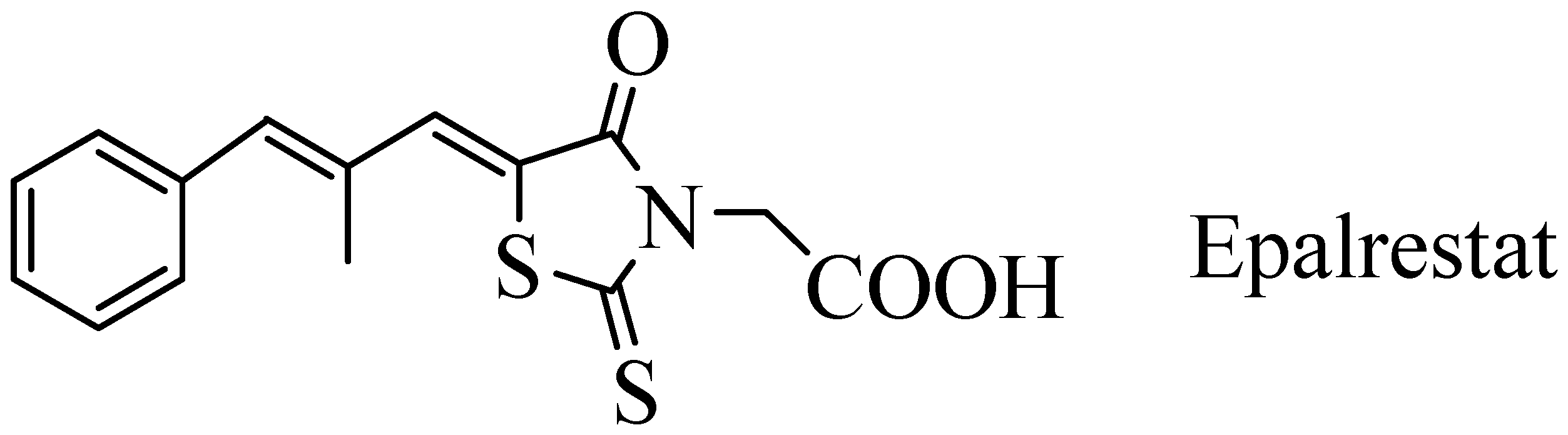
A New Method of Synthesis of Epalrestat


Abstract

1. Introduction
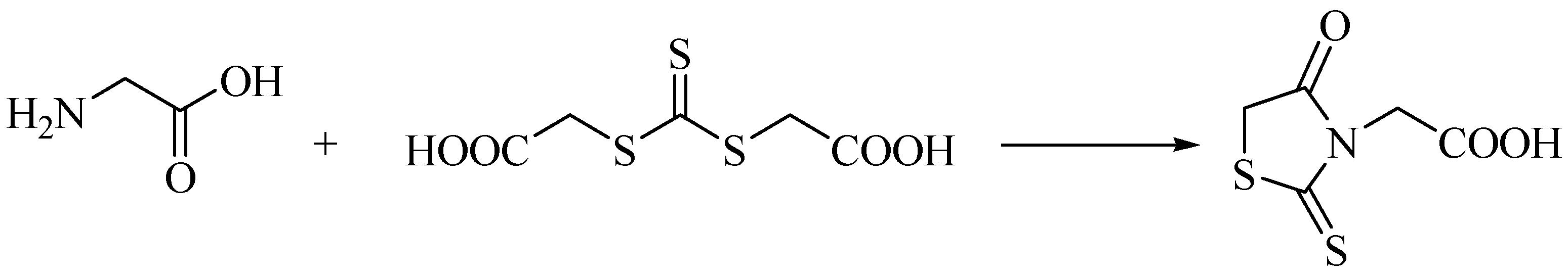
2. Methods and Discussion
3. Materials and Results
3.1. The Comprehensive Experimental Section
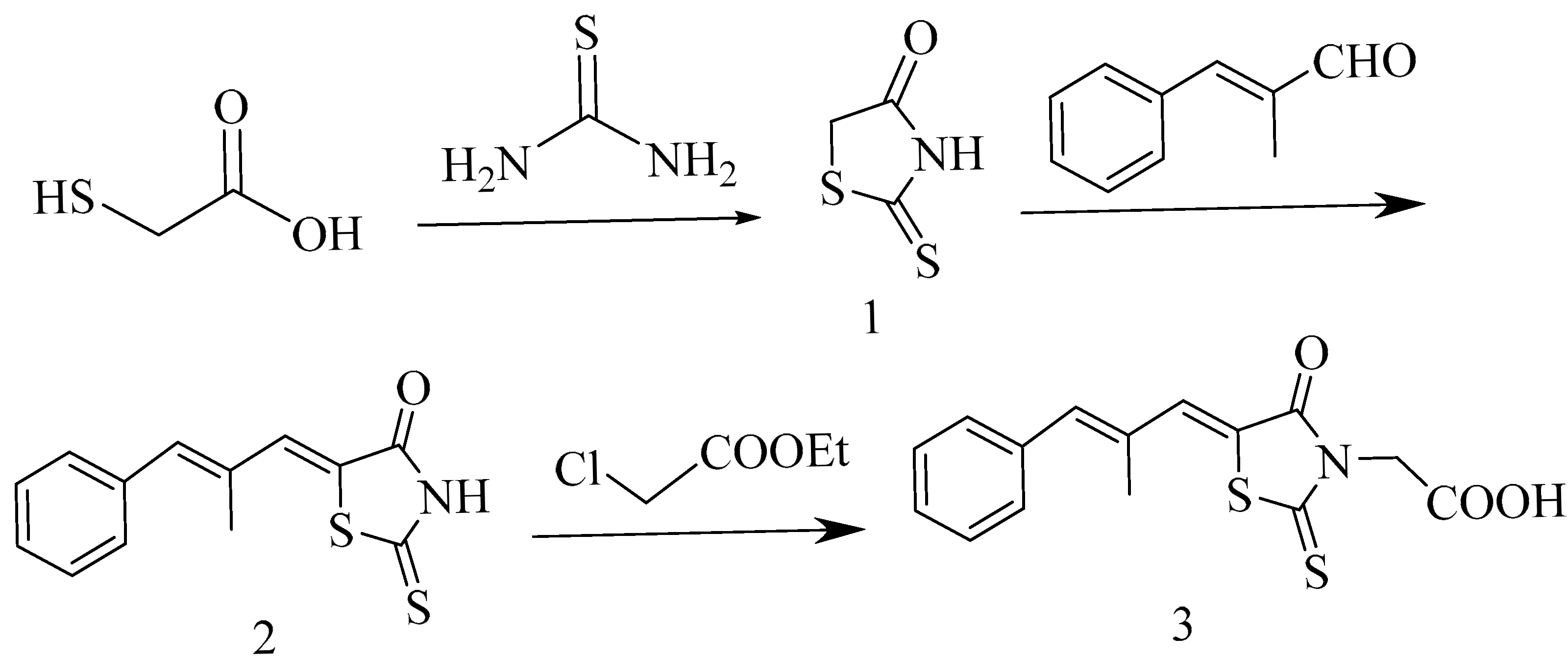
3.2. Rhodanine Was Prepared According to Ref. [22]
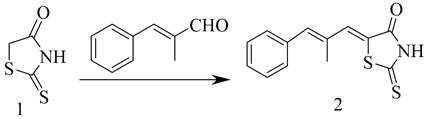
3.3. The Synthesis of 5-((1Z, 2E)-2-Methyl-3-phenylallylidene)-2-thioxothiazolidin-4-one (Compound 2)
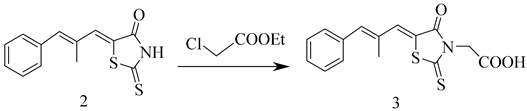
3.4. The Synthesis of Epalrestat
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kattaer, A.; Quelle-Regaldie, A.; Sánchez, L.; Concheiro, A.; Alvarez-Lorenzo, C. Formulation and characterization of epalrestat-loaded polysorbate 60 cationic niosomes for ocular delivery. Pharmaceutics 2023, 15, 1247. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, U.D.; Kamalkishore, M.K.; Vittalrao, A.M.; Eshwaraiah, P.K.S. Cognition enhancing abilities of vitamin D, epalrestat and their combination in diabetic rats with and without scopolamine induced amnesia. Cogn. Neurodyn. 2022, 16, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Yang, Y.; Zhou, X.; Chen, H.G. Advances in polysaccharides of natural source of anti-diabetes effect and mechanism. Mol. Biol. Rep. 2024, 51, 101. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, S.; Shivakumar, M.S.; Manivasagam, T.; Somasundaram, S.T.; Seedevi, P. Neuroprotective effect of epalrestat on hydrogen peroxide-induced neurodegeneration in SH-SY5Y cellular model. J. Microbiol. Biotechnol. 2021, 31, 867–874. [Google Scholar] [CrossRef]
- Alvi, Z.; Akhtar, M.; Rahman, N.U.; Hosny, K.M.; Sindi, A.M.; Khan, B.A.; Nazir, I.; Sadaquat, H. Utilization of gelling polymer to formulate nanoparticles loaded with epalrestat-cyclodextrin inclusion complex: Formulation, characterization, in-silico modelling and in-vivo toxicity evaluation. Polymers 2022, 13, 4350. [Google Scholar] [CrossRef]
- Zhang, T.S.; Wu, J.R.; Yao, X.M.; Zhang, Y.; Wang, Y.; Han, Y.; Wu, Y.; Xu, Z.Y.; Lan, J.; Han, S.Y.; et al. The aldose reductase inhibitor epalrestat maintains blood-brain barrier integrity by enhancing endothelial cell function during cerebral ischemia. Mol. Neurobiol. 2023, 60, 3741–3757. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Ruiz, F.; Darmanin, C.; Chung, R.P.T. Aldose reductase structures: Implications for mechanism and inhibition. Cell. Mol. Life Sci. 2004, 61, 750–762. [Google Scholar] [CrossRef]
- Tatsunami, R.; Sato, K.; Murao, Y.; Yama, K.; Yu, Y.; Ohno, S.; Tampo, Y. Epalrestat suppresses cadmium-induced cytotoxicity through Nrf2 in endothelial cells. Exp. Ther. Med. 2021, 21, 393. [Google Scholar] [CrossRef]
- Bailly, C. Moving toward a new horizon for the aldose reductase inhibitor epalrestat to treat drug-resistant cancer. Eur. J. Pharmacol. 2022, 931, 175191. [Google Scholar] [CrossRef]
- Choudhary, S.; Kumar, M.; Silakari, O. QM/MM analysis, synthesis and biological evaluation of epalrestat based mutual-prodrugs for diabetic neuropathy and nephropathy. Bioorg. Chem. 2021, 108, 104556. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tan, X.H. Protective effect of epalrestat on peripheral nerves in rats with diabetic peripheral neuropathy via NF-KB pathway. Trop. J. Pharm. Res. 2023, 22, 239–244. [Google Scholar] [CrossRef]
- Zheng, Q.R.; Su, Q. Pharmacologic actions and clinical utility of epalrestat. Chin. J. New Drugs Clin. Remedies 2006, 25, 876–884. [Google Scholar]
- Li, D.X.; Bo, G.M.; Bu, X.H. Synthesis of epalrestat as a new drug for diabetes. J. Shanxi Univ. 1995, 18, 413–416. [Google Scholar]
- Yu, S.H.; Yang, F.L.; Zhu, Q.; He, W. Synthesis of epalrestat. Chin. J. Pharm. 1996, 27, 5–6. [Google Scholar]
- Tu, H.P.; Wang, J.; Hua, Z.M.; Yuan, Z.D.; Yang, L.P. Synthesis of new drug epalrestat. J. East China Norm. Univ. 1999, 3, 104–106. [Google Scholar]
- Jiang, Y.; Zhang, R.J.; Ma, K.X.; Ren, Y.; Wang, D.C.; Liang, S.F. Synthesis of aldose reductase inhibitor epalrestat. Chin. J. Mod. Appl. Pharm. 1999, 16, 25–26. [Google Scholar]
- Li, Y.Z.; Lai, Y.S.; Liang, C.Y. Study on synthetic technology of epalrestat. Chin. J. Med. Chem. 2001, 11, 165–167. [Google Scholar]
- Sheng, R.; Liu, T.; Hu, Y.Z. Improved method for synthesis of epalrestat. J. Zhejiang Univ. 2003, 32, 356–358. [Google Scholar]
- Yan, S.Q.; Guo, W.; He, S.W.; Cao, H.Y.; Xie, C.W.; Wang, C.Y. Improved Synthesis of epalrestat. Food Drug 2022, 24, 215–217. [Google Scholar]
- Nitsche, C.; Klein, C.D. Aqueous microwave-assisted one-pot synthesis of N-substituted rhodanines. Tetra. Lett. 2012, 53, 5197–5201. [Google Scholar] [CrossRef]
- Pan, Z.L.; An, W.K.; Wu, L.L.; Fan, L.X.; Yang, G.Y.; Xu, C.L. A New Synthesis Strategy for Rhodanine and Its Derivatives. Synlett 2021, 32, 1131–1134. [Google Scholar] [CrossRef]
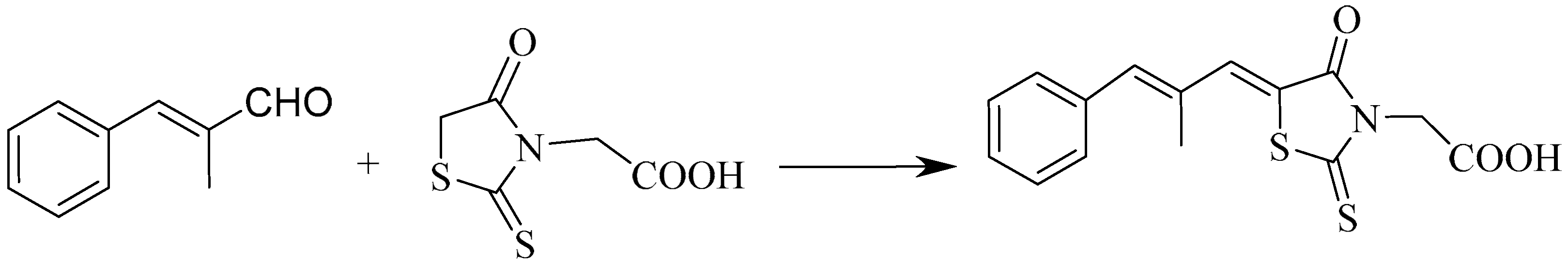


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| No. | Solvent | Temp. (°C) | Time (h) | Base | Yield (%) |
| 1 | CH2Cl2 | 50 | 1 | none | 13.6 |
| 2 | EtOAc | 50 | 1 | none | -- a |
| 3 | DMF | 50 | 1 | none | 12.8 |
| 4 | EtOH | 50 | 1 | none | 14.5 |
| 5 | EtOH | 50 | 5 | Na2CO3 (2.0 eq) | 35.2 |
| 6 | EtOH | 50 | 5 | K2CO3 (2.0 eq) | 45.2 |
| 7 | EtOH | 50 | 5 | SrCO3 (2.0 eq) | 15.1 |
| 8 | EtOH | 50 | 5 | K3PO4 (2.0 eq) | 56.3 |
| 9 | EtOH | 50 | 5 | 25%KOH (2.0 eq) | 77.7 |
| 10 | EtOH | 50 | 5 | 25%NaOH (2.0 eq) | 59.0 |
| 11 | EtOH | 50 | 5 | 25%NH4OH (2.0 eq) | 65.0 |
| 12 | EtOH | 50 | 5 | NaOAc (2.0 eq) | 42.4 |
| 13 | EtOH | 50 | 5 | Et3N (2.0 eq) | -- |
| 14 | EtOH | 50 | 5 | DBU (2.0 eq) | 60.0 |
| 15 | EtOH | 50 | 5 | DMAP (2.0 eq) | 63.2 |
| 16 | EtOH | 50 | 5 | 8%KOH (2.0 eq) | 56.3 |
| 17 | EtOH | 50 | 5 | 10%KOH (2.0 eq) | 59.4 |
| 18 | EtOH | 50 | 5 | 15%KOH (2.0 eq) | 64.2 |
| 19 | EtOH | 50 | 5 | 20%KOH (2.0 eq) | 71.3 |
| 20 | EtOH | 50 | 5 | 30%KOH (2.0 eq) | 77.9 |
| 21 | EtOH | 50 | 5 | 40%KOH (2.0 eq) | 77.9 |
| 22 | EtOH | 50 | 5 | 50%KOH (2.0 eq) | 78.0 |
| 23 | EtOH | 25 | 2 | 25%KOH (2.0 eq) | 56.9 |
| 24 | EtOH | 40 | 2 | 25%KOH (2.0 eq) | 77.6 |
| 25 | EtOH | 60 | 2 | 25%KOH (2.0 eq) | 77.9 |
| 26 | EtOH | 70 | 2 | 25%KOH (2.0 eq) | 77.9 |
| 27 | EtOH | 40 | 1 | 25%KOH (2.0 eq) | 77.3 |
| 28 | EtOH | 40 | 3 | 25%KOH (2.0 eq) | 77.7 |
| 29 | EtOH | 40 | 5 | 25%KOH (2.0 eq) | 77.9 |
| 30 | EtOH | 40 | 1 | 25%KOH (1.0 eq) | 46.7 |
| 31 | EtOH | 40 | 1 | 25%KOH (1.5 eq) | 59.8 |
| 32 | EtOH | 40 | 1 | 25%KOH (2.5 eq) | 77.4 |
| 33 | CH2Cl2 | 40 | 1 | 25%KOH (2.0 eq) | 51.3 |
| 34 | EtOAc | 40 | 1 | 25%KOH (2.0 eq) | 58.7 |
| 35 | DMF | 40 | 1 | 25%KOH (2.0 eq) | 56.3 |
 | |||||
|---|---|---|---|---|---|
| No. | Solvent | Temp. (°C) | Time (h) | Base | Yield (%) |
| 1 | CH2Cl2 | 50 | 1 | none | -- a |
| 2 | EtOAc | 50 | 1 | none | -- |
| 3 | DMF | 50 | 1 | none | 2.9 |
| 4 | EtOH | 50 | 1 | none | 7.5 |
| 5 | DMSO | 50 | 1 | none | 5.6 |
| 6 | EtOH | 50 | 5 | 50%KOH (2.0 eq) | 52.8 |
| 7 | EtOH | 50 | 5 | 50%NaOH (2.0 eq) | 39.5 |
| 8 | EtOH | 50 | 5 | SrCO3 (2.0 eq) | -- |
| 9 | EtOH | 50 | 5 | K2CO3 (2.0 eq) | -- |
| 10 | EtOH | 50 | 5 | Et3N (2.0 eq) | -- |
| 11 | EtOH | 50 | 5 | NaOAc (2.0 eq) | 26.3 |
| 12 | EtOH | 50 | 5 | DBU (2.0 eq) | 34.9 |
| 13 | EtOH | 50 | 5 | DMAP (2.0 eq) | 39.8 |
| 14 | EtOH | 50 | 5 | 10%KOH (2.0 eq) | 10.3 |
| 15 | EtOH | 50 | 5 | 20%KOH (2.0 eq) | 19.4 |
| 16 | EtOH | 50 | 5 | 30%KOH (2.0 eq) | 28.2 |
| 17 | EtOH | 50 | 5 | 40%KOH (2.0 eq) | 35.3 |
| 18 | EtOH | 50 | 5 | 60%KOH (2.0 eq) | 52.9 |
| 19 | EtOH | 25 | 2 | 50%KOH (2.0 eq) | 30.3 |
| 20 | EtOH | 40 | 2 | 50%KOH (2.0 eq) | 41.6 |
| 21 | EtOH | 60 | 2 | 50%KOH (2.0 eq) | 36.8 |
| 22 | EtOH | 70 | 2 | 50%KOH (2.0 eq) | 30.8 |
| 23 | EtOH | 40 | 3 | 50%KOH (2.0 eq) | 45.8 |
| 24 | EtOH | 40 | 5 | 50%KOH (2.0 eq) | 52.7 |
| 25 | EtOH | 40 | 7 | 50%KOH (2.0 eq) | 52.6 |
| 26 | EtOH | 40 | 5 | 50%KOH (1.0 eq) | 31.5 |
| 27 | EtOH | 40 | 5 | 50%KOH (1.5 eq) | 40.0 |
| 28 | EtOH | 40 | 5 | 50%KOH (2.5 eq) | 52.7 |
| 29 | CH2Cl2 | 40 | 5 | 50%KOH (2.0 eq) | 27.3 |
| 30 | EtOAc | 40 | 5 | 50%KOH (2.0 eq) | 32.7 |
| 31 | DMF | 40 | 5 | 50%KOH (2.0 eq) | 45.6 |
| 32 | DMSO | 40 | 5 | 50%KOH (2.0 eq) | 41.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Z.; Wu, L.; Fan, L.; An, W.; Yang, G.; Xu, C. A New Method of Synthesis of Epalrestat. Reactions 2025, 6, 37. https://doi.org/10.3390/reactions6020037
Pan Z, Wu L, Fan L, An W, Yang G, Xu C. A New Method of Synthesis of Epalrestat. Reactions. 2025; 6(2):37. https://doi.org/10.3390/reactions6020037
Chicago/Turabian StylePan, Zhenliang, Lulu Wu, Liangxin Fan, Wankai An, Guoyu Yang, and Cuilian Xu. 2025. "A New Method of Synthesis of Epalrestat" Reactions 6, no. 2: 37. https://doi.org/10.3390/reactions6020037
APA StylePan, Z., Wu, L., Fan, L., An, W., Yang, G., & Xu, C. (2025). A New Method of Synthesis of Epalrestat. Reactions, 6(2), 37. https://doi.org/10.3390/reactions6020037