
Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
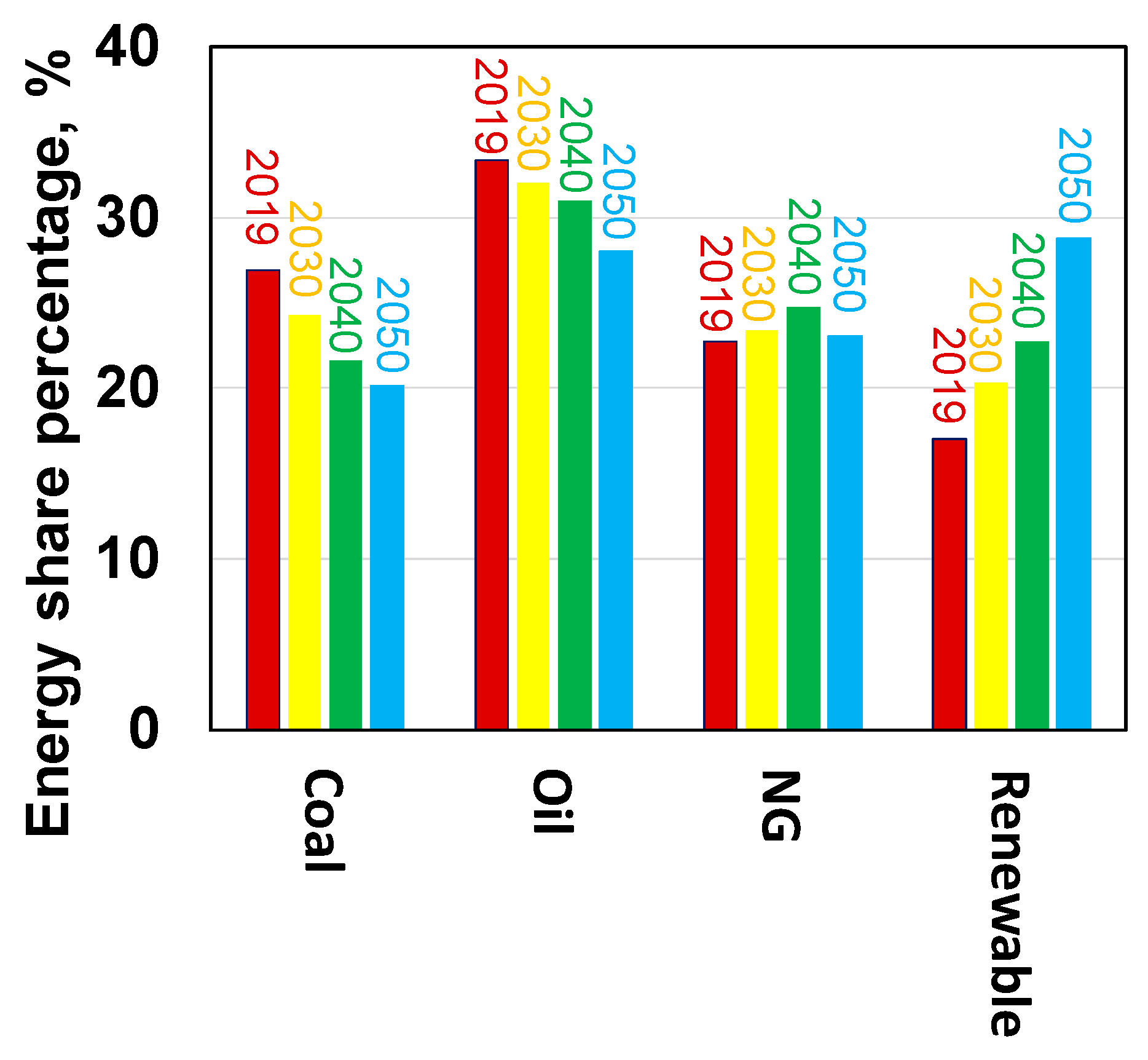
1. Introduction
2. Results and Discussion
2.1. Catalyst Crystal Structure and Catalytic Performance
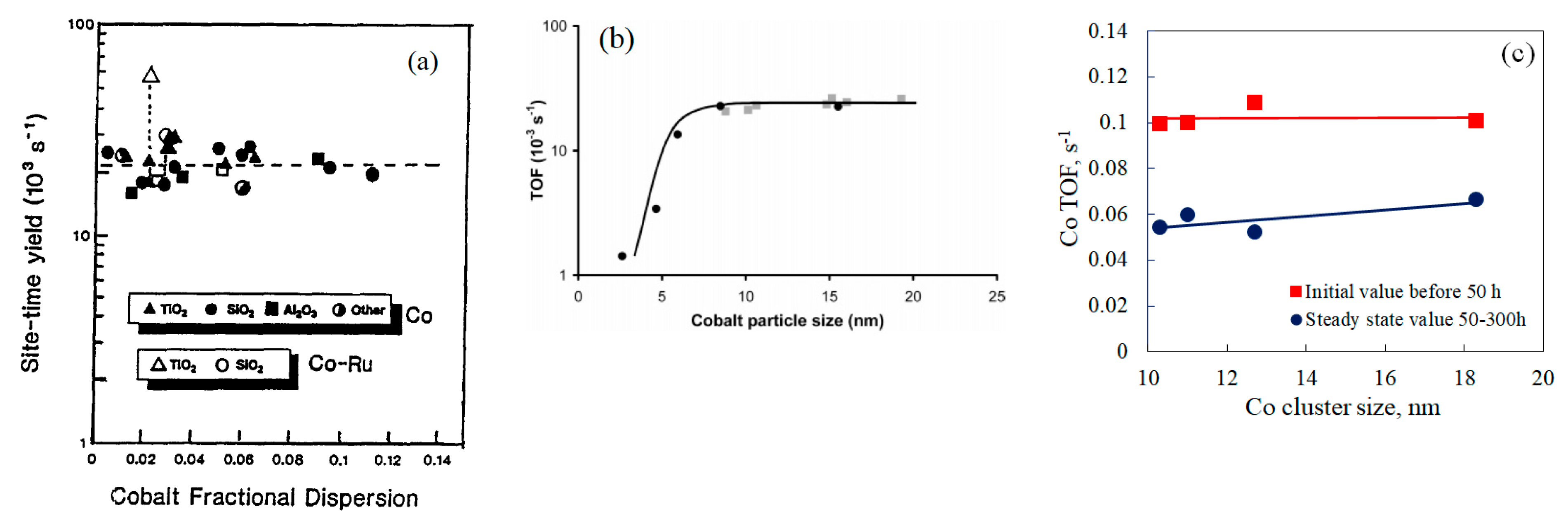
2.2. Metal Particle Size Effects on Catalyst Performance
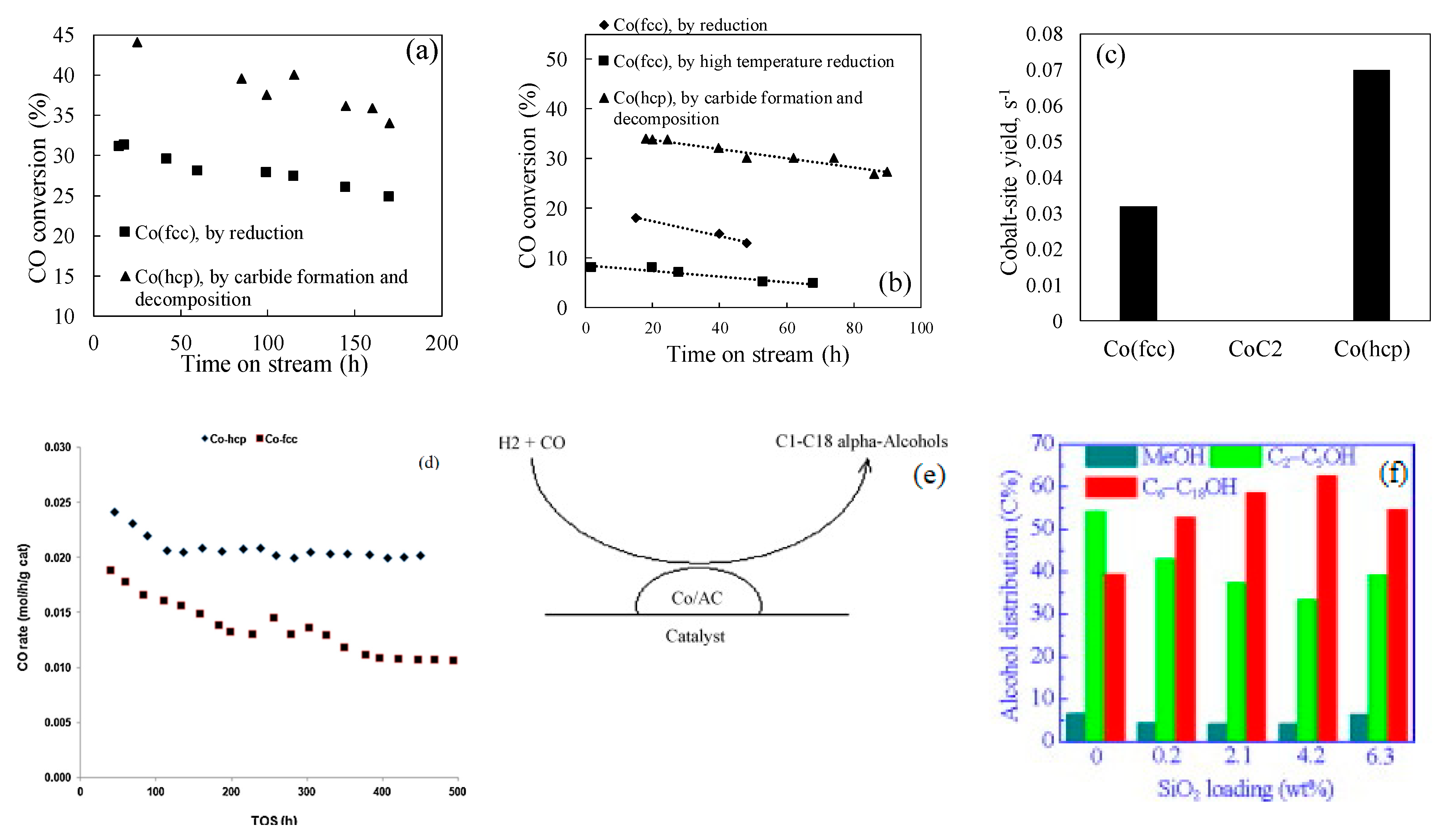
2.2.1. Examples of Cobalt Catalyst
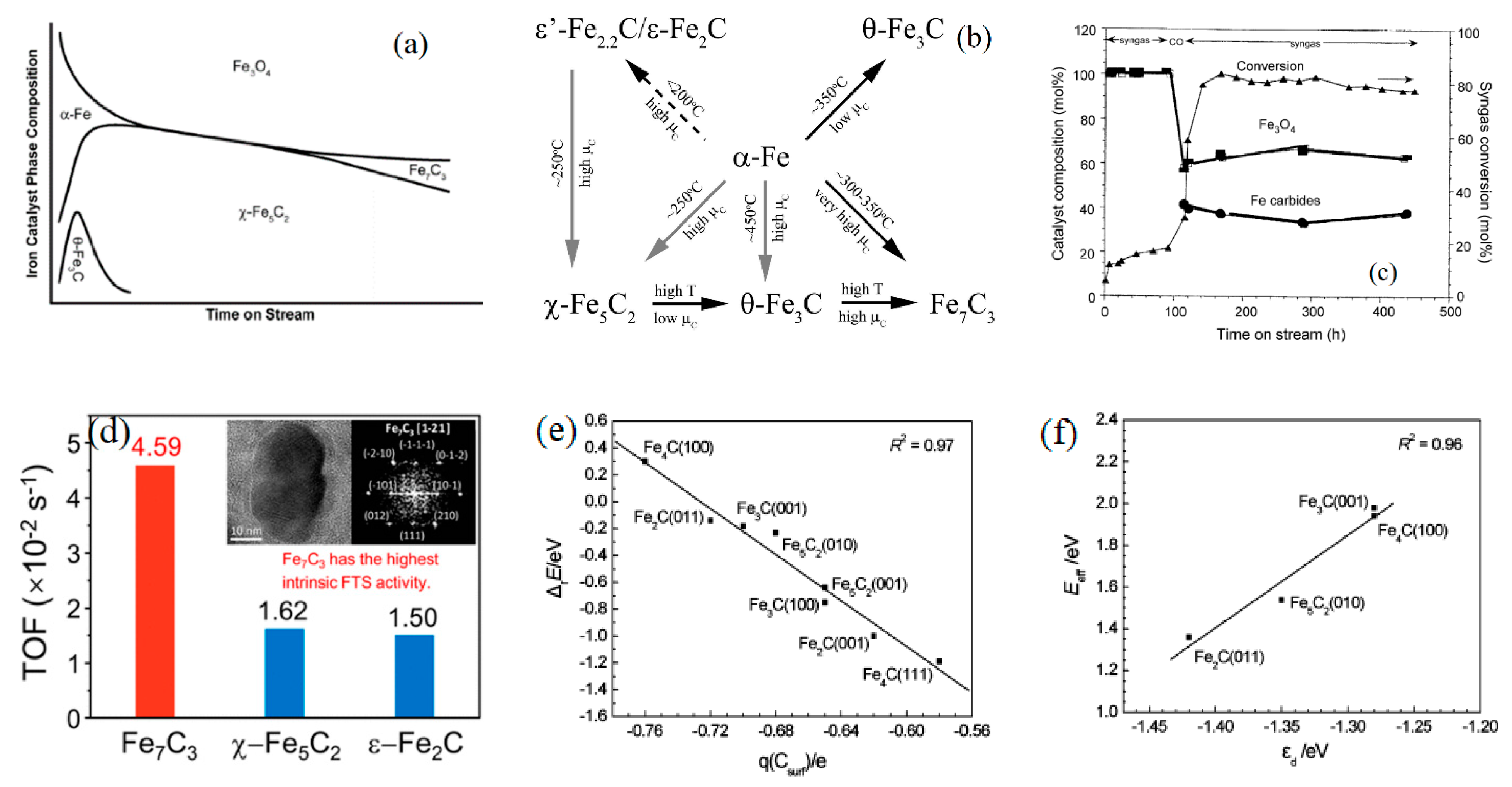
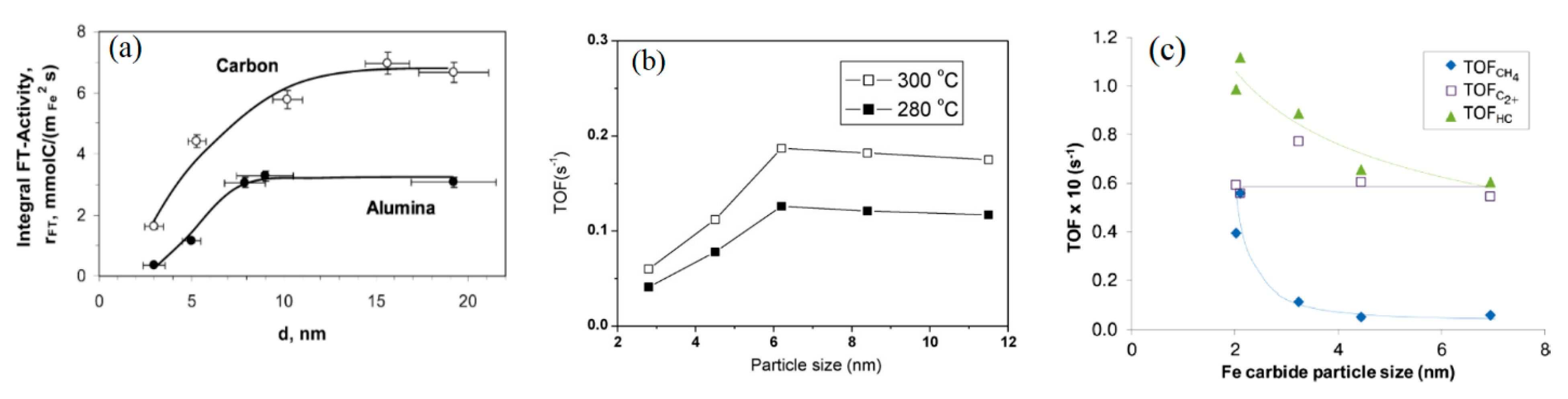
2.2.2. Examples of Iron Catalyst
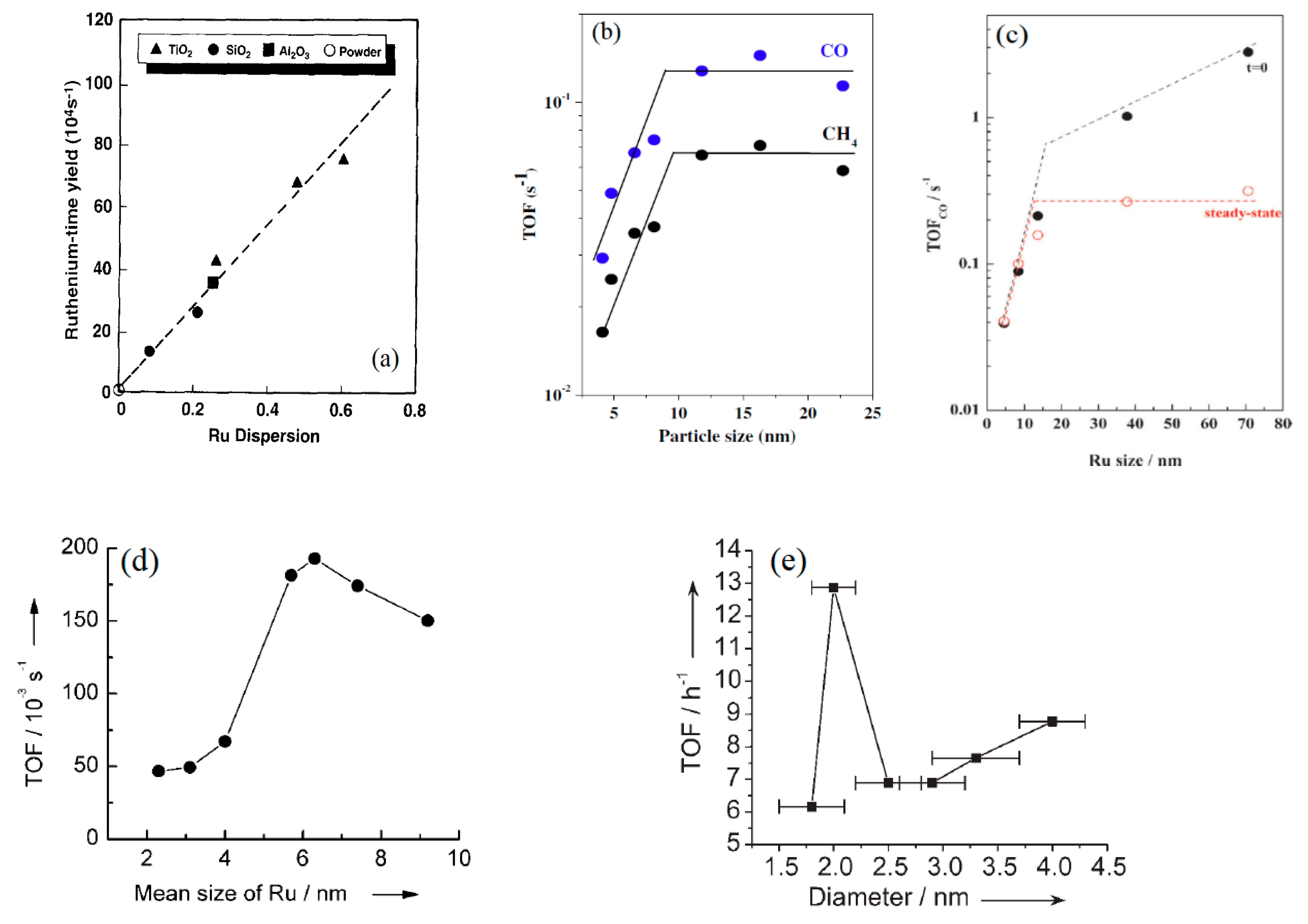
2.2.3. Examples of Ruthenium Catalyst
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Energy Information Administration’s International Energy Outlook 2020 (IEO2020). Available online: https://www.eia.gov/outlooks/ieo/pdf/ieo2020.pdf (accessed on 30 October 2020).
- Dry, M. The Fischer–Tropsch Process: 1950–2000; Elsevier: Amsterdam, The Netherlands, 2002; Volume 71, pp. 227–241. [Google Scholar] [CrossRef]
- Xiang, H.W.; Yang, Y.; Li, Y.W. Indirect coal-to-liquids technology from fundamental research to commercialization. Sci. Sin. Chim. 2014, 14, 1876–1892. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer-Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Jacobs, G.; Spark, D.E.; Todic, B.; Bukur, D.B. Quantitative comparison of iron and cobalt based catalysts for the Fischer-Tropsch synthesis under clean and poisoning conditions. Catal. Today 2020, 343, 125–136. [Google Scholar] [CrossRef]
- Carballo, J.M.G.; Finocchio, E.; García, S.; Rojas, S.; Ojeda, M.; Busca, G.; Fierro, J.L.G. Support effects on the structure and performance of ruthenium catalysts for the Fischer–Tropsch synthesis. Catal. Sci. Technol. 2011, 1, 1013–1023. [Google Scholar] [CrossRef]
- Carballo, J.M.G.; Yang, J.; Holmen, A.; Garcia-Rodriguez, S.; Rojas, S.; Ojeda, M.; Fierro, J.L.G. Catalytic effects of ruthenium particle size on the Fischer–Tropsch Synthesis. J. Catal. 2011, 284, 102–108. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, R.S.L.; Fiato, A. Fischer-Tropsch synthesis on cobalt and ruthenium. Metal dispersion and support effects on reaction rate and selectivity. J. Catal. 1992, 137, 212–224. [Google Scholar] [CrossRef]
- Yang, J.; Shafer, W.D.; Pendyala, V.R.R.; Jacobs, G.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch Synthesis: Deuterium Kinetic Isotopic Effect for a 2.5 % Ru/NaY Catalyst. Top. Catal. 2014, 57, 508–517. [Google Scholar] [CrossRef]
- Ma, W.P.; Shafer, W.D.; Martinelli, M.; Sparks, D.E.; Davis, B.H. Fischer-Tropsch Synthesis: Using deuterium tracer coupled with kinetic approach to study the kinetic isotopic effects of iron, cobalt and ruthenium catalysts. Catal. Today 2020, 343, 137–145. [Google Scholar] [CrossRef]
- Dry, M.E. Fischer-Tropsch Synthesis. In Catalysis—Science and Technology; Anderson, J.R., Boudart, M., Eds.; Springer: New York, NY, USA, 1981; Chapter 4; Volume 1, p. 196. [Google Scholar]
- De Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and Reactivity of ϵ−χ−θ Iron Carbide Catalyst Phases in Fischer−Tropsch Synthesis: Controlling μC. J. Am. Chem. Soc. 2010, 132, 14928–14941. [Google Scholar] [CrossRef]
- Davis, B.H. Fischer–Tropsch Synthesis: Reaction mechanisms for iron catalysts. Catal. Today 2009, 141, 25–33. [Google Scholar] [CrossRef]
- Chang, Q.; Zhang, C.; Liu, C.; Wei, Y.; Cheruvathur, A.V.; Dugulan, A.I.; Niemantsverdriet, J.W.; Liu, X.; He, Y.; Qing, M.; et al. Relationship between Iron Carbide Phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and Catalytic Performances of Fe/SiO2 Fischer–Tropsch Catalysts. ACS Catal. 2018, 8, 3304–3316. [Google Scholar] [CrossRef]
- Lu, F.X.; Chen, X.; Lei, Z.G.; Wen, L.X.; Zhang, Y. Revealing the activity of different iron carbides for Fischer-Tropsch synthesis. Appl. Catal. B Environ. 2021, 281, 119521. [Google Scholar] [CrossRef]
- Huo, C.F.; Li, Y.W.; Wang, J.; Jiao, H. Insight into CH4 Formation in Iron-Catalyzed Fischer−Tropsch Synthesis. J. Am. Chem. Soc. 2009, 131, 14713–14721. [Google Scholar] [CrossRef]
- Van Santen, R.A.; Ghouri, M.M.; Shetty, S.; Hensen, E.M.H. Structure sensitivity of the Fischer–Tropsch reaction: Molecular kinetics simulations. Catal. Sci. Technol. 2011, 1, 891–911. [Google Scholar] [CrossRef]
- Ralston, W.T.; Melaet, G.; Saephan, T.; Somorjai, G.A. Evidence of Structure Sensitivity in the Fischer–Tropsch Reaction on Model Cobalt Nanoparticles by Time-Resolved Chemical Transient. Angew. Chem. Int. Ed. 2017, 56, 7415–7419. [Google Scholar] [CrossRef]
- Ducreux, O.; Rebours, B.; Lynch, J.; Roy-Auberger, M.; Bazin, D. Microstructure of Supported cobalt Fischer-Tropsch catalysts. Oil Gas Sci. Tech. 2009, 64, 49–62. [Google Scholar] [CrossRef]
- Khodakov, A.Y. Fischer-Tropsch synthesis: Relations between structure of cobalt catalysts and their catalytic performance. Catal. Today 2009, 144, 251–257. [Google Scholar] [CrossRef]
- Sadeqzadeh, M.; Karaca, H.; Safonova, O.V.; Fongarland, P.; Chambrey, S.; Roussel, P.; Griboval-Constant, A.; Lacroix, M.; Curulla-Ferre, D.; Luck, F.; et al. Identification of the active species in the working alumina-supported cobalt catalyst under various conditions of Fischer–Tropsch synthesis. Catal. Today 2011, 164, 62–67. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Fischer–Tropsch synthesis: Activity of metallic phases of cobalt supported on silica. Catal. Today 2013, 215, 13–17. [Google Scholar] [CrossRef]
- Dinega, D.P.; Bawendi, M.G. A Solution-Phase Chemical Approach to a New Crystal Structure of Cobalt. Angew. Chem. Int. Ed. 1999, 38, 1788–1791. [Google Scholar] [CrossRef]
- Enache, D.I.; Rebours, B.; Roy-Auberger, M.; Revel, R. In Situ XRD Study of the Influence of Thermal Treatment on the Characteristics and the Catalytic Properties of Cobalt-Based Fischer–Tropsch Catalysts. J. Catal. 2002, 205, 346–353. [Google Scholar] [CrossRef]
- Liu, J.; Su, H.; Sun, D.; Zhang, B.; Li, W. Crystallographic dependence of CO activation on cobalt catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef]
- Mohandas, J.C.; Gnanamani, M.K.; Jacobs, G.; Ma, W.P.; Ji, Y.; Khalid, S.; Davis, B.H. Fischer Tropsch Synthesis: Characterization and Reaction Testing of Cobalt Carbide. ACS Catal. 2011, 1, 1581–1588. [Google Scholar] [CrossRef]
- Jalama, K.; Ma, W.P.; Jacob, G.; Sparks, D.; Qian, D.L.; Davis, B.H. Fischer-Tropsch synthesis over Pt/Co/Al2O3 catalyst: Improvement in catalyst stability by activation with diluted CO. Appl. Catal. 2020, 602, 117645. [Google Scholar] [CrossRef]
- Jiao, G.P.; Ding, Y.J.; Zhu, H.J.; Li, X.; Li, J.; Lin, R.; Dong, W.; Gong, L.; Pei, Y.; Lu, Y. Effect of La2O3 doping on syntheses of C1–C18 mixed linear α-alcohols from syngas over the Co/AC catalysts. Appl. Catal. 2009, 364, 137–142. [Google Scholar] [CrossRef]
- Pei, Y.P.; Ding, Y.J.; Zhu, H.J.; Du, H. One-step production of C1–C18 alcohols via Fischer-Tropsch reaction over activated carbon-supported cobalt catalysts: Promotional effect of modification by SiO2. Chin. J. Catal. 2015, 36, 355–361. [Google Scholar] [CrossRef]
- Li, W.Z.; Liu, J.X.; Gu, J.; Zhou, W.; Yao, S.Y.; Si, R.; Guo, Y.; Su, H.Y.; Yan, C.H.; Li, W.X.; et al. Chemical Insights into the Design and Development of Face-Centered Cubic Ruthenium Catalysts for Fischer–Tropsch Synthesis. J. Am. Chem. Soc. 2017, 139, 2267–2276. [Google Scholar] [CrossRef]
- Yang, Y.; Xiang, H.W.; Xu, Y.Y.; Bai, L.; Li, Y.W. Effect of potassium promoter on precipitated iron-manganese catalyst for Fischer-Tropsch synthesis. Appl. Catal. 2004, 266, 181–194. [Google Scholar] [CrossRef]
- Tian, Z.P.; Wang, C.H.; Yue, J.; Zhang, X.H.; Ma, L.L. Effect of a potassium promoter on the Fischer–Tropsch synthesis of light olefins over iron carbide catalysts encapsulated in graphene-like carbon. Catal. Sci. Technol. 2019, 9, 2728–2741. [Google Scholar] [CrossRef]
- Tao, Z.; Yang, Y.; Wan, H.; Li, Y.W. Effect of manganese on a potassium-promoted iron-based Fischer-Tropsch synthesis catalyst. Catal. Lett. 2007, 114, 161–168. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Jacobs, G.; Pendyala, R.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer−Tropsch Synthesis: Influence of Mn on the Carburization Rates and Activities of Fe-Based Catalysts by TPR-EXAFS/XANES and Catalyst Testing. J. Phys. Chem. C 2011, 115, 4783–4792. [Google Scholar] [CrossRef]
- Yang, Y.; Xiang, H.W.; Tian, L.; Wang, H.; Zhang, C.H.; Tao, Z.C.; Xu, Y.Y.; Zhong, B.; Li, Y.W. Structure and Fischer–Tropsch performance of iron–manganese catalyst incorporated with SiO2. Appl. Catal. A Gen. 2005, 284, 105–122. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I. Performance and characterization of Ru/Al2O3 and Ru/SiO2 catalysts modified with Mn for Fischer-Tropsch synthesis. Appl. Catal. 2008, 340, 203–211. [Google Scholar] [CrossRef]
- Chen, Y.W.; Wang, H.T.; Goodwin, J.G.; Shiflett, W.K., Jr. Fischer-tropsch synthesis over zeolite-supported ruthenium catalysts derived from Ru3(CO)12. Appl. Catal. 1983, 8, 303–314. [Google Scholar] [CrossRef]
- Boudart, M. Advances in Catalysis and Related Subjects; Eley, D.D., Ed.; Academic Press: New York, NY, USA, 1969; Volume 20, p. 85. [Google Scholar]
- Li, J.L.; Coville, N.J. The effect of boron on the catalyst reducibility and activity of Co/TiO2 Fischer–Tropsch catalysts. Appl. Catal. 1999, 181, 201–208. [Google Scholar] [CrossRef]
- Iglesia, E.; Reyes, S.C.; Madon, R.J.; Soled, S.L. Selectivity Control and Catalyst Design in the Fischer-Tropsch Synthesis: Sites, Pellets, and Reactors. Adv. Catal. 1993, 39, 221–302. [Google Scholar] [CrossRef]
- Iglesia, E. Design, synthesis, and use of cobalt-based Fischer-Tropsch synthesis catalysts. Appl. Catal. A Gen. 1997, 161, 59–78. [Google Scholar] [CrossRef]
- Vannice, M.A. CO hydrogenation over well-dispersed carbon-supported iron catalysts. J. Catal. 1983, 75, 416–422. [Google Scholar]
- Jones, V.K.; Neubauer, L.R.; Bartholomew, C.H. Effects of crystallite size and support on the CO hydrogenation activity/selectivlty properties of Fe/Carbon. J. Phys. Chem. 1986, 90, 4832–4839. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Den Breejen, J.P.; Radstake, P.B.; Bezemer, G.L.; Bitter, J.H.; Frøseth, V.; Holmen, A.; de Jong, K.P. On the origin of the cobalt particle size effects in Fischer-Tropsch catalysis. J. Am. Chem. Soc. 2009, 131, 7197–7203. [Google Scholar] [CrossRef]
- Borg, Ø.; Dietzel, P.D.; Spjelkavik, A.I.; Tveten, E.Z.; Walmsley, J.C.; Diplas, S.; Eri, S.; Holmen, A.; Rytter, E. Fischer-Tropsch synthesis: Cobalt particle size and support effects on intrinsic activity and product distribution. J. Catal. 2008, 259, 161–164. [Google Scholar] [CrossRef]
- Prieto, G.; Martínez, A.; Concepción, P.; Moreno-Tost, R. Cobalt particle size effects in Fischer–Tropsch synthesis: Structural and in situ spectroscopic characterisation on reverse micelle-synthesised Co/ITQ-2 model catalysts. J. Catal. 2009, 266, 129–144. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Keogh, R.A.; Bukur, D.B.; Davis, B.H. Fischer-Tropsch synthesis: Effect of Pd, Pt, Re, and Ru noble metal promoters on the activity and selectivity of a 25%Co/Al2O3 catalyst. Appl. Catal. 2012, 437, 1–9. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Jacobs, G.; Ma, W.; Klettlinger, J.L.S.; Yen, C.H.; Davis, B.H. Fischer-Tropsch synthesis: Effect of catalyst particle (sieve) size range on activity, selectivity, and aging of a Pt promoted Co/Al2O3 catalyst. Chem. Eng. J. 2014, 249, 279–284. [Google Scholar] [CrossRef]
- Saib, A.M.; Borgna, A.; de Loosdrecht, J.V.; van Berge, P.J.; Niemantsverdriet, J.W. XANES study of the susceptibility of nano-sized cobalt crystallites to oxidation during realistic Fischer–Tropsch synthesis. Appl. Catal. A 2006, 312, 12–19. [Google Scholar] [CrossRef]
- Barkhuizen, D.; Mabaso, E.I.; Viljoen, E.; Welker, C.; Claeys, M.; van Steen, E.; Fletcher, J.C.Q. Experimental approaches to the preparation of supported metal nanoparticles. Pure Appl. Chem. 2006, 78, 1759–1769. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, Y.J.; Khanna, P.K.; Jun, K.W.; Bae, J.W.; Kim, Y.H. Alumina-supported iron oxide nanoparticles as Fischer–Tropsch catalysts:Effect of particle size of iron oxide. J. Mol. Catal. A Chem. 2010, 323, 84. [Google Scholar] [CrossRef]
- Torres, G.H.M.; Bitter, J.H.; Davidian, T.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Iron particle size effects for direct production of lower olefins from synthesis gas. J. Am. Chem. Soc. 2012, 134, 16207–16215. [Google Scholar] [CrossRef] [PubMed]
- Keyvanloo, K.; Mardkhe, M.K.; Alam, T.M.; Bartholomew, C.H.; Woodfield, B.F.; Hecker, W.C. Supported Iron Fischer–Tropsch Catalyst: Superior Activity and Stability Using a Thermally Stable Silica-Doped Alumina Support. ACS Catal. 2014, 4, 1071–1077. [Google Scholar] [CrossRef]
- Kellner, C.S.; Bell, A.T. Effects of dispersion on the activity and selectivity of alumina-supported ruthenium catalysts for carbon monoxide hydrogenation. J. Catal. 1982, 75, 251–261. [Google Scholar] [CrossRef]
- Eslava, J.L.; Sun, X.H.; Gascon, J.; Kapteijnb, F.; Ramos, I. Ruthenium particle size and cesium promotion effects in Fischer–Tropsch synthesis over high surface-area graphite supported catalysts. Catal. Sci. Technol. 2017, 7, 1235–1244. [Google Scholar] [CrossRef]
- Carballo, J.M.G.; Alonso, F.J.P.; Ojeda, M.; Garcia-Garcia, F.J.; Fierro, J.L.G.; Rojas, S. Evidences of Two-Regimes in the Measurement of Ru Particle Size Effect for CO Dissociation during Fischer–Tropsch Synthesis. ChemCatChem 2014, 6, 2084–2094. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, S.; Zhang, Q.; Wang, Y. Ruthenium nanoparticles supported on carbon nanotubes as efficient catalysts for selective conversion of synthesis gas to diesel fuel. Angew. Chem. Int. Ed. 2009, 48, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.X.; Cai, Z.P.; Wang, T.; Kou, Y.; Yan, N. Aqueous-Phase Fischer–Tropsch Synthesis with a Ruthenium Nanocluster Catalyst. Angew. Chem. 2008, 120, 758–761. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Yang, X.; Duan, H.; Qi, H.; Su, Y.; Liang, B.; Tao, H.; Liu, B.; Chen, D.; et al. Tuning reactivity of Fischer–Tropsch synthesis by regulating TiOx overlayer over Ru/TiO2 nanocatalysts. Nat. Commun. 2020, 11, 3185. [Google Scholar] [CrossRef]
- Fu, T.J.; Li, Z.H. Review of recent development in Co-based catalysts supported on carbon materials for Fischer–Tropsch synthesis. Chem. Eng. Sci. 2015, 135, 3–20. [Google Scholar] [CrossRef]
- Gavrilović, L.; Save, J.; Blekkan, E.A. The effect of potassium on cobalt-based Fischer–Tropsch catalysts with different cobalt particle sizes. Catalysts 2019, 9, 351. [Google Scholar] [CrossRef]
- Ghogia, A.C.; Nzihou, A.; Serp, P.; Soulantica, K.; Pham Minh, D. Cobalt catalysts on carbon-based materials for Fischer-Tropsch synthesis: A review. Appl. Catal. 2021, 609, 117906. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, Y.J.; Karandikar, P.R.; Jun, K.W.; Ha, K.S.; Park, H.G. Fischer–Tropsch catalysts deposited with size-controlled Co3O4 nanocrystals: Effect of Co particle size on catalytic activity and stability. Appl. Catal. 2012, 411, 15–23. [Google Scholar] [CrossRef]
- Qi, Z.; Chen, L.; Zhang, S.; Su, J.; Somorjai, G.A. A mini review of cobalt-based nanocatalyst in Fischer-Tropsch synthesis. Appl. Catal. 2020, 602, 17701. [Google Scholar] [CrossRef]
- Yang, J.; Frøseth, V.; Chen, D.; Holmen, A. Particle size effect for cobalt Fischer–Tropsch catalysts based on in situ CO chemisorption. Surf. Sci. 2016, 648, 67–73. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, W.P.; Wang, Y. Recent advances in understanding the key catalyst factors for Fischer-Tropsch synthesis. J. Energy Chem. 2013, 22, 27–38. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.P.; Gao, P.; Todic, B.; Bhatelia, T.; Bukur, D.B.; Khalid, S.; Davis, B.H. Fischer-Tropsch Synthesis: Differences Observed in Local Atomic Structure and Selectivity with Pd Compared to Typical Promoters (Pt, Re, Ru) of Co/Al2O3 Catalysts. Top. Catal. 2012, 55, 811–817. [Google Scholar] [CrossRef]
- Ghasvareh, P.; Smith, K.J. Effects of Co particle size on the stability of Co/Al2O3 and Re-Co/Al2O3 catalysts in a slurry-phase Fischer-Tropsch reactor. Energy Fuels 2016, 30, 9721–9729. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.P.; Davis, B.H. Influence of Reduction Promoters on Stability of Cobalt/γ-Alumina Fischer-Tropsch Synthesis Catalysts. Catalysts 2014, 4, 49–76. [Google Scholar] [CrossRef]
- Liu, J.X.; Wang, P.; Xu, W.; Hensen, E.J. Particle size and crystal phase effects in Fischer-Tropsch catalysts. Engineering 2017, 3, 467–476. [Google Scholar] [CrossRef]
- Gu, B.; Zhou, C.; He, S.; Moldovan, S.; Chernavskii, P.A.; Ordomsky, V.V.; Khodakov, A.Y. Size and promoter effects on iron nanoparticles confined in carbon nanotubes and their catalytic performance in light olefin synthesis from syngas. Catal. Today 2020, 357, 203–213. [Google Scholar] [CrossRef]
- Xie, J.; Torres Galvis, H.M.; Koeken, A.C.J.; Kirilin, A.; Dugulan, A.I.; Ruitenbeek, M.; de Jong, K.P. Size and Promoter Effects on Stability of Carbon-Nanofiber-Supported Iron-Based Fischer–Tropsch Catalysts. ACS Catal. 2016, 6, 4017–4024. [Google Scholar] [CrossRef]
- Wang, H.W.; Lu, J.L. A review on particle size effect in metal-catalyzed heterogeneous reactions. Chin. Chem. 2020, 38, 1422–1444. [Google Scholar] [CrossRef]
- Kang, J.; Deng, W.P.; Zhang, Q.H.; Wang, Y. Ru particle size effect in Ru/CNT-catalyzed Fischer-Tropsch synthesis. J. Energy Chem. 2013, 22, 321–328. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Dalai, A.K. Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis. Reactions 2021, 2, 62-77. https://doi.org/10.3390/reactions2010006
Ma W, Dalai AK. Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis. Reactions. 2021; 2(1):62-77. https://doi.org/10.3390/reactions2010006
Chicago/Turabian StyleMa, Wenping, and Ajay K. Dalai. 2021. "Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis" Reactions 2, no. 1: 62-77. https://doi.org/10.3390/reactions2010006
APA StyleMa, W., & Dalai, A. K. (2021). Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis. Reactions, 2(1), 62-77. https://doi.org/10.3390/reactions2010006