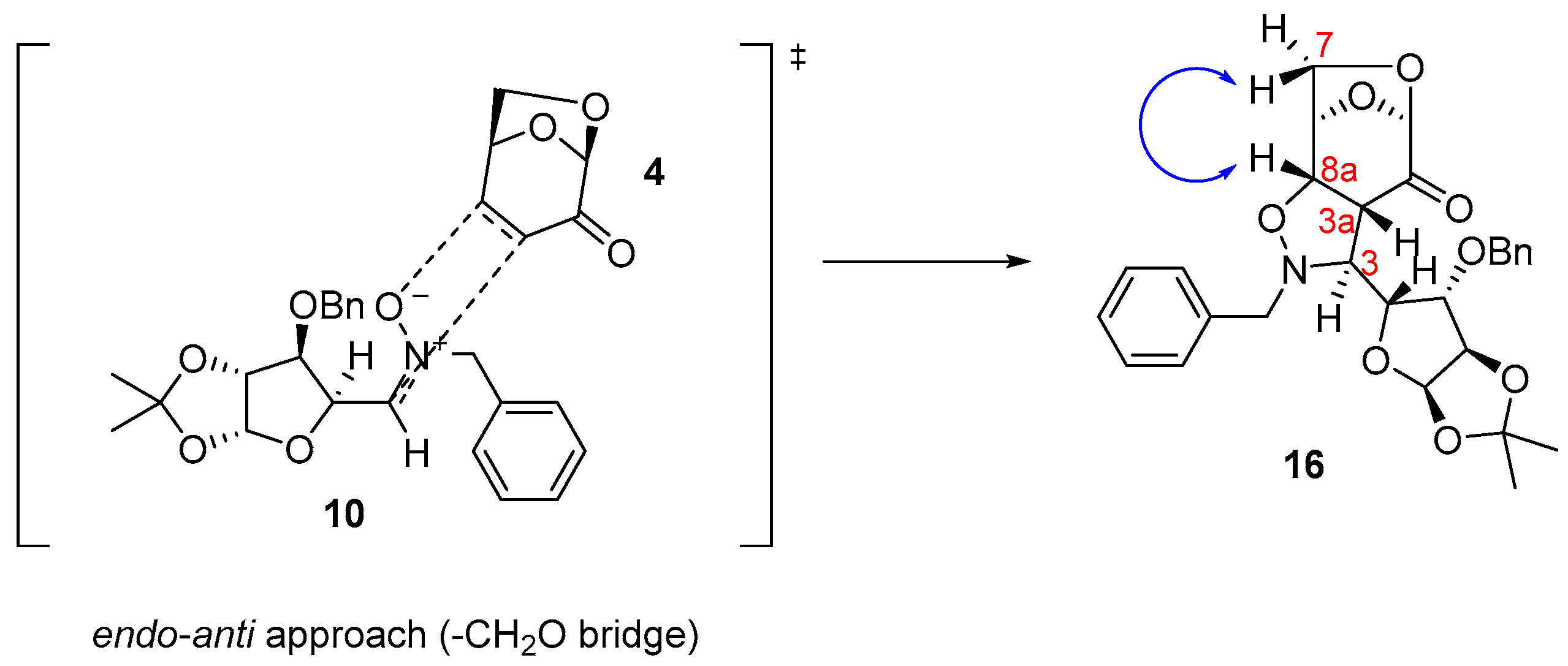
3. Discussion
4. Materials and Methods
4.1. General Details
4.2. General Procedure for Solvent-Free 1,3-Dipolar Cycloaddition of Nitrones and Dipolarophiles
4.3. Synthesis of Cycloadduct 16
4.4. Synthesis of Cycloadducts 20a, 20b and 20c
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padwa, A.; Pearson, W.H. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Wiley: New York, NY, USA, 2002. [Google Scholar]
- Huisgen, R. Adventures with heterocycles. Chem. Pharm. Bull. 2000, 48, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Kissane, M.; Maguire, A.R. Asymmetric 1,3-dipolar cycloadditions of acrylamides. Chem. Soc. Rev. 2010, 39, 845–883. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Asymmetric 1,3-dipolar cycloadditions. Tetrahedron 2007, 63, 3235–3285. [Google Scholar] [CrossRef]
- Gothelf, K.V.; Jorgensen, K.A. Asymmetric 1,3-Dipolar Cycloaddition Reactions. Chem. Rev. 1998, 98, 863–909. [Google Scholar] [CrossRef]
- Frühauf, H.W. Metal-Assisted Cycloaddition Reactions in Organotransition Metal Chemistry. Chem. Rev. 1997, 97, 523–596. [Google Scholar] [CrossRef]
- Broggini, G.; Molteni, G.; Terraneo, A.; Zecchi, G. Transition metal complexation in 1,3-dipolar cycloadditions. Heterocycles 2003, 59, 823–858. [Google Scholar] [CrossRef]
- Karlsson, S.; Högberg, H.E. Asymmetric 1,3-dipolar cycloadditions for the construction of enantiomerically pure heterocycles. A review. Org. Prep. Proced. Int. 2001, 33, 103–172. [Google Scholar] [CrossRef]
- Osborn, H.M.I.; Gemmell, N.; Harwood, L.M. 1,3-Dipolar cycloaddition reactions of carbohydrate derived nitrones and oximes. J. Chem. Soc. Perkin Trans. 2002, 1, 2419–2438. [Google Scholar] [CrossRef]
- Zhu, J.; Lines, B.M.; Ganton, M.D.; Kerr, M.A.; Workentin, M.S. Efficient Synthesis of Isoxazolidine-Tethered Monolayer Protected Gold Nanoparticles (MPGNs) via 1,3-Dipolar Cycloadditions under High-Pressure Conditions. J. Org. Chem. 2008, 73, 1099–1105. [Google Scholar] [CrossRef]
- Krasinski, A.; Radic, Z.; Manetsch, R.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J. Am. Chem. Soc. 2005, 127, 6686–6692. [Google Scholar] [CrossRef]
- Hoffmann, R.; Woodward, R.B. Selection rules for concerted cycloaddition reactions. J. Am. Chem. Soc. 1965, 87, 2046–2048. [Google Scholar] [CrossRef]
- Padwa, A. 1,3-Dipolar Cycloaddition Chemistry; Wiley: New York, NY, USA, 1984. [Google Scholar]
- Breugst, M.; Reissig, H.U. The Huisgen reaction: Milestones of the 1,3-dipolar cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley-Interscience: London, UK, 1976. [Google Scholar]
- Frederickson, M. Optically active isoxazolidines via asymmetric cycloaddition reactions of nitrones with alkenes: Applications in organic synthesis. Tetrahedron 1997, 53, 403–425. [Google Scholar] [CrossRef]
- Lait, S.M.; Rankic, D.A.; Keay, B.A. 1,3-Aminoalcohols and Their Derivatives in Asymmetric Organic Synthesis. Chem. Rev. 2007, 107, 767–796. [Google Scholar] [CrossRef] [PubMed]
- Koumbis, A.E.; Gallos, J.K. 1,3-Dipolar Cycloadditions in the Synthesis of Carbohydrate Mimics. Part 2: Nitrones and Oximes. Curr. Org. Chem. 2003, 7, 585–628. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Wiley VCH: New York, NY, USA, 2007. [Google Scholar]
- Confalone, P.N.; Huie, E.M. The [3 + 2] Nitrone–Olefin Cycloaddition Reaction. Org. React. 1988, 36, 1. [Google Scholar]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. [3 + 2] Dipolar cycloadditions of cyclic nitrones with alkenes. Org. React. 2017, 94, 1. [Google Scholar]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. Stereocontrolled Cyclic Nitrone Cycloaddition Strategy for the Synthesis of Pyrrolizidine and Indolizidine Alkaloids. Chem. Eur. J. 2009, 15, 7808–7821. [Google Scholar] [CrossRef]
- Cycloaddition Reactions in Carbohydrate Chemistry; Giuliano, R.M., Ed.; ACS Symposium Series 494; American Chemical Society: Washington, DC, USA, 1992. [Google Scholar]
- Herczegh, P.; Kovács, I.; Erdösi, G.; Varga, T.; Agócs, A.; Szilágyi, L.; Sztaricskai, F.; Berecibar, A.; Lukacs, G.; Olesker, A. Stereoselective cycloaddition reactions of carbohydrate derivatives. Pure Appl. Chem. 1997, 69, 519–524. [Google Scholar] [CrossRef]
- Kanemasa, S.; Tsuge, O. Recent advances in synthetic applications of nitrile oxide cycloaddition. Heterocycles 1990, 30, 719–736. [Google Scholar] [CrossRef]
- Broggini, G.; Zecchi, G. Pyrrolizidine and indolizidine syntheses involving 1,3-Dipolar cycloadditions. Synthesis 1999, 6, 905–917. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Cardona, F.; Salanski, P.; Chmielewski, M.; Valenza, S.; Goti, A.; Brandi, A. Remarkable High-pressure Enhancement of Enantiopure Nitrone Cycloadditions to Glycals: General Access to (1 → 2)-Linked Pseudo Aza-C-disaccharides. Synlett 1998, 12, 1444–1446. [Google Scholar] [CrossRef]
- Martina, K.; Tagliapietra, S.; Veselov, V.V.; Cravotto, G. Green Protocols in Heterocycle Syntheses via 1,3-Dipolar Cycloadditions. Chem. Front. 2019, 7, 95. [Google Scholar] [CrossRef]
- Mabrour, M.; Bougrin, K.; Benhida, R.; Loupy, A.; Soufiaoui, M. An efficient one-step regiospecific synthesis of novel isoxazolines and isoxazoles of N-substituted saccharin derivatives through solvent-free microwave-assisted [3+2] cycloaddition. Tetrahedron Lett. 2007, 48, 443–447. [Google Scholar] [CrossRef]
- Pospíšil, J.; Potáček, M. Microwave-assisted solvent-free intramolecular 1,3-dipolar cycloaddition reactions leading to hexahydrochromeno[4,3-b]pyrroles: Scope and limitations. Tetrahedron 2007, 63, 337–346. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Martel, A.; Dhal, R.; Dujardin, G. 1,3-Dipolar Cycloaddition of N-Substituted Dipolarophiles and Nitrones: Highly Efficient Solvent-Free Reaction. J. Org. Chem. 2008, 73, 2621–2632. [Google Scholar] [CrossRef]
- Almansour, A.I.; Suresh Kumar, R.; Arumugam, N.; Sriram, D. A solvent free, four-component synthesis and 1,3-dipolar cycloaddition of 4(H)-pyrans with nitrile oxides: Synthesis and discovery of antimycobacterial activity of enantiomerically pure 1,2,4-oxadiazoles. Eur. J. Med. Chem. 2012, 53, 416–423. [Google Scholar] [CrossRef]
- Chakraborty, B. Solvent-free synthesis and 1,3-dipolar cycloaddition reactions of N-methyl-C-(2-furyl) nitrone in a ball mill and anticancer activities of the new cycloadducts. J. Heterocycl. Chem. 2020, 57, 477–485. [Google Scholar] [CrossRef]
- Comba, M.B.; Tsai, Y.H.; Sarotti, A.M.; Mangione, M.I.; Suárez, A.G.; Spanevello, R.A. Levoglucosenone and its new applications: Valorization of cellulose residues. Eur. J. Org. Chem. 2018, 2018, 590–604. [Google Scholar] [CrossRef]
- Camp, J.E.; Greatrex, B.W. Levoglucosenone: Bio-based platform for drug discovery. Front. Chem. 2022, 10, 902239. [Google Scholar] [CrossRef]
- Liu, X.; Carr, P.; Gardiner, M.G.; Banwell, M.G.; Elbanna, A.H.; Khalil, Z.G.; Capon, R.J. Levoglucosenone and Its Pseudoenantiomer iso-Levoglucosenone as Scaffolds for Drug Discovery and Development. ACS Omega 2020, 5, 13926–13939. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, D.; Matassini, C.; Goti, A.; Angeli, A.; Carta, F.; Supuran, C.T.; Spanevello, R.; Cardona, F. Glycomimetic Based Approach toward Selective Carbonic Anhydrase Inhibitors. ACS Med. Chem. Lett. 2020, 11, 727–731. [Google Scholar] [CrossRef]
- Banwell, M.G.; Liu, X.; Connal, L.A.; Gardiner, M.G. Synthesis of Functionally and Stereochemically Diverse Polymers via Ring-Opening Metathesis Polymerization of Derivatives of the Biomass-Derived Platform Molecule Levoglucosenone Produced at Industrial Scale. Macromolecules 2020, 53, 5308–5314. [Google Scholar] [CrossRef]
- Stanfield, M.K.; Terry, R.S.; Smith, J.A.; Thickett, S.C. Levoglucosan and levoglucosenone as bio-based platforms for polymer synthesis. Polym. Chem. 2023, 14, 4949–4956. [Google Scholar] [CrossRef]
- Flourat, A.L.; Pezzana, L.; Belgacem, S.; Dosso, A.; Sangermano, M.; Fadlallah, S.; Allais, F. Levoglucosenone to 3D-printed green materials: Synthesizing sustainable and tunable monomers for eco-friendly photo-curing. Green Chem. 2023, 25, 7571–7581. [Google Scholar] [CrossRef]
- Blake, A.J.; Cook, T.A.; Forsyth, A.C.; Gould, R.O.; Paton, R.M. l,3-Dipolar Cycloaddition Reactions of Levoglucosenone. Tetrahedron 1992, 48, 8053–8064. [Google Scholar] [CrossRef]
- Coşkun, N.; Parlar, A. One-Pot Synthesis and Hydroxylaminolysis of Asymmetrical Acyclic Nitrones. Synth. Commun. 2005, 35, 2445–2451. [Google Scholar] [CrossRef]
- Muller, C.; Gomez-Zurita Frau, M.A.; Ballinari, D.; Colombo, S.; Bitto, A.; Martegani, E.; Airoldi, C.; van Neuren, A.S.; Stein, M.; Weiser, J.; et al. Design, Synthesis, and Biological Evaluation of Levoglucosenone-Derived Ras Activation Inhibitors. Chem. Med. Chem. 2009, 4, 524–528. [Google Scholar] [CrossRef]
- Wang, C.; Wang, D.; Yan, H.; Wang, H.; Pan, B.; Xin, X.; Li, X.; Wu, F.; Wan, B. Rhodium-Catalyzed Cyclization of Diynes with Nitrones: A Formal [2+2+5] Approach to Bridged Eight-Membered Heterocycles. Angew. Chem. Int. Ed. 2014, 53, 11940–11943. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Du, H.; Liang, W.; Lou, C.; Wu, Y.; Liu, B. Synthesis of 1,4-epoxy-2-aryltetrahydro-1-benzazepines via rhodium(III)-catalyzed C–H allylation/intramolecular 1,3-dipolar cycloaddition. Chem. Commun. 2024, 60, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Djahieche, M.; Boufenaya, H.; Boukhari, A. Regio-and stereoselectivity of [3+2] cycloaddition reaction of methacrolein with diarylnitrones catalyzed by chiral rhodium complex. Rev. Roum. Chim. 2019, 64, 973–980. [Google Scholar] [CrossRef]
- Parmeggiani, C.; Matassini, C.; Cardona, F.; Goti, A. On the Oxidation of Hydroxylamines with o-Iodoxybenzoic Acid (IBX). Synthesis 2017, 49, 2890–2900. [Google Scholar]
- Matassini, C.; Mirabella, S.; Goti, A.; Cardona, F. Double Reductive Amination and Selective Strecker Reaction of a d-Lyxaric Aldehyde: Synthesis of Diversely Functionalized 3,4,5-Trihydroxypiperidines. Eur. J. Org. Chem. 2012, 2012, 3920–3924. [Google Scholar] [CrossRef]
- Chen, F.-E.; Zhao, J.-F.; Xiong, F.-J.; Xie, B.; Zhang, P. An improved synthesis of a key intermediate for (+)-biotin from d-mannose. Carbohydr. Res. 2007, 342, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F.L.; Merino, P.; Tejero, T. Synthesis of N-Benzyl Nitrones. Synth. Commun. 1994, 24, 2537–2550. [Google Scholar] [CrossRef]
- Cardona, F.; Faggi, E.; Liguori, M.; Cacciarini, M.; Goti, A. Total syntheses of hyacinthacine A2 and 7-deoxycasuarine by cycloaddition to a carbohydrate derived nitron. Tetrahedron Lett. 2003, 44, 2315–2318. [Google Scholar] [CrossRef]
- Martella, D.; D’Adamio, G.; Parmeggiani, C.; Cardona, F.; Moreno-Clavijo, E.; Robina, I.; Goti, A. Cycloadditions of Sugar-Derived Nitrones Targeting Polyhydroxylated Indolizidines. Eur. J. Org. Chem. 2016, 2016, 1588–1598. [Google Scholar] [CrossRef]
- Revuelta, J.; Cicchi, S.; Goti, A.; Brandi, A. Enantiopure Cyclic Nitrones: A Useful Class of Building Blocks for Asymmetric Syntheses. Synthesis 2007, 4, 485–504. [Google Scholar] [CrossRef]
- Cardona, F.; Lalli, D.; Faggi, C.; Goti, A.; Brandi, A. Quasienantiomeric levoglucosenone and isolevoglucosenone allow the parallel kinetic resolution of a racemic nitrone. J. Org. Chem. 2008, 73, 1999–2002. [Google Scholar] [CrossRef]
- Cicchi, S.; Höld, I.; Brandi, A. New synthesis of five-membered cyclic nitrones from tartaric acid. J. Org. Chem. 1993, 58, 5274–5275. [Google Scholar] [CrossRef]
- Li, M.-Y.; Gua, A.; Li, J.; Liu, Y. Advanced green synthesis: Solvent-free and catalyst-free reaction. Green Synth. Catal. 2025, 6, 36–66. [Google Scholar] [CrossRef]
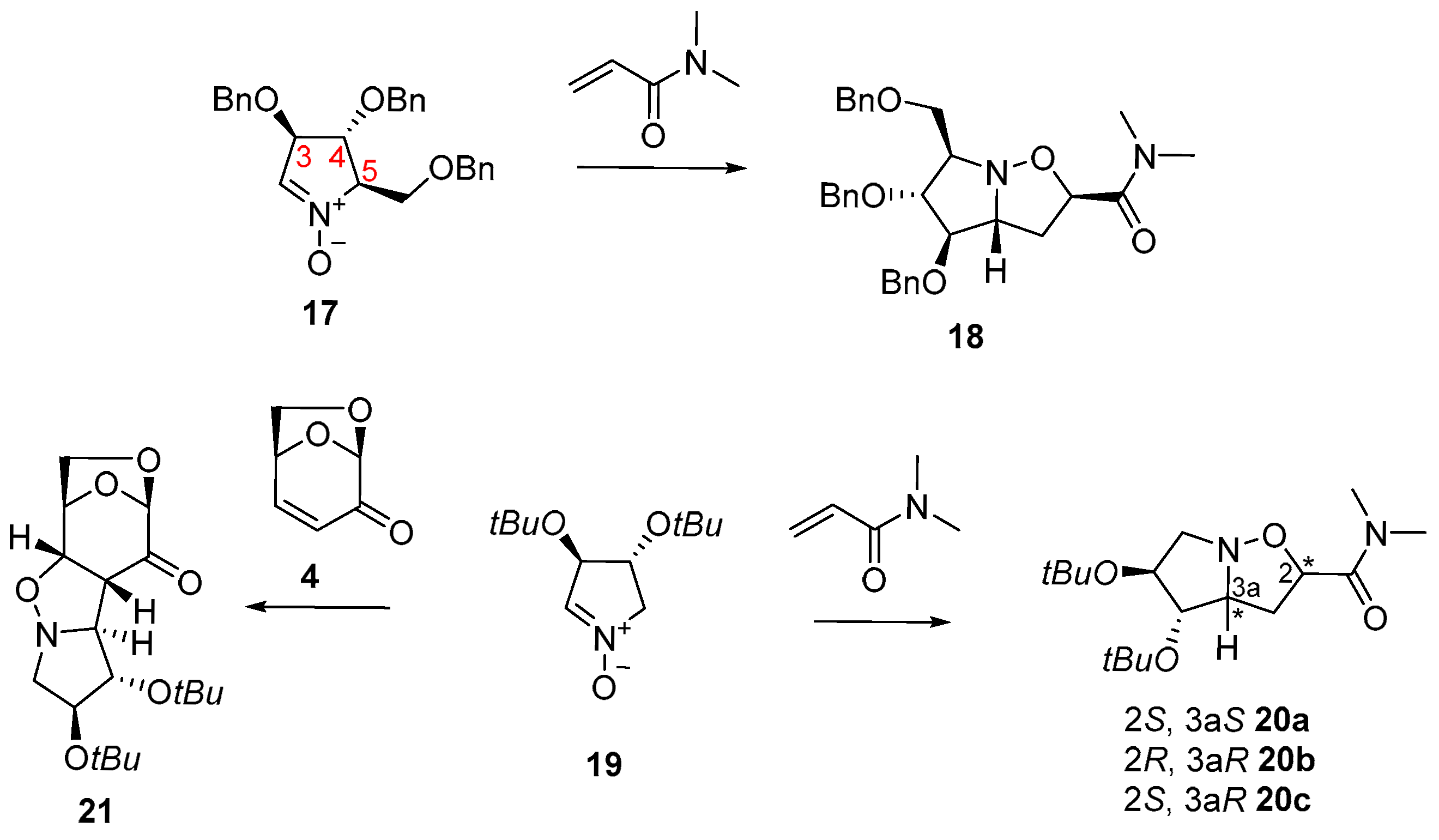
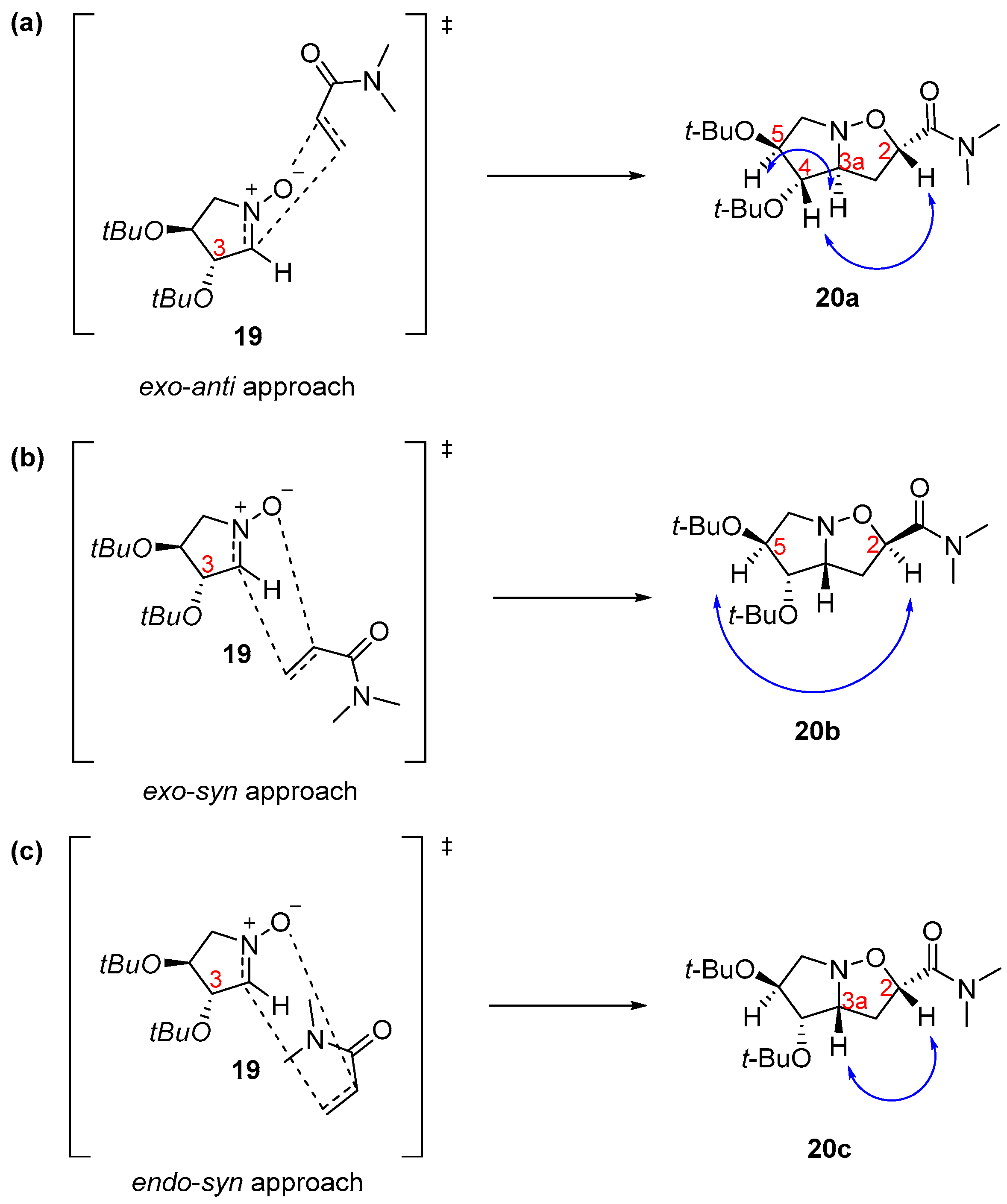
| Entry | Nitrones | Dipolarophile | Reaction Conditions | Heating | Temperature | Time | Product (Yield) |
|---|---|---|---|---|---|---|---|
| 1 a | 17 | N,N-dimethylacrylamide | CH2Cl2 | - | r.t. | 72 h | 18 (85%) |
| 2 | 17 | N,N-dimethylacrylamide | solvent-free | - | r.t. | 54 h | 18 (60%) |
| 3 | 17 | N,N-dimethylacrylamide | solvent-free | - (orbital shaker) | r.t. | 15 h | 18 (71%) |
| 4 | 17 | N,N-dimethylacrylamide | solvent-free | standard | 40 °C | 18 h | 18 (59%) |
| 5 | 17 | levoglucosenon | dry toluene | standard | 60 °C | 96 h | - |
| 6 | 19 | N,N-dimethylacrylamide | toluene | - | r. t. | 2.5 h | 20 (65%) |
| 7 | 19 | N,N-dimethylacrylamide | solvent-free | - (orbital shaker) | r.t. | 1 h | 20 (70%) |
| 8 b | 19 | levoglucosenone | dry toluene | - | r.t. | 2 h | 21 (88%) |
| 9 | 19 | levoglucosenone | solvent-free | standard | 80 °C | 2 h | 21 (80%) |
| 10 | 19 | levoglucosenone | solvent-free | microwaves (150 W) | 80 °C | 45 min | 21 (76%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratesi, D.; Morano, A.; Goti, A.; Cardona, F.; Matassini, C. Solvent-Free 1,3-Dipolar Cycloadditions of Nitrones for a More Sustainable Synthesis of Glycomimetics. Reactions 2025, 6, 36. https://doi.org/10.3390/reactions6020036
Pratesi D, Morano A, Goti A, Cardona F, Matassini C. Solvent-Free 1,3-Dipolar Cycloadditions of Nitrones for a More Sustainable Synthesis of Glycomimetics. Reactions. 2025; 6(2):36. https://doi.org/10.3390/reactions6020036
Chicago/Turabian StylePratesi, Debora, Alessio Morano, Andrea Goti, Francesca Cardona, and Camilla Matassini. 2025. "Solvent-Free 1,3-Dipolar Cycloadditions of Nitrones for a More Sustainable Synthesis of Glycomimetics" Reactions 6, no. 2: 36. https://doi.org/10.3390/reactions6020036
APA StylePratesi, D., Morano, A., Goti, A., Cardona, F., & Matassini, C. (2025). Solvent-Free 1,3-Dipolar Cycloadditions of Nitrones for a More Sustainable Synthesis of Glycomimetics. Reactions, 6(2), 36. https://doi.org/10.3390/reactions6020036